
Key Points
-
Glioblastoma is the most frequent and most aggressive malignant primary brain tumour and remains almost universally incurable in both children and adults.
-
Comprehensive molecular profiling studies have greatly broadened our knowledge of the underlying genomic and epigenomic aberrations that are associated with glioblastoma initiation and progression.
-
Genetic lesions result in disrupted epigenetic control mechanisms by altering histone modifications, DNA methylation and gene expression patterns in a large proportion of glioblastomas.
-
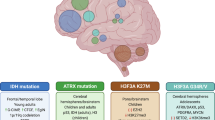
Based on recurrent combinations of genomic and/or epigenomic features with distinct patient characteristics, glioblastomas across all ages are being divided into meaningful biological subgroups, which are likely to guide the design of future clinical trials.
-
The complex interplay between the glioblastoma genome and epigenome opens the way for the development of novel innovative therapeutic strategies that are urgently needed to tackle this deadly brain tumour.
Abstract
We have extended our understanding of the molecular biology that underlies adult glioblastoma over many years. By contrast, high-grade gliomas in children and adolescents have remained a relatively under-investigated disease. The latest large-scale genomic and epigenomic profiling studies have yielded an unprecedented abundance of novel data and provided deeper insights into gliomagenesis across all age groups, which has highlighted key distinctions but also some commonalities. As we are on the verge of dissecting glioblastomas into meaningful biological subgroups, this Review summarizes the hallmark genetic alterations that are associated with distinct epigenetic features and patient characteristics in both paediatric and adult disease, and examines the complex interplay between the glioblastoma genome and epigenome.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

References
Louis, D. N., Ohgaki, H., Wiestler, O. D. & Cavenee, W. K. WHO Classification of Tumors of the Central Nervous System (International Agency for Research on Cancer, 2007).
Dolecek, T., Propp, J., Stroup, N. & Kruchko, C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol. 14 (Suppl. 5), 49 (2012).
Ohgaki, H. & Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 170, 1445–1453 (2007).
Puget, S. et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS ONE 7, e30313 (2012).
Khuong-Quang, D. A. et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 124, 439–447 (2012).
Warren, K. Diffuse intrinsic pontine glioma: poised for progress. Front. Oncol. 2, 205 (2012).
Cage, T. et al. Feasibility, safety, and indications for surgical biopsy of intrinsic brainstem tumors in children. Child. Nerv. Syst. 29, 1313–1319 (2013).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). This was the first study to show that the addition of temozolomide to radiotherapy results in a statistically significant survival benefit for patients with newly diagnosed GBM.
Stupp, R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009).
Cohen, K. J. et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol. 13, 317–323 (2011).
Hargrave, D., Bartels, U. & Bouffet, E. Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol. 7, 241–248 (2006).
Janssens, G. et al. Hypofractionation versus conventional radiation therapy for newly diagnosed diffuse intrinsic pontine glioma: a matched-cohort analysis. Int. J. Radiat. Oncol. 85, 315–320 (2013).
Jansen, M. H., van Vuurden, D. G., Vandertop, W. P. & Kaspers, G. J. Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer Treat. Rev. 38, 27–35 (2012).
Godard, S. et al. Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res. 63, 6613–6625 (2003).
Shai, R. et al. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene 22, 4918–4923 (2003).
Maher, E. et al. Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res. 66, 11502–11513 (2006).
Tso, C.-L. et al. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res. 66, 159–167 (2006).
Faury, D. et al. Molecular profiling identifies prognostic subgroups of pediatric glioblastoma and shows increased YB-1 expression in tumors. J. Clin. Oncol. 25, 1196–1208 (2007).
Nutt, C. et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 63, 1602–1607 (2003).
Freije, W. et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 64, 6503–6510 (2004).
Shirahata, M. et al. Gene expression-based molecular diagnostic system for malignant gliomas is superior to histological diagnosis. Clin. Cancer Res. 13, 7341–7356 (2007).
Liang, Y. et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc. Natl Acad. Sci. USA 102, 5814–5819 (2005).
Phillips, H. et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 (2006). This was one of the first studies to classify GBM into three distinct molecular subclasses on the basis of gene expression profiling signatures that were associated with differences in patient survival.
Gravendeel, L. et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 69, 9065–9072 (2009).
Verhaak, R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). This TCGA study discovered four distinct subtypes of GBM that were distinguished by gene expression patterns associated with distinct genetic aberrations and clinical characteristics.
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). This most recent TCGA study describes the landscape of somatic genomic alterations on the basis of multi-dimensional and comprehensive characterization of more than 500 GBM tumours.
Noushmehr, H. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 (2010). This was the first TCGA study to apply genome-wide DNA methylation profiling, and it identified a G–CIMP that was linked to a less severe outcome.
Huse, J., Phillips, H. & Brennan, C. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia 59, 1190–1199 (2011).
Zheng, S., Chheda, M. G. & Verhaak, R. G. Studying a complex tumor: potential and pitfalls. Cancer J. 18, 107–114 (2012).
Carro, M. S. et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325 (2010).
Bhat, K. et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 25, 2594–2609 (2011).
Frattini, V. et al. The integrated landscape of driver genomic alterations in glioblastoma. Nature Genet. 45, 1141–1149 (2013). This manuscript reports the genomic landscape of driver genes that are targeted by both mutations and CNAs through an integrated computational and experimental pipeline. It also reviews the landscape of gene fusions in GBM.
Danussi, C. et al. RHPN2 drives mesenchymal transformation in malignant glioma by triggering RhoA activation. Cancer Res. 73, 5140–5150 (2013).
Bhat, K. P. et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346 (2013).
Haque, T. et al. Gene expression profiling from formalin-fixed paraffin-embedded tumors of pediatric glioblastoma. Clin. Cancer Res. 13, 6284–6292 (2007).
Paugh, B. S. et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 28, 3061–3068 (2010).
Paugh, B. S. et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J. Clin. Oncol. 29, 3999–4006 (2011).
Bax, D. A. et al. A distinct spectrum of copy number aberrations in paediatric high grade gliomas. Clin. Cancer Res. 16, 3368–3377 (2010).
Qu, H. Q. et al. Genome-wide profiling using single-nucleotide polymorphism arrays identifies novel chromosomal imbalances in pediatric glioblastomas. Neuro Oncol. 12, 153–163 (2010).
Sturm, D. et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437 (2012). This study applied genome-wide DNA methylation profiling to a combined cohort of paediatric and adult patients and described biological subgroups of GBMs that were associated with distinct molecular aberrations and clinical characteristics.
Schwartzentruber, J. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012).
Fontebasso, A., Liu, X.-Y., Sturm, D. & Jabado, N. Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain Pathol. 23, 210–216 (2013).
Bjerke, L. et al. Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 3, 512–519 (2013).
Bender, S. et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672 (2013).
Costello, J., Berger, M., Huang, H. & Cavenee, W. Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer Res. 56, 2405–2410 (1996).
Baeza, N., Weller, M., Yonekawa, Y., Kleihues, P. & Ohgaki, H. PTEN methylation and expression in glioblastomas. Acta Neuropathol. 106, 479–485 (2003).
Nakamura, M., Yonekawa, Y., Kleihues, P. & Ohgaki, H. Promoter hypermethylation of the RB1 gene in glioblastomas. Lab. Invest. 81, 77–82 (2001).
Amatya, V., Naumann, U., Weller, M. & Ohgaki, H. TP53 promoter methylation in human gliomas. Acta Neuropathol. 110, 178–184 (2005).
Alaminos, M. et al. EMP3, a myelin-related gene located in the critical 19q13.3 region, is epigenetically silenced and exhibits features of a candidate tumor suppressor in glioma and neuroblastoma. Cancer Res. 65, 2565–2571 (2005).
Bruna, A. et al. High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11, 147–160 (2007).
Waha, A. et al. Epigenetic silencing of the protocadherin family member PCDH-γ-A11 in astrocytomas. Neoplasia 7, 193–199 (2005).
Zhou, H. et al. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin. Cancer Res. 13, 2344–2353 (2007).
Zardo, G. et al. Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors. Nature Genet. 32, 453–458 (2002).
Esteller, M., Hamilton, S., Burger, P., Baylin, S. & Herman, J. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 59, 793–797 (1999). This was the first study to show inactivation by promoter hypermethylation of the MGMT gene in samples from patients with GBM.
Felsberg, J. et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 129, 659–670 (2011).
Wick, W. et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 13, 707–715 (2012).
Malmström, A. et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 13, 916–926 (2012). References 56 and 57 showed that MGMT promoter methylation is a predictive biomarker for the benefit from alkylating agent chemotherapy, particularly in elderly patients with GBM.
Costello, J., Futscher, B., Tano, K., Graunke, D. & Pieper, R. Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J. Biol. Chem. 269, 17228–17237 (1994). This was the first study to report on the hypermethylation of the MGMT gene in human glioma cells.
Costello, J., Futscher, B., Kroes, R. & Pieper, R. Methylation-related chromatin structure is associated with exclusion of transcription factors from and suppressed expression of the O-6-methylguanine DNA methyltransferase gene in human glioma cell lines. Mol. Cell. Biol. 14, 6515–6521 (1994).
Esteller, M. et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 343, 1350–1354 (2000).
Hegi, M. E. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003 (2005). This study showed that the presence of an inactivated MGMT gene confers a benefit from alkylating agent chemotherapy using temozolomide.
Olson, R., Brastianos, P. & Palma, D. Prognostic and predictive value of epigenetic silencing of MGMT in patients with high grade gliomas: a systematic review and meta-analysis. J. Neuro-Oncol. 105, 325–335 (2011).
Donson, A., Addo-Yobo, S., Handler, M., Gore, L. & Foreman, N. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatr. Blood Cancer 48, 403–407 (2007).
Buttarelli, F. et al. Evaluation status and prognostic significance of O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation in pediatric high grade gliomas. Child. Nerv. Syst. 26, 1051–1056 (2010).
Srivastava, A. et al. MGMT gene promoter methylation in pediatric glioblastomas. Child. Nerv. Syst. 26, 1613–1618 (2010).
Lee, J. et al. MGMT promoter gene methylation in pediatric glioblastoma: analysis using MS-MLPA. Child. Nerv. Syst. 27, 1877–1883 (2011).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). This comprehensive TCGA study on 206 GBM samples reported three core signalling pathways (RTK–RAS–PI3K, P53 and RB) that are implicated in GBM development and treatment resistance.
Cadieux, B., Ching, T.-T., VandenBerg, S. & Costello, J. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 66, 8469–8476 (2006).
Hovestadt, V. et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 125, 913–916 (2013).
Malhotra, A. et al. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res. 23, 762–776 (2013).
Bozdag, S. et al. Age-specific signatures of glioblastoma at the genomic, genetic, and epigenetic levels. PLoS ONE 8, e62982 (2013).
Jones, C., Perryman, L. & Hargrave, D. Paediatric and adult malignant glioma: close relatives or distant cousins? Nature Rev. Clin. Oncol. 9, 400–413 (2012).
Zheng, S. et al. A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev. 27, 1462–1472 (2013).
Bax, D. et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin. Cancer Res. 15, 5753–5761 (2009).
Cho, J. et al. Glioblastoma-derived epidermal growth factor receptor carboxyl-terminal deletion mutants are transforming and are sensitive to EGFR-directed therapies. Cancer Res. 71, 7587–7596 (2011).
Gan, H., Cvrljevic, A. & Johns, T. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 280, 5350–5370 (2013).
Biernat, W., Huang, H., Yokoo, H., Kleihues, P. & Ohgaki, H. Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol. 14, 131–136 (2004).
Ozawa, T. et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 24, 2205–2218 (2010).
Paugh, B. et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 73, 6219–6229 (2013).
Sanborn, J. et al. Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res. 73, 6036–6045 (2013).
Vogt, N. et al. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc. Natl Acad. Sci. USA 101, 11368–11373 (2004).
Singh, D. et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337, 1231–1235 (2012). This is the first report of a recurrent gene fusion ( FGFR–TACC ) in GBM. This study has paved the way to a personalized, tailored therapeutic approach with FGFR inhibitors for GBM that harbours FGFR–TACC fusions.
Parker, B. et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J. Clin. Invest. 123, 855–865 (2013).
Phillips, J. et al. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol. 23, 565–573 (2013).
Zarghooni, M. et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor α and poly (ADP-ribose) polymerase as potential therapeutic targets. J. Clin. Oncol. 28, 1337–1344 (2010).
Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008). This comprehensive analysis discovered various genes that were not previously known to be altered in GBM; most notably, recurrent hotspot mutations in IDH1.
Liu, X.-Y. et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 124, 615–625 (2012).
Jiao, Y. et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3, 709–722 (2012).
Dawson, M. & Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 (2012).
Shen, H. & Laird, P. Interplay between the cancer genome and epigenome. Cell 153, 38–55 (2013).
Killela, P. et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl Acad. Sci. USA 110, 6021–6026 (2013). This study was the first to report recurrent somatic mutations in the TERT gene promoter as a probable mechanism of telomerase activation in a large proportion of GBMs.
Arita, H. et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 126, 267–276 (2013).
Nonoguchi, N. et al. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 126, 931–937 (2013).
Vinagre, J. et al. Frequency of TERT promoter mutations in human cancers. Nature Commun. 4, 2185 (2013).
Koelsche, C. et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 126, 907–915 (2013).
Boldrini, L. et al. Telomerase activity and hTERT mRNA expression in glial tumors. Int. J. Oncol. 28, 1555–1560 (2006).
Heaphy, C. et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425 (2011). This was the first study to report the presence of an ALT phenotype that occurred in tumours with mutations in ATRX or DAXX.
Hunter, C. et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 66, 3987–3991 (2006).
Cahill, D. et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 13, 2038–2045 (2007).
Yip, S. et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 15, 4622–4629 (2009).
Wu, G. et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature Genet. 44, 251–253 (2012). References 41 and 101 were the first studies to identify recurrent H3F3A or HIST1H3B mutations in paediatric HGGs and the first reports on somatic histone mutations in human cancer.
Downing, J. et al. The Pediatric Cancer Genome Project. Nature Genet. 44, 619–622 (2012).
Zhang, J. et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nature Genet. 45, 602–612 (2013).
Jones, D. T. W. et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nature Genet. 45, 927–932 (2013).
Venneti, S. et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 23, 558–564 (2013).
Lewis, P. et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). This was the first study to show the functional inhibition of PRC2 by a gain-of-function mutation at position K27 of histone variant H3.3, which led to globally reduced levels of H3K27me3.
Chan, K.-M. et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 27, 985–990 (2013).
Swartling, F. J. et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 21, 601–613 (2012).
Puissant, A. et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 3, 308–323 (2013).
Fontebasso, A. et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 125, 659–669 (2013).
Li, F. et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 153, 590–600 (2013).
Chen, Y.-J. et al. Association of mutant TP53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res. 66, 6473–6476 (2006).
Yan, H. et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009). This study identified IDH1 and IDH2 mutations in most lower grade gliomas and secondary GBMs that were associated with better outcome than those with wild-type IDH genes.
Ichimura, K. et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 11, 341–347 (2009).
Zhang, C., Moore, L., Li, X., Yung, W. & Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro Oncol. 15, 1114–1126 (2013).
Ohgaki, H. & Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 19, 764–772 (2013).
Larjavaara, S. et al. Incidence of gliomas by anatomic location. Neuro Oncol. 9, 319–325 (2007).
Turcan, S. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012). This study was the first to show a causal link between mutations in IDH1 and the G–CIMP.
Kim, W. & Liau, L. IDH mutations in human glioma. Neurosurg. Clin. N. Am. 23, 471–480 (2012).
Ichimura, K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol. 29, 131–139 (2012).
Lass, U. et al. Clonal analysis in recurrent astrocytic, oligoastrocytic and oligodendroglial tumors implicates IDH1- mutation as common tumor initiating event. PLoS ONE 7, e41298 (2012).
Hartmann, C. et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 120, 707–718 (2010).
Hartmann, C. et al. Longterm survival in primary glioblastoma with versus without isocitrate dehydrogenase mutations. Clin. Cancer Res. 19, 5146–5157 (2013).
Ward, P. S. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 (2010).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
Figueroa, M. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011).
Chowdhury, R. et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 (2011).
Guilhamon, P. et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nature Commun. 4, 2166 (2013).
Koso, H. et al. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc. Natl Acad. Sci. USA 109, E2998–E3007 (2012).
Alcantara Llaguno, S. et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15, 45–56 (2009).
Liu, C. et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 (2011).
Sugiarto, S. et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell 20, 328–340 (2011).
Friedmann-Morvinski, D. et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084 (2012).
Monje, M. et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl Acad. Sci. USA 108, 4453–4458 (2011).
Lai, A. et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 29, 4482–4490 (2011).
Kang, H. et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011).
Ohnishi, A. et al. Expression of the oligodendroglial lineage-associated markers Olig1 and Olig2 in different types of human gliomas. J. Neuropathol. Exp. Neurol. 62, 1052–1059 (2003).
Lu, Q. R. et al. Oligodendrocyte lineage genes (OLIG) as molecular markers for human glial brain tumors. Proc. Natl Acad. Sci. USA 98, 10851–10856 (2001).
Marie, Y. et al. OLIG2 as a specific marker of oligodendroglial tumour cells. Lancet 358, 298–300 (2001).
Barrett, L. E. et al. Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell 21, 11–24 (2012).
Appolloni, I. et al. Antagonistic modulation of gliomagenesis by Pax6 and Olig2 in PDGF-induced oligodendroglioma. Int. J. Cancer 131, E1078–E1087 (2012).
Wang, Y. et al. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell 15, 514–526 (2009).
Prados, M. D. et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J. Clin. Oncol. 27, 579–584 (2009).
Brown, P. D. et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J. Clin. Oncol. 26, 5603–5609 (2008).
van den Bent, M. J. et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 27, 1268–1274 (2009).
Lassman, A. B. et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01–03 and 00–01. Clin. Cancer Res. 11, 7841–7850 (2005).
Franceschi, E. et al. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br. J. Cancer 96, 1047–1051 (2007).
Neyns, B. et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 20, 1596–1603 (2009).
Raymond, E. et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J. Clin. Oncol. 26, 4659–4665 (2008).
Wen, P. Y. et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99–08. Clin. Cancer Res. 12, 4899–4907 (2006).
Reardon, D. A. et al. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br. J. Cancer 101, 1995–2004 (2009).
Dresemann, G. et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J. Neurooncol. 96, 393–402 (2010).
Friedman, H. S. et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 27, 4733–4740 (2009).
Lai, A. et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J. Clin. Oncol. 29, 142–148 (2011).
Batchelor, T. T. et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 31, 3212–3218 (2013).
Tanaka, S., Louis, D. N., Curry, W. T., Batchelor, T. T. & Dietrich, J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nature Rev. Clin. Oncol. 10, 14–26 (2013).
Drappatz, J. et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J. Neurooncol. 107, 133–138 (2012).
Friday, B. B. et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro. Oncol. 14, 215–221 (2012).
Barker, C. A., Bishop, A. J., Chang, M., Beal, K. & Chan, T. A. Valproic acid use during radiation therapy for glioblastoma associated with improved survival. Int. J. Radiat. Oncol. 86, 504–509 (2013).
Weller, M. et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 77, 1156–1164 (2011).
Rohle, D. et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630 (2013).
Davis, M. et al. ML309: A potent inhibitor of R132H mutant IDH1 capable of reducing 2-hydroxyglutarate production in U87 MG glioblastoma cells. 2012 Apr 16 [Updated 2013 May 8]. In Probe Reports from the NIH Molecular Libraries Program [Internet] (Bethesda (MD): National Center for Biotechnology Information (US), 2010–).
Wang, F. et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626 (2013).
Tonjes, M. et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nature Med. 19, 901–908 (2013).
Yang, W. et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature Cell Biol. 14, 1295–1304 (2012).
Yang, W. et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150, 685–696 (2012).
McCabe, M. T. et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl Acad. Sci. USA 109, 2989–2994 (2012).
Ryan, R. J. et al. EZH2 codon 641 mutations are common in BCL2-rearranged germinal center B cell lymphomas. PLoS ONE 6, e28585 (2011).
Sneeringer, C. J. et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl Acad. Sci. USA 107, 20980–20985 (2010).
Marian, C. et al. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin. Cancer Res. 16, 154–163 (2010).
Cheng, Z. et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 19, 1748–1759 (2013).
Hegi, M. et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib — a phase II trial. Mol. Cancer Ther. 10, 1102–1112 (2011).
Mellinghoff, I. et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 353, 2012–2024 (2005).
Fenton, T. et al. Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240. Proc. Natl Acad. Sci. USA 109, 14164–14169 (2012).
Jun, H., Bronson, R. & Charest, A. Inhibition of EGFR induces a c-MET driven stem cell population in glioblastoma. Stem Cells http://dx.doi.org/10.1002/stem.1554 (2013).
Bielen, A. et al. Enhanced efficacy of IGF1R inhibition in pediatric glioblastoma by combinatorial targeting of PDGFRα/β. Mol. Cancer Ther. 10, 1407–1418 (2011).
Szerlip, N. et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl Acad. Sci. USA 109, 3041–3046 (2012).
Little, S. et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 72, 1614–1620 (2012).
Marusyk, A. & Polyak, K. Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta 1805, 105–117 (2010).
Harada, K. et al. Intratumoral cytogenetic heterogeneity detected by comparative genomic hybridization and laser scanning cytometry in human gliomas. Cancer Res. 58, 4694–4700 (1998).
Jung, V. et al. Evidence of focal genetic microheterogeneity in glioblastoma multiforme by area-specific CGH on microdissected tumor cells. J. Neuropath. Exp. Neur. 58, 993–999 (1999).
Nobusawa, S. et al. Intratumoral patterns of genomic imbalance in glioblastomas. Brain Pathol. 20, 936–944 (2010).
Ren, Z.-P. et al. Molecular genetic analysis of p53 intratumoral heterogeneity in human astrocytic brain tumors. J. Neuropath. Exp. Neur. 66, 944–954 (2007).
Hegi, M. et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 26, 4189–4199 (2008).
Sottoriva, A. et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl Acad. Sci. USA 110, 4009–4014 (2013).
Snuderl, M. et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20, 810–817 (2011).
Inda, M.-d.-M. et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 24, 1731–1745 (2010).
Stommel, J. et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318, 287–290 (2007).
Greaves, M. & Maley, C. Clonal evolution in cancer. Nature 481, 306–313 (2012).
Jordan, C., Guzman, M. & Noble, M. Cancer stem cells. N. Engl. J. Med. 355, 1253–1261 (2006).
Hambardzumyan, D., Parada, L., Holland, E. & Charest, A. Genetic modeling of gliomas in mice: new tools to tackle old problems. Glia 59, 1155–1168 (2011).
Chen, L., Zhang, Y., Yang, J., Hagan, J. & Li, M. Vertebrate animal models of glioma: understanding the mechanisms and developing new therapies. Biochim. Biophys. Acta 1836, 158–165 (2013).
Lee, J. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9, 391–403 (2006).
Radaelli, E. et al. Immunohistopathological and neuroimaging characterization of murine orthotopic xenograft models of glioblastoma multiforme recapitulating the most salient features of human disease. Histol. Histopathol. 24, 879–891 (2009).
Zhao, Y. et al. An extensive invasive intracranial human glioblastoma xenograft model: role of high level matrix metalloproteinase 9. Am. J. Pathol. 176, 3032–3049 (2010).
Luchman, H. et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 14, 184–191 (2012).
Jacques, T. et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 29, 222–235 (2010).
Chow, L. et al. Cooperativity within and among Pten, 53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 19, 305–316 (2011).
Holland, E., Hively, W., DePinho, R. & Varmus, H. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 12, 3675–3685 (1998).
Ding, H. et al. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res. 63, 1106–1113 (2003).
Holland, E. A mouse model for glioma: biology, pathology, and therapeutic opportunities. Toxicol. Pathol. 28, 171–177 (2000).
Weiss, W. et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res. 63, 1589–1595 (2003).
Wei, Q. et al. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res. 66, 7429–7437 (2006).
Zhu, H. et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc. Natl Acad. Sci. USA 106, 2712–2716 (2009).
Hambardzumyan, D., Amankulor, N. M., Helmy, K. Y., Becher, O. J. & Holland, E. C. Modeling adult gliomas using RCAS/t-va technology. Transl. Oncol. 2, 89–95 (2009).
Hede, S.-M. et al. GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia 57, 1143–1153 (2009).
Lei, L. et al. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS ONE 6, e20041 (2011).
Becher, O. & Holland, E. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 66, 3355 (2006).
Becher, O. et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 70, 2548–2557 (2010).
Reilly, K., Loisel, D., Bronson, R., McLaughlin, M. & Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nature Genet. 26, 109–113 (2000).
Bajenaru, M. et al. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol. Cell. Biol. 22, 5100–5113 (2002).
Reilly, K. et al. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc. Natl Acad. Sci. USA 101, 13008–13013 (2004).
Zhu, Y. et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 8, 119–130 (2005).
Kwon, C.-H. et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 68, 3286–3294 (2008).
Sasaki, M. et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 26, 2038–2049 (2012).
Barrow, J. et al. Homozygous loss of ADAM3A revealed by genome-wide analysis of pediatric high-grade glioma and diffuse intrinsic pontine gliomas. Neuro Oncol. 13, 212–222 (2011).
Raffel, C. et al. Analysis of oncogene and tumor suppressor gene alterations in pediatric malignant astrocytomas reveals reduced survival for patients with PTEN mutations. Clin. Cancer Res. 5, 4085–4090 (1999).
Pollack, I. F. et al. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: results from the Children's Cancer Group 945 cohort. J. Neurosurg. 105, 418–424 (2006).
Pollack, I. et al. Age and TP53 mutation frequency in childhood malignant gliomas: results in a multi-institutional cohort. Cancer Res. 61, 7404–7407 (2001).
Pollack, I. et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Child. Nerv. Syst. 27, 87–94 (2011).
Gallia, G. et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol. Cancer Res. 4, 709–714 (2006).
Schiffman, J. et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 70, 512–519 (2010).
Nicolaides, T. et al. Targeted therapy for BRAFV600E malignant astrocytoma. Clin. Cancer Res. 17, 7595–7604 (2011).
Acknowledgements
This work was principally supported by the PedBrain Tumor Project contributing to the International Cancer Genome Consortium, funded by German Cancer Aid (109252) and by the German Federal Ministry of Education and Research (BMBF; grants #01KU1201A, MedSys #0315416C and NGFNplus #01GS0883). This work was further performed within the context of the I-CHANGE consortium supported by Genome Canada, Genome Quebec, the Canadian Institutes for Health Research (CIHR), McGill University, Montreal, Quebec, Canada, and the Montreal Children's Hospital Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- World Health Organization classification of tumours of the central nervous system
-
(WHO classification of tumours of the CNS). A classification system in which histological grading is applied as a means of predicting the biological behaviour of a tumour. It ranges from benign tumours (grade I) to highly aggressive, rapidly progressing tumours with frequently fatal outcome (grade IV).
- Gliomas
-
Tumours that have histological features that are similar to normal glial cells; that is, astrocytes (astrocytoma), oligodendrocytes (oligodendroglioma), or ependymal cells (ependymoma). However, the term is often used to imply only astrocytic or oligodendroglial tumours.
- Diffuse intrinsic pontine gliomas
-
(DIPGs). Highly infiltrative glial tumours that occur in the pons. They occur almost exclusively in children, with a peak age at diagnosis of between 5 and 9 years.
- Temozolomide
-
An alkylating chemotherapeutic agent that is used for the treatment of glioblastoma. It triggers tumour cell death through extensive DNA damage.
- CpG island methylator phenotype
-
(CIMP). A DNA methylation pattern of widespread CpG island promoter methylation. CIMP is frequently reported to be associated with distinct tumour subgroups, patient prognosis and response to treatment.
- Chromothripsis
-
Clustered chromosomal rearrangements in one or a few chromosomes during cancer development, which are thought to occur through a one-step catastrophic genomic event.
- Double-minute chromosomes
-
Small circular fragments of extrachromosomal DNA that frequently harbour one or more oncogenes.
- High-amplitude focal copy-number aberrations
-
Small fragments (typically ∼3 megabases or smaller in size) of amplified or homozygously deleted DNA. Such aberrations often result in numerous copies of oncogenes or the deletion of both copies of tumour suppressor genes.
- Alternative lengthening of telomeres
-
(ALT). A mechanism (or mechanisms) by which 5–10% of human cancers maintain or increase the overall length of their telomeres without the need of increased telomerase activity. The exact molecular mechanism (or mechanisms) of ALT remain elusive, but they may rely on recombination-mediated elongation.
- Polycomb repressive complex 2
-
(PRC2). One of two classes of Polycomb-group proteins. PRC2 has methyltransferase activity and primarily trimethylates histone H3 on lysine 27 (that is, H3K27me3), which is a mark of transcriptionally silent chromatin.
- Warburg effect
-
A predominant production of energy by a high rate of glycolysis followed by lactic acid fermentation in the cytosol that is observed in most cancer cells in the presence of oxygen.
Rights and permissions
About this article
Cite this article
Sturm, D., Bender, S., Jones, D. et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 14, 92–107 (2014). https://doi.org/10.1038/nrc3655
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrc3655
This article is cited by
-
Current and future therapeutic strategies for high-grade gliomas leveraging the interplay between epigenetic regulators and kinase signaling networks
Journal of Experimental & Clinical Cancer Research (2024)
-
Role of Histone Deacetylase Inhibitor in Diabetic Painful Neuropathy
Molecular Neurobiology (2024)
-
Integrin signaling in cancer: bidirectional mechanisms and therapeutic opportunities
Cell Communication and Signaling (2023)
-
Spatially controlled construction of assembloids using bioprinting
Nature Communications (2023)
-
Polymorphisms in autophagy genes are genetic susceptibility factors in glioblastoma development
BMC Cancer (2022)
