
Key Points
-
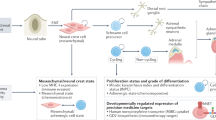
Neuroblastoma is the most common extracranial tumour of childhood. This tumour originates from precursor cells of the peripheral (sympathetic) nervous system and usually arises in a paraspinal location in the abdomen or chest.
-
The aetiology of neuroblastoma is unknown, but it seems unlikely that environmental exposures are important. A subset of patients inherits a genetic predisposition to neuroblastoma, and these patients usually have multifocal primary tumours that arise at an early age. A predisposition locus has been mapped to the short arm of chromosome 16.
-
Neuroblastomas can be classified into subtypes that are predictive of clinical behaviour based on the patterns of genetic change. This information can be useful in the selection of therapy.
-
Favourable tumours are characterized by near-triploid karyotypes with whole chromosome gains. These tumours rarely have structural rearrangements, and they usually express the TrkA neurotrophin receptor. Patients with these tumours are more likely to be less than 1 year of age, have localized tumours and a good prognosis.
-
Unfavourable tumours are characterized by structural changes, including deletions of 1p or 11q, unbalanced gain of 17q and/or amplification of the MYCN protooncogene. They might also express the TrkB neurotrophin receptor and its ligand, brain-derived neurotrophic factor (BDNF). These patients are usually older than 1 year of age, have more advanced stages of disease and a much worse prognosis, even with aggressive treatment.
-
Mass screening for neuroblastoma at 6–12 months of age led to an increased prevalence of neuroblastoma detected in the screened populations, but no decrease in mortality from this disease. The tumours detected have overwhelmingly been of the favourable genetic subtype.
-
Novel, biologically based therapies are being developed that would specifically target the genes, proteins and pathways that are responsible for malignant transformation and progression in neuroblastomas. These approaches are likely to be more effective and less toxic than conventional therapy.
-
In the future, it is likely that more extensive molecular profiling of the genetic changes and expression patterns of neuroblastoma will lead to an even more precise subclassification system that will be predictive of outcome, as well as therapies to which the tumour is most likely to be responsive.
Abstract
Neuroblastoma is a tumour derived from primitive cells of the sympathetic nervous system and is the most common solid tumour in childhood. Interestingly, most infants experience complete regression of their disease with minimal therapy, even with metastatic disease. However, older patients frequently have metastatic disease that grows relentlessly, despite even the most intensive multimodality therapy. Recent advances in understanding the biology and genetics of neuroblastomas have allowed classification into low-, intermediate- and high-risk groups. This allows the most appropriate intensity of therapy to be selected — from observation alone to aggressive, multimodality therapy. Future therapies will focus increasingly on the genes and biological pathways that contribute to malignant transformation or progression.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


References
Knudson, A. G. J. & Strong, L. C. Mutation and cancer: neuroblastoma and pheochromocytoma. Am. J. Hum. Genet. 24, 514–522 (1972).
Kushner, B. H., Gilbert, F. & Helson, L. Familial neuroblastoma: case reports, literature review, and etiologic considerations. Cancer 57, 1887–1893 (1986).
Maris, J. M. & Matthay, K. K. Molecular biology of neuroblastoma. J. Clin. Oncol. 17, 2264–2279 (1999).
Kushner, B. H. & Helson, L. Monozygotic siblings discordant for neuroblastoma: etiologic implications. J. Pediatr. 107, 405–409 (1985).
Kushner, B. H., Hajdu, S. I. & Helson, L. Synchronous neuroblastoma and von Recklinghausen's disease: a review of the literature. J. Clin. Oncol. 3, 117–120 (1985).
Maris, J. M. et al. Familial predisposition to neuroblastoma does not map to chromosome band 1p36. Cancer Res. 56, 3421–3425 (1996).
Weiss, M. J. et al. Localization of a hereditary neuroblastoma predisposition gene to 16p12-p13. Med. Pediatr. Oncol. 35, 526–530 (2000).
Bown, N. P., Pearson, A. D. J. & Reid, M. M. High incidence of constitutional balanced translocations in neuroblastoma. Cancer Genet. Cytogenet. 69, 166–167 (1993).
Biegel, J. A. et al. Constitutional 1p36 deletion in a child with neuroblastoma. Am. J. Hum. Genet. 52, 176–182 (1993).
Laureys, G. et al. Constitutional translocation t(1;17)(p36.31-p36.13;q11.2-q12.1) in a neuroblastoma patient. Establishment of somatic cell hybrids and identification of PND/A12M2 on chromosome 1 and NF1/SCYA7 on chromosome 17 as breakpoint flanking single copy markers. Oncogene 10, 1087–1093 (1995).
White, P. S. et al. Detailed molecular analysis of 1p36 in neuroblastoma. Med. Pediatr. Oncol. 36, 37–41 (2001).
Maris, J. M. et al. Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12-13. Cancer Res. 62, 6651–6658 (2002). The first report of linkage analysis, identifying a candidate locus on 16p12-13.
Kaneko, Y. et al. Different karyotypic patterns in early and advanced stage neuroblastomas. Cancer Res. 47, 311–318 (1987). The first report to associate karyotypic pattern with stage and prognosis, and the first to show the association of near-triploid tumours in infants with whole chromosome gains.
Kaneko, Y. et al. Current urinary mass screening or catecholamine metabolites at 6 months of age may be detecting only a small portion of high-risk neuroblastomas: a chromosome and N-myc amplification study. J. Clin. Oncol. 8, 2005–2013 (1990).
Look, A. T., Hayes, F. A., Nitschke, R., McWilliams, N. B. & Green, A. A. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N. Engl. J. Med. 311, 231–235 (1984). The first report to show the prognostic significance of tumour-cell DNA content in infants with neuroblastoma.
Look, A. T. et al. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma. A Pediatric Oncology Group Study. J. Clin. Oncol. 9, 581–591 (1991).
Kaneko, Y. & Knudson, A. G. Mechanism and relevance of ploidy in neuroblastoma. Genes Chromosom. Cancer 29, 89–95 (2000).
Schwab, M. et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 305, 245–248 (1983). Reports the cloning of the MYCN proto-oncogene as the gene amplified in neuroblastoma cell lines and a primary tumour.
Schwab, M. et al. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature 308, 288–291 (1984).
Corvi, R., Amler, L. C., Savelyeva, L., Gehring, M. & Schwab, M. MYCN is retained in single copy at chromosome 2 band p23-24 during amplification in human neuroblastoma cells. Proc. Natl Acad. Sci. USA 91, 5523–5527 (1994).
Schneider, S. S. et al. Isolation and structural analysis of a 1.2-megabase N-myc amplicon from a human neuroblastoma. Mol. Cell. Biol. 12, 5563–5570 (1992).
Brodeur, G. M. & Fong, C. T. Molecular biology and genetics of human neuroblastoma. Cancer Genet. Cytogenet. 41, 153–174 (1989).
Reiter, J. L. & Brodeur, G. M. High-resolution mapping of a 130-kb core region of the MYCN amplicon in neuroblastomas. Genomics 32, 97–103 (1996).
Reiter, J. L. & Brodeur, G. M. MYCN is the only highly expressed gene from the core amplified domain in human neuroblastomas. Genes Chromosom. Cancer 23, 134–140 (1998).
Brodeur, G. M., Seeger, R. C., Schwab, M., Varmus, H. E. & Bishop, J. M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224, 1121–1124 (1984). Amplification of the MYCN oncogene is strongly associated with advanced stages of disease in neuroblastoma.
Seeger, R. C. et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 313, 1111–1116 (1985). The first report to show the adverse prognostic significance of MYCN amplification in neuroblastoma patients.
Brodeur, G. M., Maris, J. M., Yamashiro, D. J., Hogarty, M. D. & White, P. S. Biology and genetics of human neuroblastomas. J. Pediatr. Hematol. Oncol. 19, 93–101 (1997).
Brodeur, G. M. & Ambros, P. F. in Neuroblastoma (eds Brodeur, G. M., Sawada, T., Tsuchida, Y. & Voûte, P. A.) 355–369 (Elsevier Science B. V., Amsterdam, 2000).
Brodeur, G. M. & Maris, J. M. in Principles and Practice of Pediatric Oncology (eds Pizzo, P. & Poplack, D.) 895–937 (2002).
Brodeur, G. M. in The Genetic Basis of Human Cancer (eds Vogelstein, B. & Kinzler, K. W.) 751–772 (McGraw–Hill, Inc., New York, 2002).
Brodeur, G. M. et al. Consistent N-myc copy number in simultaneous or consecutive neuroblastoma samples from sixty individual patients. Cancer Res. 47, 4248–4253 (1987).
Seeger, R. C. et al. Expression of N-myc by neuroblastomas with one or multiple copies of the oncogene. Prog. Clin. Biol. Res. 271, 41–49 (1988).
Norris, M. D. et al. Evidence that the MYCN oncogene regulates MRP gene expression in neuroblastoma. Eur. J. Cancer 33, 1911–1916 (1997).
Shohet, J. M. et al. Minichromosome maintenance protein MCM7 is a direct target of the MYCN transcription factor in neuroblastoma. Cancer Res. 62, 1123–1128 (2002).
Nakagawara, A., Arima, M., Azar, C. G., Scavarda, N. J. & Brodeur, G. M. Inverse relationship between TRK expression and N-MYC amplification in human neuroblastomas. Cancer Res. 52, 1364–1368 (1992).
Wada, R. K. et al. Human neuroblastoma cell lines that express N-myc without gene amplification. Cancer 72, 3346–3354 (1993).
Cohn, S. L. et al. High levels of N-myc protein in a neuroblastoma cell line lacking N-myc amplification. Prog. Clin. Biol. Res. 366, 21–27 (1991).
Sivak, L. E. et al. Autoregulation of the human N-myc oncogene is disrupted in amplified but not single-copy neuroblastoma cell lines. Oncogene 15, 1937–1946 (1997).
Chan, H. S. et al. MYCN protein expression as a predictor of neuroblastoma prognosis. Clin. Cancer Res. 3, 1699–1706 (1997).
Bordow, S. B., Norris, M. D., Haber, P. S., Marshall, G. M. & Haber, M. Prognostic significance of MYCN oncogene expression in childhood neuroblastoma. J. Clin. Oncol. 16, 3286–3294 (1998).
Cohn, S. L. et al. MYCN expression is not prognostic of adverse outcome in advanced-stage neuroblastoma with nonamplified MYCN. J. Clin. Oncol. 18, 3604–3613 (2000).
Fong, C. T. et al. Loss of heterozygosity for the short arm of chromosome 1 in human neuroblastomas: correlation with N-myc amplification. Proc. Natl Acad. Sci. USA 86, 3753–3757 (1989).
Gehring, M., Berthold, F., Edler, L., Schwab, M. & Amler, L. C. The 1p deletion is not a reliable marker for the prognosis of patients with neuroblastoma. Cancer Res. 55, 5366–5369 (1995).
Caron, H. et al. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N. Engl. J. Med. 334, 225–230 (1996).
Maris, J. M. et al. Loss of heterozygosity at 1p36 independently predicts for disease progression but not decreased overall survival probability in neuroblastoma patients: a Children's Cancer Group study. J. Clin. Oncol. 18, 1888–1899 (2000).
Jinbo, T., Iwamura, Y., Kaneko, M. & Sawaguchi, S. Coamplification of the L-myc and N-myc oncogenes in a neuroblastoma cell line. Jpn. J. Cancer Res. 80, 299–301 (1989).
Corvi, R. et al. Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene 10, 1081–1086 (1995).
Van Roy, N. et al. Identification of two distinct chromosome 12-derived amplification units in neuroblastoma cell line NGP. Cancer Genet. Cytogenet. 82, 151–154 (1995).
Brinkschmidt, C. et al. Comparative genomic hybridization (CGH) analysis of neuroblastomas — an important methodological approach in paediatric tumour pathology. J. Pathol. 181, 394–400 (1997).
Lastowska, M. et al. Comparative genomic hybridization study of primary neuroblastoma tumors. United Kingdom Children's Cancer Study Group. Genes Chromosom. Cancer 18, 162–169 (1997).
Vandesompele, J. et al. Genetic heterogeneity of neuroblastoma studied by comparative genomic hybridization. Genes Chromosom. Cancer 23, 141–152 (1998).
Caron, H. Allelic loss of chromosome 1 and additional chromosome 17 material are both unfavourable prognostic markers in neuroblastoma. Med. Pediatr. Oncol. 24, 215–221 (1995).
Bown, N. et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 340, 1954–1961 (1999). Definitive report of the prevalence and clinical significance of unbalanced 17q gain in neuroblastomas.
Van Roy, N. et al. Analysis of 1;17 translocation breakpoints in neuroblastoma: implications for mapping of neuroblastoma genes. Eur. J. Cancer 33, 1974–1978 (1997).
Lastowska, M. et al. Breakpoint position on 17q identifies the most aggressive neuroblastoma tumors. Genes Chromosom. Cancer 34, 428–436 (2002).
Islam, A. et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 19, 617–623 (2000).
Ireland, C. M. Activated N-ras oncogenes in human neuroblastoma. Cancer Res. 49, 5530–5533 (1989).
Moley, J. F. et al. Low frequency of ras gene mutations in neuroblastomas, pheochromocytomas and medullary thyroid cancers. Cancer Res. 51, 1596–1599 (1991).
Tanaka, T. et al. Expression of Ha-ras oncogene products in human neuroblastomas and the significant correlation with a patient's prognosis. Cancer Res. 48, 1030–1034 (1988).
Brodeur, G. M. et al. Cytogenetic features of human neuroblastomas and cell lines. Cancer Res. 41, 4678–4686 (1981). Definitive report of distal 1p deletions as a genetic change characteristic of neuroblastomas. The clinical significance of deletion of 1p was shown subsequently in large studies.
White, P. S. et al. A region of consistent deletion in neuroblastoma maps within 1p36.2-3. Proc. Natl Acad. Sci. USA 92, 5520–5524 (1995).
Martinsson, T., Shoberg, P. -M., Hedborg, F. & Kogner, P. Deletion of chromosome 1p loci and microsatellite instability in neuroblastomas analyzed with short-tandem repeat polymorphisms. Cancer Res. 55, 5681–5686 (1995).
Ejeskar, K. et al. Fine mapping of a tumour suppressor candidate gene region in 1p36.2-3, commonly deleted in neuroblastomas and germ cell tumours. Med. Pediatr. Oncol. 36, 61–66 (2001).
Caron, H. et al. Chromosome bands 1p35-36 contain two distinct neuroblastoma tumor suppressor loci, one of which is imprinted. Genes Chromosom. Cancer 30, 168–174 (2001).
Bauer, A. et al. Smallest region of overlapping deletion in 1p36 in human neuroblastoma: a 1 Mbp cosmid and PAC contig. Genes Chromosom. Cancer 31, 228–239 (2001).
Hogarty, M. D. et al. Identification of a 1-megabase consensus region of deletion at 1p36.3 in primary neuroblastomas. Med. Pediatr. Oncol. 35, 512–515 (2000).
Maris, J. M. et al. Comprehensive analysis of chromosome 1p deletions in neuroblastoma. Med. Pediatr. Oncol. 36, 32–36 (2001).
Ohira, M. et al. Identification and characterization of a 500-kb homozygously deleted region at 1p36.2-p36.3 in a neuroblastoma cell line. Oncogene 19, 4302–4307 (2000).
Chen, Y. Z. et al. Homozygous deletion in a neuroblastoma cell line defined by a high-density STS map spanning human chromosome band 1p36. Genes Chromosom. Cancer 31, 326–332 (2001).
Srivatsan, E. S., Ying, K. L. & Seeger, R. C. Deletion of chromosome 11 and of 14q sequences in neuroblastoma. Genes Chromosom. Cancer 7, 32–37 (1993).
Plantaz, D. et al. Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int. J. Cancer 91, 680–686 (2001).
Guo, C. et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 18, 4948–4957 (1999).
Suzuki, T. et al. Frequent loss of heterozygosity on chromosome 14q in neuroblastoma. Cancer Res. 49, 1095–1098 (1989).
Hoshi, M. et al. Detailed deletion mapping of chromosome band 14q32 in human neuroblastoma defines a 1.1-Mb region of common allelic loss. Br. J. Cancer 82, 1801–1807 (2000).
Thompson, P. M. et al. Loss of heterozygosity for chromosome 14q in neuroblastoma. Med. Pediatr. Oncol. 36, 28–31 (2001).
Vogan, K. et al. Absence of p53 gene mutations in primary neuroblastomas. Cancer Res. 53, 5269–5273 (1993).
Hosoi, G. et al. Low frequency of the p53 gene mutations in neuroblastoma. Cancer 73, 3087–3093 (1994).
Keshelava, N. et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 61, 6185–6193 (2001).
Tweddle, D. A., Malcolm, A. J., Bown, N., Pearson, A. D. & Lunec, J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. 61, 8–13 (2001).
Moll, U. M., LaQuaglia, M., Benard, J. & Riou, G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc. Natl Acad. Sci. USA 92, 4407–4411 (1995).
Moll, U. M. et al. Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol. Cell. Biol. 16, 1126–1137 (1996).
Goldman, S. C., Chen, C. Y., Lansing, T. J., Gilmer, T. M. & Kastan, M. B. The p53 signal transduction pathway is intact in human neuroblastoma despite cytoplasmic localization. Am. J. Pathol. 148, 1381–1385 (1996).
Beltinger, C. P., White, P. S., Sulman, E. P., Maris, J. M. & Brodeur, G. M. No CDKN2 mutations in neuroblastomas. Cancer Res. 55, 2053–2055 (1995).
Iolascon, A. et al. Structural and functional analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, and CDKN2C) in neuroblastoma. Pediatr. Res. 43, 139–144 (1998).
Kawamata, N., Seriu, T., Koeffler, H. P. & Bartram, C. R. Molecular analysis of the cyclin-dependent kinase inhibitor family: p16(CDKN2/MTS1/INK4A), p18(INK4C) and p27(Kip1) genes in neuroblastomas. Cancer 77, 570–575 (1996).
Thompson, P. M. et al. Homozygous deletion of CDKN2A (p16INK4a/p14ARF) but not within 1p36 or at other tumor suppressor loci in neuroblastoma. Cancer Res. 61, 679–686 (2001).
Johnson, M. R., Look, A. T., DeClue, J. E., Valentine, M. B. & Lowy, D. R. Inactivation of the NF1 gene in human melanoma and neuroblastoma cell lines without impaired regulation of GTP Ras. Proc. Natl Acad. Sci. USA 90, 5539–5543 (1993).
The, I. et al. Neurofibromatosis type 1 gene mutations in neuroblastoma. Nature Genet. 3, 62–66 (1993).
Yano, H. & Chao, M. V. Neurotrophin receptor structure and interactions. Pharm. Acta Helv. 74, 253–260 (2000).
Patapoutian, A. & Reichardt, L. F. Trk receptors: mediators of neurotrophin action. Curr. Opin. Neurobiol. 11, 272–280 (2001).
Nakagawara, A. et al. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N. Engl. J. Med. 328, 847–854 (1993). The first report to show the favourable prognostic value of TrkA expression in neuroblastomas. Other reports were reported independently with similar results.
Suzuki, T., Bogenmann, E., Shimada, H., Stram, D. & Seeger, R. C. Lack of high-affinity nerve growth factor receptors in aggressive neuroblastomas. J. Natl Cancer Inst. 85, 377–384 (1993).
Kogner, P. et al. Coexpression of messenger RNA for TRK protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer Res. 53, 2044–2050 (1993).
Ambros, I. M. et al. Role of ploidy, chromosome 1p, and Schwann cells in the maturation of neuroblastoma. N. Engl. J. Med. 334, 1505–1511 (1996).
Ambros, I. M. et al. Neuroblastoma cells provoke Schwann cell proliferation in vitro. Med. Pediatr. Oncol. 36, 163–168 (2001).
Nakagawara, A. & Brodeur, G. M. Role of neurotrophins and their receptors in human neuroblastomas: a primary culture study. Eur. J. Cancer 33, 2050–2053 (1997).
Nakagawara, A., Azar, C. G., Scavarda, N. J. & Brodeur, G. M. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol. Cell. Biol. 14, 759–767 (1994). The first report to associate TrkB and BDNF expression with high-risk neuroblastomas that have MYCN amplification.
Acheson, A. et al. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature 374, 450–453 (1995).
Matsumoto, K., Wada, R. K., Yamashiro, J. M., Kaplan, D. R. & Thiele, C. J. Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res. 55, 1798–1806 (1995).
Eggert, A. et al. Expression of neurotrophin receptor TrkA inhibits angiogenesis in neuroblastoma. Med. Pediatr. Oncol. 35, 569–572 (2000).
Ho, R. et al. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res. 62, 6462–6466 (2002).
Yamashiro, D. J., Nakagawara, A., Ikegaki, N., Liu, X. -G. & Brodeur, G. M. Expression of TrkC in favorable human neuroblastomas. Oncogene 12, 37–41 (1996).
Ryden, M. et al. Expression of mRNA for the neurotrophin receptor TrkC in neuroblastomas with favourable tumour stage and good prognosis. Br. J. Cancer 74, 773–779 (1996).
Casaccia-Bonnefil, P., Gu, C. & Chao, M. V. Neurotrophins in cell survival/death decisions. Adv. Exp. Med. Biol. 468, 275–282 (1999).
Hempstead, B. L. The many faces of p75NTR. Curr. Opin. Neurobiol. 12, 260–267 (2002).
Goldstein, L. J. et al. Expression of the multidrug resistance, MDR1, gene in neuroblastomas. J. Clin. Oncol. 8, 128–136 (1990).
Chan, H. S. et al. P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N. Engl. J. Med. 325, 1608–1614 (1991).
Norris, M. D. et al. Expression of the gene for multidrug-resistance-associated protein and outcome in patients with neuroblastoma. N. Engl. J. Med. 334, 231–238 (1996).
Hiyama, E. et al. Correlating telomerase activity levels with human neuroblastoma outcomes. Nature Med. 1, 249–255 (1995).
Brodeur, G. M. Do the ends justify the means? Nature Med. 1, 203–205 (1995).
Brodeur, G. M. & Castle, V. P. in Apoptosis and Cancer Chemotherapy (eds Hickman, J. A. & Dive, C.) 305–318 (Humana, New Jersey, 1999).
Bunone, G., Mariotti, A., Compagni, A., Morandi, E. & Della Valle, G. Induction of apoptosis by p75 neurotrophin receptor in human neuroblastoma cells. Oncogene 14, 1463–1470 (1997).
Fulda, S., Sieverts, H., Friesen, C., Herr, I. & Debatin, K. M. The CD95 (APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res. 57, 3823–3829 (1997).
Castle, V. P. et al. Expression of the apoptosis-suppressing protein bcl-2, in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am. J. Pathol. 143, 1543–1550 (1993).
Oue, T. et al. In situ detection of DNA fragmentation and expression of bcl-2 in human neuroblastoma: relation to apoptosis and spontaneous regression. J. Pediatr. Surg. 31, 251–257 (1996).
Dole, M. et al. Bcl-2 inhibits chemotherapy-induced apoptosis in neuroblastoma. Cancer Res. 54, 3253–3259 (1994).
Dole, M. G. et al. Bcl-xL is expressed in neuroblastoma cells and modulates chemotherapy-induced apoptosis. Cancer Res. 55, 2576–2582 (1995).
Nakagawara, A. et al. High levels of expression and nuclear localization of interleukin-1β converting enzyme (ICE) and CPP32 in favorable human neuroblastomas. Cancer Res. 57, 4578–4584 (1997).
Westermann, F. & Schwab, M. Genetic parameters of neuroblastomas. Cancer Lett. 184, 127–147 (2002).
Brodeur, G. M. et al. Revisions of the international criteria for neuroblastoma diagnosis, staging and response to treatment. J. Clin. Oncol. 11, 1466–1477 (1993). A description of the International Neuroblastoma Staging System currently used throughout the world.
Hann, H. W. L. et al. Prognostic importance of serum ferritin in patients with stages III and IV neuroblastoma. The Children's Cancer Study Group Experience. Cancer Res. 45, 2843–2848 (1985).
Zeltzer, P. M., Marangos, P. J., Evans, A. E. & Schneider, S. L. Serum neuron-specific enolase in children with neuroblastoma. Relationship to stage and disease course. Cancer 57, 1230–1234 (1986).
Ladisch, S. & Wu, Z. L. Detection of a tumour-associated ganglioside in plasma of patients with neuroblastoma. Lancet 1, 136–138 (1985).
Quinn, J. J., Altman, A. J. & Frantz, C. N. Serum lactic dehydrogenase, an indicator of tumor activity in neuroblastoma. J. Pediatr. 97, 89–91 (1980).
Shuster, J. J. et al. Serum lactate dehydrogenase in childhood neuroblastoma. A Pediatric Oncology Group recursive partitioning study. Am. J. Clin. Oncol. 15, 295–303 (1992).
Shimada, H. et al. Histopathologic prognostic factors in neuroblastic tumors: definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J. Natl Cancer Inst. 73, 405–413 (1984). The original report of the popular histopathological classification for predicting outcome of neuroblastoma patients. This was subsequently revised into the International Neuroblastoma Pathology Classification.
Shimada, H. et al. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer 86, 349–363 (1999).
Shimada, H. et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 86, 364–372 (1999).
Combaret, V. et al. Clinical relevance of CD44 cell-surface expression and N-myc gene amplification in a multicentric analysis of 121 pediatric neuroblastomas. J. Clin. Oncol. 14, 25–34 (1996).
Castleberry, R. P. et al. The International Neuroblastoma Risk Groups (INRG): a preliminary report. Eur. J. Cancer 33, 2113–2116 (1997). First report of an international consensus on neuroblastoma risk groups using a combination of clinical and biological variables.
Beckwith, J. & Perrin, E. In situ neuroblastomas: a contribution to the natural history of neural crest tumors. Am. J. Pathol. 43, 1089–1104 (1963).
Turkel, S. B. & Itabashi, H. H. The natural history of neuroblastic cells in the fetal adrenal gland. Am. J. Pathol. 76, 225–243 (1975).
Ikeda, Y., Lister, J., Bouton, J. M. & Buyukpamukcu, M. Congenital neuroblastoma, neuroblastoma in situ, and the normal fetal development of the adrenal. J. Pediatr. Surg. 16, 636–644 (1981).
Evans, A. E., Gerson, J. & Schnaufer, L. Spontaneous regression of neuroblastoma. Natl Cancer Inst. Monogr. 44, 49–54 (1976).
Sawada, T. et al. Neuroblastoma. Mass screening for early detection and its prognosis. Cancer 53, 2731–2735 (1984). A seminal paper that indicates the potential value of mass screening for early detection of disease to improve the prognosis of neuroblastoma. Subsequent reports from mass screening programmes in Quebec and Germany indicate that there is no impact on mortality.
Takeda, T. et al. Japanese experience of screening. Med. Pediatr. Oncol. 17, 368–372 (1989).
Schilling, F. H. et al. Screening for neuroblastoma. Lancet 344, 1157–1158 (1994).
Woods, W. G. et al. A population-based study of the usefulness of screening for neuroblastoma. Lancet 348, 1682–1687 (1996).
Kaneko, Y. et al. Chromosomes and screening for neuroblastoma. Lancet 1, 174–175 (1988).
Hayashi, Y., Inaba, T., Hanada, R. & Yamamoto, K. Chromosome findings and prognosis in 15 patients with neuroblastoma found by VMA mass screening. J. Pediatr. 112, 567–571 (1988).
Hayashi, Y., Hanada, R. & Yamamoto, K. Biology of neuroblastomas in Japan found by screening. Am. J. Pediatr. Hematol. Oncol. 14, 342–347 (1992).
Brodeur, G. M. et al. Biological aspects of neuroblastomas identified by mass screening in Quebec. Med. Pediatr. Oncol. 36, 157–159 (2001).
Woods, W. G. et al. Screening of infants and mortality due to neuroblastoma. N. Engl. J. Med. 346, 1041–1046 (2002).
Schilling, F. H. et al. Neuroblastoma screening at one year of age. N. Engl. J. Med. 346, 1047–1053 (2002).
Kaneko, Y., Kobayashi, H., Maseki, N., Nakagawara, A. & Sakurai, M. Disomy 1 with terminal 1p deletion is frequent in mass-screening-negative/late-presenting neuroblastomas in young children, but not in mass-screening-positive neuroblastomas in infants. Int. J. Cancer 80, 54–59 (1999).
Tajiri, T. et al. Clinical and biologic characteristics for recurring neuroblastoma at mass screening cases in Japan. Cancer 92, 349–353 (2001).
van Limpt, V. et al. SAGE analysis of neuroblastoma reveals a high expression of the human homologue of the Drosophila Delta gene. Med. Pediatr. Oncol. 35, 554–558 (2000).
Spieker, N. et al. The MEIS1 oncogene is highly expressed in neuroblastoma and amplified in cell line IMR32. Genomics 71, 214–221 (2001).
Khan, J. et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nature Med. 7, 673–679 (2001).
Truckenmiller, M. E. et al. Gene expression profile in early stage of retinoic acid-induced differentiation of human SH-SY5Y neuroblastoma cells. Restor. Neurol. Neurosci. 18, 67–80 (2001).
Weiss, W. A., Aldape, K., Mohapatra, G., Feuerstein, B. G. & Bishop, J. M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 16, 2985–2995 (1997). First report of a transgenic mouse that overexpresses the MYCN proto-oncogene under the control of a tyrosine kinase promoter and develops neuroblastoma with high frequency.
Weiss, W. A., Godfrey, T., Francisco, C. & Bishop, J. M. Genome-wide screen for allelic imbalance in a mouse model for neuroblastoma. Cancer Res. 60, 2483–2487 (2000).
Norris, M. D., Burkhart, C. A., Marshall, G. M., Weiss, W. A. & Haber, M. Expression of N-myc and MRP genes and their relationship to N-myc gene dosage and tumor formation in a murine neuroblastoma model. Med. Pediatr. Oncol. 35, 585–589 (2000).
Sidell, N., Altman, A., Haussler, M. R. & Seeger, R. C. Effects of retinoic acid (RA) on the growth and phenotypic expression of several human neuroblastoma cell lines. Exp. Cell Res. 148, 21–30 (1983).
Thiele, C. J., Reynolds, C. P. & Israel, M. A. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 313, 404–406 (1985).
Reynolds, C. P. et al. Comparison of 13-cis-retinoic acid to trans-retinoic acid using human neuroblastoma cell lines. Prog. Clin. Biol. Res. 385, 237–244 (1994).
Matthay, K. K. et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N. Engl. J. Med. 341, 1165–1173 (1999). First clinical trial indicating that a survival advantage exists for the treatment of neuroblastoma patients with retinoic acid following bone-marrow transplantation.
Lovat, P. E. et al. Effector mechanisms of fenretinide-induced apoptosis in neuroblastoma. Exp. Cell Res. 260, 50–60 (2000).
Ponzoni, M. et al. Differential effects of N-(4-hydroxyphenyl)retinamide and retinoic acid on neuroblastoma cells: apoptosis versus differentiation. Cancer Res. 55, 853–861 (1995).
Reynolds, C. P. Differentiating agents in pediatric malignancies: retinoids in neuroblastoma. Curr. Oncol. Rep. 2, 511–518 (2000).
Galderisi, U., Cascino, A. & Giordano, A. Antisense oligonucleotides as therapeutic agents. J. Cell Physiol. 181, 251–257 (1999).
Evans, A. E. et al. Antitumor activity of CEP-751 (KT-6587) on human neuroblastoma and medulloblastoma xenografts. Clin. Cancer Res. 5, 3594–3602 (1999). First report of a tyrosine kinase inhibitor that is selective for Trk receptors with potential use in treating neuroblastomas.
Meitar, D., Crawford, S. E., Rademaker, A. W. & Cohn, S. L. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J. Clin. Oncol. 14, 405–414 (1996). Definitive report correlating tumour angiogenesis with high-risk features and outcome in neuroblastomas. This report serves as the rationale for anti-angiogenesis therapy in high-risk neuroblastomas.
Wassberg, E., Pahlman, S., Westlin, J. E. & Christofferson, R. The angiogenesis inhibitor TNP-470 reduces the growth rate of human neuroblastoma in nude rats. Pediatr. Res. 41, 327–333 (1997).
Katzenstein, H. M. et al. Effectiveness of the angiogenesis inhibitor TNP-470 in reducing the growth of human neuroblastoma in nude mice inversely correlates with tumor burden. Clin. Cancer Res. 5, 4273–4278 (1999).
Shusterman, S., Grupp, S. A. & Maris, J. M. Inhibition of tumor growth in a human neuroblastoma xenograft model with TNP-470. Med. Pediatr. Oncol. 35, 673–676 (2000).
Erdreich-Epstein, A. et al. Integrins α(v)β3 and α(v)β5 are expressed by endothelium of high-risk neuroblastoma and their inhibition is associated with increased endogenous ceramide. Cancer Res. 60, 712–721 (2000).
Jouanneau, E. et al. Lack of antitumor activity of recombinant endostatin in a human neuroblastoma xenograft model. J. Neurooncol. 51, 11–18 (2001).
Kim, E. S. et al. Distinct response of experimental neuroblastoma to combination antiangiogenic strategies. J. Pediatr. Surg. 37, 518–522 (2002).
Davidoff, A. M., Leary, M. A., Ng, C. Y. & Vanin, E. F. Gene therapy-mediated expression by tumor cells of the angiogenesis inhibitor flk-1 results in inhibition of neuroblastoma growth in vivo. J. Pediatr. Surg. 36, 30–36 (2001).
Hanahan, D., Bergers, G. & Bergsland, E. Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J. Clin. Invest. 105, 1045–1047 (2000).
Frost, J. D. et al. A phase I/IB trial of murine monoclonal anti-GD2 antibody 14. G2a plus interleukin-2 in children with refractory neuroblastoma: a report of the Children's Cancer Group. Cancer 80, 317–333 (1997).
Yu, A. L. et al. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J. Clin. Oncol. 16, 2169–2180 (1998).
Cheung, N. K., Kushner, B. H., Yeh, S. D. & Larson, S. M. 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: a phase II study. Int. J. Oncol. 12, 1299–1306 (1998).
De Kraker, J. et al. First line targeted radiotherapy, a new concept in the treatment of advanced stage neuroblastoma. Eur. J. Cancer 31A, 600–602 (1995).
Matthay, K. K. et al. Phase I dose escalation of 131I-metaiodobenzylguanidine with autologous bone marrow support in refractory neuroblastoma. J. Clin. Oncol. 16, 229–236 (1998).
Yanik, G. A. et al. Pilot study of iodine-131-metaiodobenzylguanidine in combination with myeloablative chemotherapy and autologous stem-cell support for the treatment of neuroblastoma. J. Clin. Oncol. 20, 2142–2149 (2002).
Fitzek, M. M. et al. Neuroendocrine tumors of the sinonasal tract. Results of a prospective study incorporating chemotherapy, surgery, and combined proton-photon radiotherapy. Cancer 94, 2623–2634 (2002).
Luttikhuis, M. E. et al. Neuroblastomas with chromosome 11q loss and single copy MYCN comprise a biologically distinct group of tumours with adverse prognosis. Br. J. Cancer 85, 531–537 (2001).
Bhattacharyya, N., Thornton, A. F., Joseph, M. P., Goodman, M. L. & Amrein, P. C. Successful treatment of esthesioneuroblastoma and neuroendocrine carcinoma with combined chemotherapy and proton radiation. Results in 9 cases. Arch. Otolaryngol. Head Neck Surg. 123, 34–40 (1997).
Gurney, J. G. et al. Infant cancer in the US: histology-specific incidence and trends, 1973 to 1992. J. Pediatr. Hematol. Oncol. 19, 428–432 (1997).
Schmidt, M. L. et al. Biologic factors determine prognosis in infants with stage IV neuroblastoma: a prospective Children's Cancer Group study. J. Clin. Oncol. 18, 1260–1268 (2000).
Acknowledgements
This work was supported in part by National Institutes of Health grants, and the Audrey E. Evans Endowed Chair. Some of this material has been published previously (see references 1–4).
Author information
Authors and Affiliations
Glossary
- PLOIDY
-
A general term that is used to describe the overall chromosome number of a cell. A normal diploid cell has a karyotype with 46 chromosomes and a DNA content of 1.0. A triploid cell with 69 chromosomes has a DNA content of 1.5.
- ALLELIC LOSS
-
(or loss of heterozygosity (LOH)). If the DNA is polymorphic in the normal constitutional DNA (two alleles identified) of a patient and only one allele is present in the tumour, then there is presumptive loss of DNA at that locus. Regions with high frequency of LOH are believed to harbour tumour-suppressor genes.
- NEUROTROPHIN
-
A protein that binds to a receptor on a nerve cell, which, in turn, activates signalling pathways that support cell survival.
- AUTOCRINE
-
A mechanism of self-activation through a ligand–receptor pathway. Autocrine activation results from a ligand that is produced by a cell binding to and activating a receptor on the same cell.
- AUTOSOMAL DOMINANT
-
A pattern of inheritance through the non-sex chromosomes, in which a gene (allele) on one chromosome in a pair results in a phenotype and is dominant over the phenotype conferred by the other allele.
- KNUDSON'S TWO-HIT HYPOTHESIS
-
Alfred Knudson proposed that familial cancers result from two rate-limiting mutations. One mutation is inherited in the constitutional DNA, and a single somatically acquired mutation in any cell of the target tissue could result in a tumour. In sporadic cases, both mutations are somatically acquired.
- NEUROFIBROMATOSIS TYPE I
-
(Or von Recklinghausen disease). An autosomal-dominant disorder that is characterized by pigmented patches of skin and by the formation of neurofibromas (tumours involving nerve tissue) in the skin, subcutaneous tissue, cranial nerves and spinal root nerves.
- HIRSCHSPRUNG DISEASE
-
A congenital condition that results from a failure to completely enervate the distal colon. This leads to obstruction of the large intestine from inadequate motility and collapse of this distal segment.
- KARYOTYPE
-
A presentation of the chromosomes of a cell organized in pairs and by size. Normal human cells have a karyotype of 46 chromosomes (23 pairs).
- SYMPATHETIC NERVOUS SYSTEM
-
The peripheral nervous system that is characterized by the neurotransmitter noradrenaline.
- ADRENAL MEDULLA
-
The centre of the adrenal gland, where ganglion cells produce chemicals such as noradrenaline and adrenaline. This is a common site from which neuroblastomas originate.
- PARASPINAL
-
Adjacent to the spine. This is a common location of sympathetic nerve cells, from which neuroblastomas arise.
- NEUROBLASTS
-
Immature nerve cells.
- SCHWANN CELLS
-
Cells that are derived from a group of embryonic cells called the neural crest, which are associated with and supportive of nerve cells. Schwann cells are the stromal cells in mature ganglioneuromas.
- COMPARATIVE GENOMIC HYBRIDIZATION
-
(CGH). A technique that is used to detect chromosome gain or loss by hybridizing DNA from a target cell and a normal cell that are differentially labelled with unique fluors to a normal karyotype.
- GANGLION CELLS
-
Mature, post-mitotic, fully differentiated nerve cells.
- PARACRINE
-
Paracrine activation results from a ligand produced by one cell binding to and activating a receptor on an adjacent cell.
- CATECHOLAMINES
-
Catecholamines are small molecules such as DOPA, dopamine and norepinephrine that function as neurotransmitters in the central and peripheral nervous systems. These compounds are broken down into urinary metabolites that can be measured in the urine.
- PROTON-BEAM THERAPY
-
Radiation therapy for local tumour control using a proton beam, as opposed to an electron or photon beam (used in more conventional radiation therapy).
Rights and permissions
About this article
Cite this article
Brodeur, G. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 3, 203–216 (2003). https://doi.org/10.1038/nrc1014
Issue Date:
DOI: https://doi.org/10.1038/nrc1014
This article is cited by
-
No-ozone cold plasma induces apoptosis in human neuroblastoma cell line via increased intracellular reactive oxygen species (ROS)
BMC Complementary Medicine and Therapies (2024)
-
LncRNA ZNF674-AS1 drives cell growth and inhibits cisplatin-induced pyroptosis via up-regulating CA9 in neuroblastoma
Cell Death & Disease (2024)
-
Predicting MYCN amplification in paediatric neuroblastoma: development and validation of a 18F-FDG PET/CT-based radiomics signature
Insights into Imaging (2023)
-
miR-210-3p enriched extracellular vesicles from hypoxic neuroblastoma cells stimulate migration and invasion of target cells
Cell & Bioscience (2023)
-
Chromosome 1p36 candidate gene ZNF436 predicts the prognosis of neuroblastoma: a bioinformatic analysis
Italian Journal of Pediatrics (2023)
