
Abstract
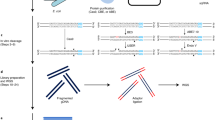
To enable low-cost, high-throughput generation of cistrome and epicistrome maps for any organism, we developed DNA affinity purification sequencing (DAP-seq), a transcription factor (TF)-binding site (TFBS) discovery assay that couples affinity-purified TFs with next-generation sequencing of a genomic DNA library. The method is fast, inexpensive, and more easily scaled than chromatin immunoprecipitation sequencing (ChIP-seq). DNA libraries are constructed using native genomic DNA from any source of interest, preserving cell- and tissue-specific chemical modifications that are known to affect TF binding (such as DNA methylation) and providing increased specificity as compared with in silico predictions based on motifs from methods such as protein-binding microarrays (PBMs) and systematic evolution of ligands by exponential enrichment (SELEX). The resulting DNA library is incubated with an affinity-tagged in vitro-expressed TF, and TF–DNA complexes are purified using magnetic separation of the affinity tag. Bound genomic DNA is eluted from the TF and sequenced using next-generation sequencing. Sequence reads are mapped to a reference genome, identifying genome-wide binding locations for each TF assayed, from which sequence motifs can then be derived. A researcher with molecular biology experience should be able to follow this protocol, processing up to 400 samples per week.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


References
Swinnen, G., Goossens, A. & Pauwels, L. Lessons from domestication: targeting cis-regulatory elements for crop improvement. Trends Plant Sci. http://dx.doi.org/10.1016/j.tplants.2016.01.014 (2016).
Deplancke, B., Alpern, D. & Gardeux, V. The genetics of transcription factor DNA binding variation. Cell http://dx.doi.org/10.1016/j.cell.2016.07.012 (2016).
Babu, M.M., Luscombe, N.M., Aravind, L., Gerstein, M. & Teichmann, S.A. Structure and evolution of transcriptional regulatory networks. Curr. Opin. Struct. Biol. 14, 283–291 (2004).
Niu, W. et al. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res. 21, 245–254 (2011).
Negre, N. et al. A cis-regulatory map of the Drosophila genome. Nature 471, 527–531 (2011).
Gerstein, M.B. et al. Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 (2012).
Celniker, S.E. et al. Unlocking the secrets of the genome. Nature 18, 927–930 (2009).
Landt, S.G. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012).
Weirauch, M.T. et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014).
Jolma, A. et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013).
Jolma, A. et al. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388 (2015).
Domcke, S. et al. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528, 575–579 (2015).
Hu, S. et al. DNA methylation presents distinct binding sites for human transcription factors. Elife 2013 (2013).
Raghav, S.K. et al. Integrative genomics identifies the corepressor SMRT as a gatekeeper of adipogenesis through the transcription factors C/EBPB and KAISO. Mol. Cell 46, 335–350 (2012).
O'Malley, R.C. et al. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell http://dx.doi.org/10.1016/j.cell.2016.04.038 (2016).
Dror, I., Golan, T., Levy, C., Rohs, R. & Mandel-Gutfreund, Y. A widespread role of the motif environment in transcription factor binding across diverse protein families. Genome Res. http://dx.doi.org/10.1101/gr.184671.114 (2015).
Los, G.V. et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. http://dx.doi.org/10.1021/cb800025k (2008).
Worsley Hunt, R. & Wasserman, W.W. Non-targeted transcription factors motifs are a systemic component of ChIP-seq datasets. Genome Biol. 15, 412 (2014).
Schultz, M.D. et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–216 (2015).
Kawakatsu, T. et al. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat. Plants 2, 16058 (2016).
Song, L. & Crawford, G.E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. http://dx.doi.org/10.1101/pdb.prot5384 (2010).
Buenrostro, J.D., Giresi, P.G., Zaba, L.C., Chang, H.Y. & Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Rizzo, J.M. & Sinha, S. Epidermal Cells: Methods and Protocols (ed. Turksen, K.) 49–59 (Springer, 2014).
Kawakatsu, T. et al. Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell 166, 492–506 (2016).
Chen, K., Zhao, B.S. & He, C. Nucleic acid modifications in regulation of gene expression. Cell Chem. Biol. 23, 74–85 (2016).
Arabidopsis Interactome Mapping Consortium. Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607 (2011).
Yazaki, J. et al. Mapping transcription factor interactome networks using HaloTag protein arrays 113, E4238–E4247.
Langmead, B. & Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Guo, Y., Mahony, S. & Gifford, D.K. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput. Biol. 8, e1002638 (2012).
Robinson, J.T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Machanick, P. & Bailey, T.L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Carroll, T.S., Liang, Z., Salama, R., Stark, R. & de Santiago, I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet. 5, 75 (2014).
Ou, J. & Zhu, L.J. motifStack: plot stacked logos for single or multiple DNA, RNA and amino acid sequence. http://bioconductor.org/packages/release/bioc/html/motifStack.html (2015).
Quinlan, A.R. & Hall, I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Harper, S. & Speicher, D.W. in Protein Chromatography (Humana Press) 681, 259–280 (2011).
Structural Genomics Consortium. et al. Protein production and purification. Nat. Methods 5, 135–146 (2008).
Urich, M.A., Nery, J.R., Lister, R., Schmitz, R.J. & Ecker, J.R. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat. Protoc. 10, 475–83 (2015).
Gallagher, S. & Chakavarti, D. Immunoblot analysis. J. Vis. Exp. 2, 2008 (2008).
Acknowledgements
This work was supported by grants from the National Science Foundation (MCB1024999) and the Gordon and Betty Moore Foundation (GBMF3034) to J.R.E., as well as from the National Science Foundation to A.G. (IOS1114484 and IOS1546873). J.R.E. is a Howard Hughes Medical Institute Investigator.
Author information
Authors and Affiliations
Contributions
R.C.O. and J.R.E. designed the original protocol. R.C.O., A.B., S.-s.C.H., M.G., and A.G. modified and updated the protocol to its current state. J.R.N. performed all the sequencing. A.B., M.G., S.-s.C.H., and J.R.E. wrote the manuscript with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Bartlett, A., O'Malley, R., Huang, Ss. et al. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat Protoc 12, 1659–1672 (2017). https://doi.org/10.1038/nprot.2017.055
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2017.055
This article is cited by
-
Identification and functional analysis of the LEAFY gene in longan flower induction
BMC Genomics (2024)
-
Snowprint: a predictive tool for genetic biosensor discovery
Communications Biology (2024)
-
Decoding the gene regulatory network of endosperm differentiation in maize
Nature Communications (2024)
-
Natural variation in OsMYB8 confers diurnal floret opening time divergence between indica and japonica subspecies
Nature Communications (2024)
-
Petal abscission is promoted by jasmonic acid-induced autophagy at Arabidopsis petal bases
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
