
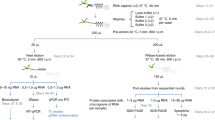
Abstract
The analysis of protein interaction networks is one of the key challenges in the study of biology. It connects genotypes to phenotypes, and disruption of such networks is associated with many pathologies. Virtually all the approaches to the study of protein complexes require cell lysis, a dramatic step that obliterates cellular integrity and profoundly affects protein interactions. This protocol starts with Virotrap, a novel approach that avoids the need for cell homogenization by fusing the protein of interest to the HIV-1 Gag protein, trapping protein complexes in virus-like particles. By using the straightforward filtering index (SFINX), which is a powerful and intuitive online tool (http://sfinx.ugent.be) that enables contaminant removal from candidate lists resulting from mass-spectrometry-based analysis, we provide a complete workflow for researchers interested in mammalian protein complexes. Given direct access to mass spectrometers, researchers can process up to 24 samples in 7 d.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Kuzmanov, U. & Emili, A. Protein-protein interaction networks: probing disease mechanisms using model systems. Genome Med. 5, 37 (2013).
Fields, S. & Song, O. A novel genetic system to detect protein-protein interactions. Nature 340, 245–246 (1989).
Eyckerman, S. et al. Design and application of a cytokine-receptor-based interaction trap. Nat. Cell Biol. 3, 1114–1119 (2001).
Gingras, A.C., Gstaiger, M., Raught, B. & Aebersold, R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654 (2007).
Rigaut, G. et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17, 1030–1032 (1999).
Malovannaya, A. et al. Analysis of the human endogenous coregulator complexome. Cell 145, 787–799 (2011).
Keilhauer, E.C., Hein, M.Y. & Mann, M. Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol. Cell Proteomics 14, 120–135 (2015).
Subbotin, R.I. & Chait, B.T. A pipeline for determining protein-protein interactions and proximities in the cellular milieu. Mol. Cell Proteomics 13, 2824–2835 (2014).
Chen, F. et al. Applying mass spectrometry to study non-covalent biomolecule complexes. Mass Spectrom. Rev. 35, 48–70 (2016).
Roux, K.J., Kim, D.I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 (2012).
Roux, K.J., Kim, D.I. & Burke, B. BioID: a screen for protein-protein interactions. Curr. Protoc. Protein Sci. 74 Unit 19.23 (2013).
Lam, S.S. et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015).
Eyckerman, S. et al. Trapping mammalian protein complexes in viral particles. Nat. Commun. 7, 11416 (2016).
Gheysen, D. et al. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 59, 103–112 (1989).
Shioda, T. & Shibuta, H. Production of human immunodeficiency virus (HIV)-like particles from cells infected with recombinant vaccinia viruses carrying the gag gene of HIV. Virology 175, 139–148 (1990).
Titeca, K. et al. SFINX: straightforward filtering index for affinity purification-mass spectrometry data analysis. J. Proteome Res. 15, 332–338 (2016).
Kutner, R.H., Zhang, X.Y. & Reiser, J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 4, 495–505 (2009).
Van Leene, J. et al. An improved toolbox to unravel the plant cellular machinery by tandem affinity purification of Arabidopsis protein complexes. Nat. Protoc. 10, 169–187 (2015).
Ning, Z., Seebun, D., Hawley, B., Chiang, C.K. & Figeys, D. From cells to peptides: 'one-stop' integrated proteomic processing using amphipols. J. Proteome Res. 12, 1512–1519 (2013).
Ning, Z., Hawley, B., Seebun, D. & Figeys, D. APols-aided protein precipitation: a rapid method for concentrating proteins for proteomic analysis. J. Membr. Biol. 247, 941–947 (2014).
Mellacheruvu, D. et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 (2013).
Sardiu, M.E. et al. Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics. Proc. Natl. Acad. Sci. USA 105, 1454–1459 (2008).
Sowa, M.E., Bennett, E.J., Gygi, S.P. & Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 (2009).
Choi, H. et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 (2011).
Meysman, P. et al. Protein complex analysis: from raw protein lists to protein interaction networks. Mass Spectrom. Rev. http://dx.doi.org/10.1002/mas.21485 (2015).
R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ (2013).
Chang, W. Shiny: Web Application Framework for R. R package version 0.11 http://CRAN.R-project.org/package=shiny (2015).
Gandrud, C., Allaire, J.J. & Lewi, B.W. NetworkD3: Tools for Creating D3 JavaScript Network Graphs from R. R package version 0.1.1 http://CRAN.R-project.org/package=networkD3 (2014).
Titeca, K. et al. sfinx: an R package for the elimination of false positives from affinity purification-mass spectrometry datasets. Bioinformatics. http://dx.doi.org/10.1093/bioinformatics/btx076 (2017).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Havugimana, P.C. et al. A census of human soluble protein complexes. Cell 150, 1068–1081 (2012).
Braun, P. et al. An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods 6, 91–97 (2009).
Couzens, A.L. et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 6, rs15 (2013).
Kim, D.I. et al. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 27, 1188–1196 (2016).
Dalby, B. et al. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods 33, 95–103 (2004).
Kingston, R.E., Chen, C.A. & Okayama, H. Calcium phosphate transfection. Curr. Protoc. Cell Biol. Chapter 20 Unit 20.23 (2003).
Nesvizhskii, A.I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 73, 2092–2123 (2010).
Shteynberg, D., Nesvizhskii, A.I., Moritz, R.L. & Deutsch, E.W. Combining results of multiple search engines in proteomics. Mol. Cell Proteomics 12, 2383–2393 (2013).
Vaudel, M. et al. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 33, 22–24 (2015).
Helsens, K. & Martens, L. Enabling computational proteomics by public and local data management systems. Circ. Cardiovasc. Genet. 5, 266 (2012).
Helsens, K. et al. ms_lims, a simple yet powerful open source laboratory information management system for MS-driven proteomics. Proteomics 10, 1261–1264 (2010).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Cline, M.S. et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2, 2366–2382 (2007).
Lingappa, J.R., Reed, J.C., Tanaka, M., Chutiraka, K. & Robinson, B.A. How HIV-1 Gag assembles in cells: putting together pieces of the puzzle. Virus Res. 193, 89–107 (2014).
Sundquist, W.I. & Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2, a006924 (2012).
Klein, K.C., Reed, J.C. & Lingappa, J.R. Intracellular destinies: degradation, targeting, assembly, and endocytosis of HIV Gag. AIDS Rev. 9, 150–161 (2007).
Jouvenet, N., Bieniasz, P.D. & Simon, S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 454, 236–240 (2008).
Votteler, J. & Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 14, 232–241 (2013).
Lin, Y.C. et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 5, 4767 (2014).
Kall, L., Storey, J.D., MacCoss, M.J. & Noble, W.S. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7, 29–34 (2008).
Stumpf, M.P. et al. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 105, 6959–6964 (2008).
Acknowledgements
K.T. is a PhD student with the Agency for Innovation by Science and Technology (IWT). K.G. acknowledges support from the Fund for Scientific Research-Flanders (FWO-Vlaanderen, grant G011312N) and the Ghent University Concerted Research Actions (grant BOF14/GOA/013). J.T. was supported by grants from IUAP P6/36, the GROUP-ID MRP-UGent and the Fund for Scientific Research-Flanders (grants G.0747.10N and G.0864.10), and is a recipient of a European Research Council (ERC) Advanced Grant (Cytokine Receptor Signaling Revisited, 340941). S.E. was supported by a Methusalem grant to J.T.
Author information
Authors and Affiliations
Contributions
The Virotrap protocol was developed by S.E., E.V.Q. and K.T. with excellent assistance from N.S., A.V. and D.D.S., and support and advice from K.G. and J.T. SFINX was developed by K.T. under the guidance of J.T. and S.E. The manuscript was written by K.T., E.V.Q. and S.E., with corrections from K.G. and J.T.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Home page of the SFINX web site interface.
(a) Circular SFINX button. (b) Tabs to navigate through the website.
Supplementary Figure 2 Analysis page of the SFINX web site interface before data input.
(a) Left panel with input possibilities. (b) Right panel with short explanation. (c) Check box for the use of example files. (d) Button to browse computer and upload personal “Basic data” file. (e) Button to browse computer and upload personal “Bait Identification” file. (f) Features to specify the use of alternative input formats for the “Basic data” file. (g) Features to specify the use of alternative input formats for the “Bait Identities” file.
Supplementary Figure 3 Filtered interactions tab of the analysis page of the SFINX web site interface after data input.
(a) Circular SFINX button. (b) Tabs to navigate through the website. (a) Results display. (b) Slider bar to determine the level of strictness. (c) Alternative result tabs in the analysis page. (d) Tabular display of the detailed results of the SFINX filtering. (e) Additional information about the quality of the data and results, and buttons for downloading of the filtered interactions. (f) Toggle for the amount of displayed detailed interactions. (g) General search bar for all the detailed results. (h) Search bars specific for each column of the detailed results table.
Supplementary Figure 4 Distribution tab of the analysis page of the SFINX web site interface after data input.
(a) Distribution display. (b) Indicator of the cut-off position.
Supplementary Figure 5 Bait data tab of the analysis page of the SFINX web site interface after data input.
(a) Tabular display of the original “Bait Identities” file as observed by SFINX.
Supplementary Figure 6 Original data tab of the analysis page of the SFINX web site interface after data input.
(a) Limited tabular display of the original “Basic data” file as observed by SFINX. (b) Check box for the visualization of the complete “Basic data” file as observed by SFINX.
Supplementary Figure 7 Network tab of the analysis page of the SFINX web site interface after data input.
(a) Interactive network visualization of the filtered interactions.
Supplementary information
Supplementary Figures and Tables
Supplementary Figures 1–7. (PDF 966 kb)
Supplementary Data 1
Example file for 'Basic data' input (semicolon-separated). (CSV 263 kb)
Supplementary Data 2
Example file for 'Bait identities' input. (TXT 0 kb)
Rights and permissions
About this article

Cite this article
Titeca, K., Van Quickelberghe, E., Samyn, N. et al. Analyzing trapped protein complexes by Virotrap and SFINX. Nat Protoc 12, 881–898 (2017). https://doi.org/10.1038/nprot.2017.014
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2017.014
This article is cited by
-
Recombinant extracellular vesicles as biological reference material for method development, data normalization and assessment of (pre-)analytical variables
Nature Protocols (2021)
-
Ring finger protein 213 assembles into a sensor for ISGylated proteins with antimicrobial activity
Nature Communications (2021)
-
Monitoring protein communities and their responses to therapeutics
Nature Reviews Drug Discovery (2020)
-
A protein-protein interaction map of the TNF-induced NF-κB signal transduction pathway
Scientific Data (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
