
Abstract
The genomes of cancer cells constantly change during pathogenesis. This evolutionary process can lead to the emergence of drug-resistant mutations in subclonal populations, which can hinder therapeutic intervention in patients. Data derived from massively parallel sequencing can be used to infer these subclonal populations using tumor-specific point mutations. The accurate determination of copy-number changes and tumor impurity is necessary to reliably infer subclonal populations by mutational clustering. This protocol describes how to use Sclust, a copy-number analysis method with a recently developed mutational clustering approach. In a series of simulations and comparisons with alternative methods, we have previously shown that Sclust accurately determines copy-number states and subclonal populations. Performance tests show that the method is computationally efficient, with copy-number analysis and mutational clustering taking <10 min. Sclust is designed such that even non-experts in computational biology or bioinformatics with basic knowledge of the Linux/Unix command-line syntax should be able to carry out analyses of subclonal populations.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
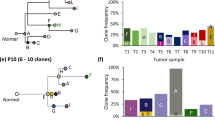

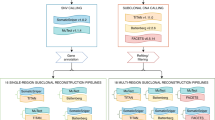
References
Beerenwinkel, N., Schwarz, R.F., Gerstung, M. & Markowetz, F. Cancer evolution: mathematical models and computational inference. Syst. Biol. 64, e1–25 (2015).
Greaves, M. & Maley, C.C. Clonal evolution in cancer. Nature 481, 306–313 (2012).
Nowell, P.C. The clonal evolution of tumor cell populations. Science 194, 23–28 (1976).
Stratton, M.R., Campbell, P.J. & Futreal, P.A. The cancer genome. Nature 458, 719–724 (2009).
Yates, L.R. & Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 13, 795–806 (2012).
Meyerson, M., Gabriel, S. & Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 11, 685–696 (2010).
Carter, S.L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
Fischer, A., Vazquez-Garcia, I., Illingworth, C.J. & Mustonen, V. High-definition reconstruction of clonal composition in cancer. Cell Rep. 7, 1740–1752 (2014).
Gusnanto, A., Wood, H.M., Pawitan, Y., Rabbitts, P. & Berri, S. Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics 28, 40–47 (2012).
Ha, G. et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 24, 1881–1893 (2014).
Oesper, L., Mahmoody, A. & Raphael, B.J. THetA: inferring intra-tumor heterogeneity from high-throughput DNA sequencing data. Genome Biol. 14, R80 (2013).
Van Loo, P. et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 107, 16910–16915 (2010).
Deshwar, A.G. et al. PhyloWGS: reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol. 16, 35 (2015).
Jiao, W., Vembu, S., Deshwar, A.G., Stein, L. & Morris, Q. Inferring clonal evolution of tumors from single nucleotide somatic mutations. BMC Bioinformatics 15, 35 (2014).
Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012).
Roth, A. et al. PyClone: statistical inference of clonal population structure in cancer. Nat. Methods 11, 396–398 (2014).
Malikic, S., McPherson, A.W., Donmez, N. & Sahinalp, C.S. Clonality inference in multiple tumor samples using phylogeny. Bioinformatics 31, 1349–1356 (2015).
Sengupta, S. et al. Bayclone: Bayesian nonparametric inference of tumor subclones using NGS data. Pacific Symposium on Biocomputing 467–478, https://www.worldscientific.com/doi/pdf/10.1142/9789814644730_0044 (2015).
Zare, H. et al. Inferring clonal composition from multiple sections of a breast cancer. PLoS Comput. Biol. 10, e1003703 (2014).
Jiang, Y., Qiu, Y., Minn, A.J. & Zhang, N.R. Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing. Proc. Natl. Acad. Sci. USA 113, E5528–E5537 (2016).
Andor, N., Harness, J.V., Muller, S., Mewes, H.W. & Petritsch, C. EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics 30, 50–60 (2014).
George, J. et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015).
Peifer, M. et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 44, 1104–1110 (2012).
Peifer, M. et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700–704 (2015).
Schramm, A. et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 47, 872–877 (2015).
Gerstung, M. et al. The evolutionary history of 2,658 cancers. Preprint at bioRxiv, http://dx.doi.org/10.1101/161562 (2017).
Seidel, D. et al. A genomics-based classification of human lung tumors. Sci. Transl. Med. 5, 209ra153 (2013).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 (2012).
Acknowledgements
We thank the Evolution and Heterogeneity Working Group of the PCAWG initiative for fruitful discussions; P. van Loo, D. Wedge, and S. Dentro for making the Battenberg calls for PD4120a available; and L. Maas for proofreading. The computation was performed on the DFG-funded CHEOPS Cluster of the Regional Computing Centre of Cologne. This work was supported by German Cancer Aid (Deutsche Krebshilfe, grant ID: 109679), the German Ministry of Science and Education (BMBF) as part of the e:Med program (grant nos. 01ZX1303A and 01ZX1406), and the Deutsche Forschungsgemeinschaft (CRU-286, CP2).
Author information
Authors and Affiliations
Contributions
Y.C. and M.P. conceived the project. Y.C., T.-P.Y., V.A., and M.P. wrote the manuscript. Y.C., T.-P.Y., V.A., and M.P. developed and optimized the algorithm. Y.C., T.-P.Y., V.A., and M.P. performed computational analysis. V.A. and U.L. provided and optimized computing and data infrastructure. All co-authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note. (PDF 662 kb)
Rights and permissions
About this article

Cite this article
Cun, Y., Yang, TP., Achter, V. et al. Copy-number analysis and inference of subclonal populations in cancer genomes using Sclust. Nat Protoc 13, 1488–1501 (2018). https://doi.org/10.1038/nprot.2018.033
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2018.033
This article is cited by
-
Computational validation of clonal and subclonal copy number alterations from bulk tumor sequencing using CNAqc
Genome Biology (2024)
-
scAbsolute: measuring single-cell ploidy and replication status
Genome Biology (2024)
-
Evolutionary trajectories of small cell lung cancer under therapy
Nature (2024)
-
APOBEC3B stratifies ovarian clear cell carcinoma with distinct immunophenotype and prognosis
British Journal of Cancer (2023)
-
Genetic analysis of low-grade adenosquamous carcinoma of the breast progressing to high-grade metaplastic carcinoma
Breast Cancer Research and Treatment (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
