
Abstract
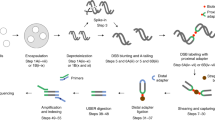
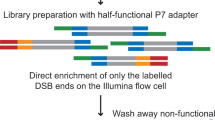
Unbiased, high-throughput assays for detecting and quantifying DNA double-stranded breaks (DSBs) across the genome in mammalian cells will facilitate basic studies of the mechanisms that generate and repair endogenous DSBs. They will also enable more applied studies, such as those to evaluate the on- and off-target activities of engineered nucleases. Here we describe a linear amplification–mediated high-throughput genome-wide sequencing (LAM-HTGTS) method for the detection of genome-wide 'prey' DSBs via their translocation in cultured mammalian cells to a fixed 'bait' DSB. Bait-prey junctions are cloned directly from isolated genomic DNA using LAM-PCR and unidirectionally ligated to bridge adapters; subsequent PCR steps amplify the single-stranded DNA junction library in preparation for Illumina Miseq paired-end sequencing. A custom bioinformatics pipeline identifies prey sequences that contribute to junctions and maps them across the genome. LAM-HTGTS differs from related approaches because it detects a wide range of broken end structures with nucleotide-level resolution. Familiarity with nucleic acid methods and next-generation sequencing analysis is necessary for library generation and data interpretation. LAM-HTGTS assays are sensitive, reproducible, relatively inexpensive, scalable and straightforward to implement with a turnaround time of <1 week.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Nussenzweig, A. & Nussenzweig, M.C. Origin of chromosomal translocations in lymphoid cancer. Cell 141, 27–38 (2010).
Alt, F.W., Zhang, Y., Meng, F.-L., Guo, C. & Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 152, 417–429 (2013).
Hendel, A., Fine, E.J., Bao, G. & Porteus, M.H. Quantifying on- and off-target genome editing. Trends Biotechnol. 33, 132–140 (2015).
Boboila, C., Alt, F.W. & Schwer, B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 116, 1–49 (2012).
Symington, L.S. & Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 (2011).
Frock, R.L. et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 33, 179–186 (2015).
Wei, P.C. et al. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell 164, 644–655.
Meng, F.-L. et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 159, 1538–1548 (2014).
Hu, J. et al. Chromosomal loop domains direct the recombination of antigen receptor genes. Cell 163, 947–959 (2015).
Dong, J. et al. Orientation-specific joining of AID-initiated DNA breaks promotes antibody class switching. Nature 525, 134–139 (2015).
Chiarle, R. et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119 (2011).
Gostissa, M. et al. IgH class switching exploits a general property of two DNA breaks to be joined in cis over long chromosomal distances. Proc. Natl. Acad. Sci. USA 111, 2644–2649 (2014).
Zhang, Y. et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148, 908–921 (2012).
O′Malley, R.C., Alonso, J.M., Kim, C.J., Leisse, T.J. & Ecker, J.R. An adapter ligation-mediated PCR method for high-throughput mapping of T-DNA inserts in the Arabidopsis genome. Nat. Protoc. 2, 2910–2917 (2007).
Williams, R. et al. Amplification of complex gene libraries by emulsion PCR. Nat. Methods 3, 545–550 (2006).
Schmidt, M. et al. High-resolution insertion-site analysis by linear amplification–mediated PCR (LAM-PCR). Nat. Methods 4, 1051–1057 (2007).
Paruzynski, A. et al. Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat. Protoc. 5, 1379–1395 (2010).
Zhou, Z.-X. et al. Mapping genomic hotspots of DNA damage by a single-strand-DNA-compatible and strand-specific ChIP-seq method. Genome Res. 23, 705–715 (2013).
Langmead, B. & Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Faust, G.G. & Hall, I.M. YAHA: fast and flexible long-read alignment with optimal breakpoint detection. Bioinformatics 28, 2417–2424 (2012).
Schwer, B. et al. Transcription-associated processes cause DNA double-strand breaks and translocations in neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 113, 2258–2263 (2012).
Kim, Y.G., Cha, J. & Chandrasegaran, S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA 93, 1156–1160 (1996).
Jinek, M. et al. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Kim, H. & Kim, J.-S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 15, 321–334 (2014).
Tsai, S.Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2014).
Wang, X. et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 33, 175–178 (2015).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Hu, J., Tepsuporn, S., Meyers, R.M., Gostissa, M. & Alt, F.W. Developmental propagation of V(D)J recombination-associated DNA breaks and translocations in mature B cells via dicentric chromosomes. Proc. Natl. Acad. Sci. USA 111, 10269–10274 (2014).
Boboila, C. et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J. Exp. Med. 207, 417–427 (2010).
Teng, G. et al. RAG represents a widespread threat to the lymphocyte genome. Cell 162, 751–765 (2015).
Klein, I.A. et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147, 95–106 (2011).
Barlow, J.H. et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632 (2013).
Khair, L., Baker, R.E., Linehan, E.K., Schrader, C.E. & Stavnezer, J. Nbs1 ChIP-seq identifies off-target DNA double-strand breaks induced by AID in activated splenic B cells. PLoS Genet. 11, e1005438 (2015).
Baranello, L. et al. DNA break mapping reveals topoisomerase II activity genome-wide. Int. J. Mol. Sci. 15, 13111–13122 (2014).
Crosetto, N. et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 10, 361–365 (2013).
Ran, F.A. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015).
Asaithamby, A. & Chen, D.J. Cellular responses to DNA double-strand breaks after low-dose -irradiation. Nucleic Acids Res. 37, 3912–3923 (2009).
Roukos, V. et al. Spatial dynamics of chromosome translocations in living cells. Science 341, 660–664 (2013).
Judo, M.S., Wedel, A.B. & Wilson, C. Stimulation and suppression of PCR-mediated recombination. Nucleic Acids Res. 26, 1819–1825 (1998).
Clepet, C. Improved full-length cDNA production based on RNA tagging by T4 DNA ligase. Nucleic Acids Res. 32, 6e–6 (2004).
Ran, F.A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Kim, D. et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 12, 237–243 (2015).
Gabriel, R. et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 29, 816–823 (2011).
Veres, A. et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 15, 27–30 (2014).
Acknowledgements
We thank members of the Alt laboratory for discussions about improving LAM-HTGTS, and we thank Z. Herbert from the Molecular Biology Core Facilities at Dana-Farber Cancer Institute for discussions on transitioning HTGTS to Illumina Miseq. This work is supported by National Institutes of Health Grant nos. R01AI020047 and R01AI077595 to F.W.A. R.L.F. was supported by the National Institutes of Health National Research Service Award no. T32AI007512. J.H. is supported by a Robertson Foundation/Cancer Research Institute Irvington Fellowship. F.W.A. is an investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
J.H., R.M.M., F.W.A. and R.L.F. wrote the manuscript with additional comments from J.D. and R.A.P. J.H. and R.L.F. designed and experimentally developed the LAM-HTGTS approach and R.M.M. wrote the translocation pipeline program. J.H., R.L.F., J.D. and R.A.P. performed experiments quoted in the manuscript that used the LAM-HTGTS approach, and all authors analyzed data that contributed to the development and/or application of the approach.
Corresponding authors
Ethics declarations
Competing interests
A patent application has been filed relating to the current LAM-HTGTS method (International Application No. PCT/US2015/061758).
Rights and permissions
About this article

Cite this article
Hu, J., Meyers, R., Dong, J. et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification–mediated high-throughput genome-wide translocation sequencing. Nat Protoc 11, 853–871 (2016). https://doi.org/10.1038/nprot.2016.043
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2016.043
This article is cited by
-
DNA polymerases in precise and predictable CRISPR/Cas9-mediated chromosomal rearrangements
BMC Biology (2023)
-
ATR kinase supports normal proliferation in the early S phase by preventing replication resource exhaustion
Nature Communications (2023)
-
Shifted PAMs generate DNA overhangs and enhance SpCas9 post-catalytic complex dissociation
Nature Structural & Molecular Biology (2023)
-
A dual function for the chromatin organizer Special A-T rich Binding Protein 1 in B-lineage cells
Cellular & Molecular Immunology (2023)
-
Multiscale reorganization of the genome following DNA damage facilitates chromosome translocations via nuclear actin polymerization
Nature Structural & Molecular Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
