
Abstract
Physiological responses to ligands such as peptides, proteins, pharmaceutical drugs or whole pathogens are generally mediated through interactions with specific cell surface protein receptors. Here we describe the application of TRICEPS, a specifically designed chemoproteomic reagent that can be coupled to a ligand of interest for the subsequent ligand-based capture of corresponding receptors on living cells and tissues. This is achieved by three orthogonal functionalities in TRICEPS—one that enables conjugation to an amino group containing ligands, a second for the ligand-based capture of glycosylated receptors on gently oxidized living cells and a biotin tag for purifying receptor peptides for analysis by quantitative mass spectrometry (MS). Specific receptors for the ligand of interest are identified through quantitative comparison of the identified peptides with a sample generated by a control probe with known (e.g., insulin) or no binding preferences (e.g., TRICEPS quenched with glycine). In combination with powerful statistical models, this ligand-based receptor capture (LRC) technology enables the unbiased and sensitive identification of one or several specific receptors for a given ligand under near-physiological conditions and without the need for genetic manipulations. LRC has been designed for applications with proteins but can easily be adapted for ligands ranging from peptides to intact viruses. In experiments with small ligands that bind to receptors with comparatively large extracellular domains, LRC can also reveal approximate ligand-binding sites owing to the defined spacer length of TRICEPS. Provided that sufficient quantities of the ligand and target cells are available, LRC can be carried out within 1 week.
Similar content being viewed by others

Introduction
Plasma membrane proteins are responsible for key biological functions, and many of these proteins act as receptors for the diverse signals that arrive at a cell and regulate its interactions with the environment. Some small hydrophobic signaling molecules are able to diffuse across the plasma membrane of a cell and activate intracellular receptors, but most biologically active ligands are hydrophilic and can activate receptor proteins only at the surface of a target cell. Thus, a typical cell in a multicellular organism is exposed to myriad different ligands in its extracellular microenvironment and must respond to this babel of signals selectively and according to its own specific character by expressing a specific set of cell surface receptors—the cellular surfaceome. In analogy, ligands have to interact exclusively with one or only a few receptors of the surfaceome in order to serve their specific biological or pharmaceutical purpose. Accordingly, the identification of a ligand-receptor interaction can often present immediate new research directions and provide invaluable information toward the understanding of mechanisms such as signal transduction, drug action, off-target effects or disease related to a ligand1,2.
Even though physical interactions of ligands with specific cell surface receptors are of particular biological and pharmacological importance, the detection of such interactions in an unbiased and direct fashion remains a difficult task. (Immuno)affinity-based techniques, such as affinity purification MS, now enable the large-scale identification of protein-protein interactions in cell lysates3,4,5,6,7. However, such approaches are usually limited to high-affinity interactions and will typically fail when involving plasma membrane proteins that are often hydrophobic and present in relatively low abundance8,9. Other technologies, such as yeast two-hybrid10,11,12 and protein array–based technologies13,14,15, are similarly unsuitable for identifying transient extracellular interactions that involve biochemically intractable membrane proteins16. An alternative in vitro technology makes use of pentamerized recombinant ectodomains of cell surface proteins produced in mammalian cells for avidity-based extracellular interaction screens (AVEXIS)17,18. This method improves the sensitivity of detection for low-affinity interactions and takes into account the fact that many cell surface proteins depend on functionally important post-translational modifications such as disulfide bonds and large hydrophilic glycans. Although very useful for interaction studies, AVEXIS and related technologies are limited to proteins with self-contained extracellular domains and may fail to detect interactions with plasma membrane proteins that need to be embedded in their natural environment, that is, within living cells or tissues, to fold into their native structures and exhibit their characteristic binding properties19,20,21,22,23,24,25.
Development of the LRC technology
In attempts to overcome the problems inherent in the analysis of plasma membrane proteins, we and others have previously used bifunctional chemoproteomic reagents to biotinylate carbohydrate-containing proteins on living cells26,27, followed by the identification of the corresponding glycopeptides using MS26,28,29,30,31. These cell surface capture (CSC) technologies rely on (i) gentle oxidation of the oligosaccharide chains of glycoproteins on living cells to generate aldehyde groups that are typically absent from cell surfaces, (ii) tagging of the aldehyde groups with bifunctional reagents that contain hydrazide and aminooxy active groups, (iii) cell lysis and trypsin proteolysis of the tagged proteins, (iv) affinity purification of tagged glycopeptides, (v) specific release of the N-linked peptides using the endoglycosidase PNGase F and (vi) analysis of the resulting samples by high mass accuracy MS. By using these technologies, several hundred plasma membrane proteins and their N-glycosites can be identified and quantified routinely in a single experiment with living cells, enabling the characterization of the surfaceome of virtually any cell line or tissue. On the basis of the insights gained from CSC strategies, we hypothesized that specifically designed trifunctional reagents would enable a ligand-based capture of cell surface glycoproteins on living cells and the subsequent identification of specific target receptors using quantitative MS. The resulting LRC technology has previously been published32 and was developed further and described in more detail for this manuscript.
Essential for the establishment of LRC was the design, synthesis and application of a novel biocompatible chemoproteomic reagent that incorporates three orthogonal functionalities: (i) a protein-reactive group enabling conjugation to ligands of interest, (ii) an aldehyde-reactive group for the spatially and temporally separated, ligand-based capture of oxidized glycoprotein receptors on living cells and (iii) an affinity handle for the purification of captured receptor glycopeptides for MS analysis. In addition to modularity in the design of the reagent, special emphasis had to be placed on water solubility, conformational flexibility, spacer length, and the gentle and efficient selective addressing of the ligand coupling and receptor capture reactions. Specifically, the final reagent contains: (i) an N-hydroxysuccinimide (NHS) ester, enabling conjugation to a ligand of interest, (ii) a trifluoroacetyl-protected hydrazine to capture oxidized glycoprotein receptors and (iii) a biotin group for the affinity purification of captured glycopeptides (Fig. 1). In analogy to its three-headed nature, we termed this novel trifunctional chemoproteomic reagent TRICEPS, which is Latin for 'three-headed'.

The reagent used for LRC experiments contains an NHS ester for coupling to ligands via primary amines, a trifluoroacetyl-protected hydrazine for the subsequent capture of glycoprotein receptors on living cells via aldehydes introduced into carbohydrates by mild oxidation and a biotin group for the affinity purification of captured glycopeptides. Flexibility is provided by the stochastic coupling of TRICEPS to topologically different sites on ligands, the long (∼48 Å) and flexible spacer between the reactive functions and the presence of typically more than one aldehyde per carbohydrate structure after oxidation. MW, molecular weight.
The NHS ester was a promising choice for the coupling of TRICEPS to ligands under physiological conditions, as it reacts readily with primary amines in pH 7–9 buffers to form stable amide bonds33. In protein and peptide ligands, several primary amines are usually present in the form of the α-amine group of the N-terminus and the ɛ-amine groups of lysine residues. These amine groups are typically exposed on the surface of proteins and enable a fast, easy and versatile coupling of TRICEPS to ligands. For the capture of oxidized glycoprotein receptors, we chose to work with aromatic hydrazines that are known to react readily and chemoselectively with aldehydes to create hydrazone linkages under slightly acidic conditions34. The crucial selective addressing of the two reactive functions in TRICEPS was achieved by protection of the nucleophilic hydrazine as the corresponding trifluoroacetamide. This precluded unwanted side reactions with electrophiles such as the reagent's own active ester group before or during ligand coupling. In contrast to other combinations of reactive or protecting groups tested, the trifluoroacetylated hydrazines were observed to undergo condensation with carbohydrate-derived aldehydes without prior deprotection. Notably, this circumvents the need for removing the protecting group under the typically prescribed harsh conditions, which are generally incompatible with coupled protein ligands.
We next established a parallel workflow for LRC using two samples (Fig. 2). In one sample, the TRICEPS-coupled ligand predominantly directs the hydrazine function of the reagents toward oxidized glycans of ligand-specific cell surface glycoprotein receptors. In parallel, an equimolar amount of TRICEPS is either quenched with glycine or coupled to a control ligand with known or no binding preferences to control for the nonspecific capture of cell surface proteins on the basis of their relative abundance on a cell line or tissue. After incubation of oxidized cells with TRICEPS-containing samples, the cells are lysed and enzymatically digested with trypsin and TRICEPS-captured peptides are isolated using streptavidin beads. Purified N-glycopeptides are specifically released from the beads through an enzymatic cleavage with PNGase F. This endoglycosidase cleaves between the proximal N-acetylglucosamine and the asparagine of the glycopeptide in the N-X-S/T glycosylation site motif (where N stands for asparagine, X stands for any amino acid except proline and S/T stands for serine or threonine, respectively). Besides releasing the glycosylated peptides, PNGase F treatment also results in the enzymatic deamidation of the glycosylated asparagine to aspartic acid. This generates a +0.984-Da mass shift and introduces the specific N[115]-X-S/T motif (where 115 is the mass of the deamidated Asn residue) in formerly N-glycosylated cell surface peptides. This LRC protocol leads to the generation of two virtually identical peptide samples that can be analyzed with a high-mass-accuracy mass spectrometer. The identified peptides are filtered for the presence of N[115]-X-S/T motifs, and the relative concentrations of cell surface glycopeptides in the ligand sample are compared with those in the control sample using MS1-based label-free quantification. Identified peptides of random cell surface glycoproteins are expected to have equal concentrations in both samples, whereas peptides of the corresponding receptors are specifically enriched in the ligand sample. The long but defined spacer between the reactive functions in TRICEPS provides flexibility for LRC but still enables an approximate localization of ligand-binding sites for small ligands that bind to receptors with comparatively large extracellular domains. TRICEPS with shorter spacers have proven less suitable in proof-of-concept applications of the technology, whereas TRICEPS with longer spacers have not been synthesized and may be suitable for applications of the technology with peptide and small-molecule ligands.

Once a sufficient amount of purified ligand and target cells have been prepared, LRC experiments can be carried out within a week provided that the produced samples can be measured immediately by MS. (a) In the first step of the procedure, the purified ligand of interest is coupled to TRICEPS and an equimolar amount of TRICEPS is quenched with glycine (or coupled to a control ligand with known or no binding preferences) in the control reaction. Living cells or tissues are activated by gentle oxidation with periodate and cells are incubated with TRICEPS-coupled ligands for LRC and stochastic biotinylation of random cell surface glycoproteins according to their abundance. (b) Cells are lysed using indirect sonication or detergent-based methods, and the proteins in the resulting lysates are digested with trypsin overnight. The resulting peptide samples are incubated with a streptavidin resin for biotin-mediated affinity enrichment of TRICEPS-captured glycopeptides. (c,d) The streptavidin resin is transferred to filter columns (c) and washed with different buffers (d). (e,f) After washing, cell surface N-glycopeptides are released by PNGase F treatment overnight and the resulting peptide samples are C18 purified (e) for analysis by MS (f). Acquired MS spectra are searched against a protein database and identified peptides are filtered for the presence of the N[115]-X-S/T motif. (g,h) Glycopeptide ion signals are quantified using an MS1-based label-free quantification (g) and statistical analysis of the resulting data sets enables the identification of specific receptor enrichments in the ligand sample compared with the control (h).
Advantages of the LRC technology
A primary advantage of the LRC strategy is that affinity-based interactions are detected in native biological systems by stabilizing ligand-receptor complexes with new covalent bonds, whereas they are present in their original cellular microenvironment. Furthermore, ligand-receptor interactions are stabilized without any washing steps in between. Thus, even weak and transient interactions that require properly folded integral plasma membrane proteins can be captured for subsequent analysis by MS. After receptor capture, the combination of an affinity handle and the chemically reactive groups in a single trifunctional reagent allows for an early tryptic digest of ligand-receptor conjugates without the loss of receptor tagging. This circumvents the problems inherent in the affinity enrichment of membrane proteins and avoids unspecific interactions of exposed hydrophobic domains during purification. Accordingly, peptide-level purification leads to clean samples with only few washing steps, and purified N-glycopeptides can be released through a specific enzymatic cleavage with the endoglycosidase PNGase F. After peptides in the samples have been identified by MS, only peptides carrying the N[115]-X-S/T motif are quantitatively compared with the ones in the control samples via a label-free quantification. In this way, only specifically captured and purified, formerly N-glycosylated cell surface peptides are considered for the detection of ligand-receptor interactions.
To detect ligand-based receptor enrichments, the identified cell surface N-glycoproteins in LRC samples have to be quantified and protein abundances have to be compared between samples. However, the primary output of an LRC experiment is not a list of quantified proteins but a list of quantified spectral features representing formerly N-glycosylated peptides. To make protein-level conclusions, one can take an average of feature abundances to obtain a single protein quantity for each sample. This approach may be sufficient to estimate protein abundances and ratios, but it neglects the independent enrichment information provided by different unique glycopeptides matching the same protein because of the peptide-level affinity purification. Therefore, this approach is inadequate when several unique peptides are identified for a given protein or when biological or technical replicates for the ligand and control samples have been obtained. To increase the informative value of such experiments, LRC data sets can be analyzed with a statistical ANOVA model35. This model assumes that the measurement errors follow Gaussian distributions and views individual features as replicate measurements of a protein's abundance and explicitly accounts for this redundancy. This analysis considerably increases the sensitivity and specificity of testing and reports false-discovery rate (FDR)-adjusted P values to rank receptor candidates in discovery-driven applications of the technology. By using this LRC protocol and analysis pipeline, ligand-receptor interactions can be detected in an unbiased fashion on virtually any cell line or even on subpopulations of cells within complex primary tissues. We have had success with virtually any type of lysine-containing ligand such as peptide ligands (e.g., apelin-17), small proteins (e.g., epidermal growth factor), medium-sized proteins (e.g., transferrin), and large proteins (e.g., antibodies against CD44, ERBB2 and PRNP), as well as with intact viruses (e.g., vaccinia virus).
General design considerations for LRC experiments
When setting up an LRC experiment, it is important to consider the number of samples one is willing to or able to produce. This usually depends on the availability of the ligand of interest (ideally 100–200 μg ligand per LRC sample) and target cells (ideally 108 cells per LRC sample). Although we have found that a single comparison of one ligand sample to one control sample can lead to the successful identification of one or several true receptors for a given ligand, we highly recommend performing discovery-driven LRC experiments in biological triplicate samples (three ligand samples versus three control samples). This setup accounts for technical and biological noise and eventually allows for a thorough statistical analysis of the resulting data and the identification of high-confidence receptor candidates. Once the scale of an LRC application is determined, experiments can be carried out with many types of ligands and target cells according to the following procedure with only minimal adaptations. However, there are a few things to consider when you are planning and carrying out the individual steps of ligand coupling, receptor capture and receptor identification.
Ligand coupling
TRICEPS. The TRICEPS reagent is a yellowish fluffy solid, and it must be desiccated owing to its hydrolytic instability (hydrolysis of the NHS ester). When kept under argon gas at −20 °C, the reagent can be stored for 1 year without considerable loss of activity. Because of the limited immediate water solubility and hydrolytic instability of TRICEPS, concentrated working stock solutions (typically 100 mM) are prepared in DMSO and stored at −20 °C. Stock solutions in dry organic solvent are stable for storage and reagents can easily be added to coupling reactions in order to obtain the desired final concentration.
Coupling buffer. TRICEPS conjugation with lysine side chains is the predominant coupling reaction and is favored under alkaline conditions (pH 7.4–pH 9). As a compromise between conditions favoring the reactivity of the NHS ester functionality and conditions compatible with protein ligands, TRICEPS coupling reactions are ideally carried out in 25 mM HEPES (pH 8.2) for 60–120 min. Buffers that contain primary amines such as Tris buffer must not be used for the coupling reaction. Other alkaline pH conditions, alternative buffers that do not contain primary amines, and/or shorter incubation times can be applied when working with delicate ligands.
Coupling ratio. When setting up a coupling reaction with TRICEPS, it is desirable to have a high TRICEPS:ligand ratio in order to increase the number of receptor-carbohydrate structures that can be captured per ligand on average. At the same time, it is important to only minimally impair the biological activity (i.e., the capability to bind and activate corresponding receptors) of a majority of coupled ligands. On the basis of the results obtained with ligands ranging from peptides to antibodies, a general coupling ratio of 50 μg of TRICEPS per 100 μg of ligand is considered appropriate. Higher coupling ratios can be applied when only a limited amount of ligand is available, but it is not recommended to use <50 μg of ligand for LRC experiments (ideally 100–200 μg). Further, it is highly recommended to use at least 40 μg of TRICEPS in every LRC sample in order to ensure sufficient capture of cell surface glycoproteins for later identification by MS.
Coupling volume. Hydrolysis of the NHS ester in TRICEPS is a major competing reaction of the coupling reaction36,37. When setting up a coupling reaction with a given TRICEPS:ligand ratio, one should take into account that hydrolysis occurs more readily in dilute protein solutions, whereas the coupling reaction is favored in more concentrated solutions. Accordingly, the actual coupling ratio (average number of TRICEPS attached to a ligand protein) is substantially lower compared with the ratio used to set up the reaction and depends on the coupling volume. LRC coupling reactions are ideally set up with 100 μg of ligand per 50–100 μl of coupling buffer. With ligands that tend to form aggregates, more dilute protein solutions can be used and higher coupling ratios (60 μg of TRICEPS per 100 μg of ligand) can be applied to compensate for excessive hydrolysis of the reagent.
Ligand functionality. In every LRC experiment, a fraction of ligands may be compromised by the attachment of TRICEPS to particular lysines that are sterically or otherwise important for the respective ligand-receptor interaction. As the coupling of TRICEPS to lysine side chains is unspecific and several lysines are typically present in protein ligands, only high TRICEPS:protein ratios will lead to an impairment of the majority of ligands. In experiments with appropriate TRICEPS:protein ratios, hydrolyzed TRICEPS and the fraction of ligands that is compromised by the coupling to reagents will simply contribute to the random capture of cell surface proteins (the vast majority of capture events in every LRC sample) so that there is no need to remove hydrolyzed TRICEPS or compromised ligands. However, we recommend testing ligands for biological activity after coupling to TRICEPS even at the recommended ratio, especially when you are working with peptide ligands with only one or few primary amines. This will assure that at least a fraction of ligands remains biologically active for successful LRC after coupling. In addition, the biotin group in TRICEPS can be used to monitor binding of coupled ligands to cells using streptavidin-conjugated fluorophores in flow cytometry or microscopy-based assays. For LRC applications with intact viruses, we recommend using at least 5 × 108 viruses (the number of infectious particles can be lower) per sample and performing preliminary infection assays after coupling viruses to TRICEPS at different ratios as described previously32.
Receptor capture
Cells. LRC experiments can be performed with 107 cells per sample, but it is highly recommended to use up to 108 cells for every sample. As different cell lines show considerable variation in cell size, the total amount of cellular material rather than the number of cells is likely to be the crucial factor. When working with adherent cells, good results have been obtained with virtually any cell line tested using four cell culture dishes (140-mm diameter) of almost confluent cells per sample. The more cells are used per sample the better, as higher cell numbers generally lead to higher numbers of identified glycopeptides, higher and more robust glycopeptide MS signals and a better sensitivity of the technology in general. Adherent cells can be left attached to cell culture dishes for the incubation with ligands, but usually cells are scraped off plates first so that all of the following steps can be done in centrifuge tubes.
Tissues. One of the advantages of the LRC protocol is the potential to detect interactions of ligands with receptors that are only present on subpopulations of cells within complex primary target tissues. Successful LRC experiments have been performed with different tissues, such as frozen human tumor tissue slices38, freshly dissected mouse adipose tissues and cultured mouse brain slices39, suggesting that the LRC technology can be applied to most if not all tissue types. We recommend mechanically disrupting tissues or performing an initial collagenase digestion to increase the direct cell surface accessibility for ligands. Collagenase digestion is recommended only if tissues are otherwise hardly accessible for soluble ligands, as this treatment can potentially affect cell surface receptors through direct or indirect mechanisms. Tissue fragments can then be processed without further dissociation to partly conserve the 3D tissue structure during the incubation with ligands.
Receptor capture. TRICEPS-coupled ligands are ideally incubated with cells in PBS (pH 6.5) for 90 min under constant agitation. The incubation is ideally performed at 4 °C but receptor capture can also be carried out at room temperature (20–25 °C) or at 37 °C in cases where temperature is thought to be important for receptor binding. Similarly, salts such as magnesium sulfate can be added if they are known to be beneficial for the stability of a ligand or its interaction with cell surface receptors. Higher pH conditions can be applied if crucial for binding of ligands to cells. However, it is notable that the LRC protocol is efficiently fine-tuned by specific pH conditions, because amide formation by the NHS ester (ligand coupling), hydrazone formation by the hydrazine (receptor capture) and stability of the hydrazone linkage (glycopeptide purification) are highly pH dependent. As described above, amide formation in the coupling reaction is efficient under alkaline conditions only. In contrast, hydrazone formation in the receptor capture reaction is efficient under acidic conditions but decreases sharply under basic conditions34. Once they have formed, hydrazones are relatively stable at pH > 7 and cells should be resuspended in a buffer (pH 8) after receptor capture.
Control reaction. In every LRC experiment, a control sample accounts for nonspecific binding of a ligand on the basis of the relative abundance of a random glycoprotein on a cell line or tissue. In the simplest conceivable LRC setup, a ligand of interest is coupled to TRICEPS and an equimolar amount of reagent is quenched with glycine and used to generate the control sample (Fig. 3a,b). However, it is highly recommended to use a ligand with known binding preferences in the control reaction so that the identification of positive control receptors can serve as a quality control for the entire experiment. By using insulin as a ligand in the control reaction, the insulin receptor and/or the insulin-like growth factor 1 receptor have been identified as positive controls on virtually any cell line or tissue tested. After the identification of a receptor for a ligand of interest, the control reaction can also be used to derive additional information about the protein domains or other structural elements of the ligand that are important for the interaction with the receptor (Fig. 3c,d).

Enrichment plots can be produced for comparisons of one ligand sample versus one control sample to detect specific enrichments of receptor candidates. (a) LRC with insulin and quenched TRICEPS on Jurkat T cells. Data are shown on the peptide level. (b) Same experiment as in a with data shown on the protein level. (c) LRC with insulin and a glycosylated ligand-IgG Fc fusion protein on Jurkat T cells. The Fc part of the fusion protein was identified as the individual protein IGHG1. Data are shown on the protein level. (d) LRC with the same ligand-IgG Fc fusion protein as in c but with the IgG Fc part of the fusion protein used as a control ligand in the control sample. The receptor candidate identified in c is still enriched, suggesting that binding is indeed mediated by the ligand and not the IgG Fc part of the fusion protein. a.u., arbitrary units. INSR, insulin receptor peptides/proteins.
Spatial flexibility. An initial concern in the development of TRICEPS was the steric accessibility of receptor carbohydrate structures in relation to the ligand-binding sites on different receptors. Accordingly, different versions of TRICEPS were synthesized that varied in their spacer length between the reactive moieties. The current reagent with a long (∼48 Å) and flexible spacer between the reactive moieties consistently proved most suitable in experiments with different ligands. Additional flexibility comes from the stochastic coupling of TRICEPS to topologically different sites on ligands and the presence of typically more than one aldehyde in a single receptor carbohydrate structure after mild oxidation. Because of the multiple degrees of freedom, ligand-coupled reagents typically reach several receptor N-glycosites, if existing. At the same time, TRICEPS experiments allow for an approximate localization of ligand-binding sites in cases where rather small ligands bind to receptors with large extracellular domains and several glycosites.
Receptor identification
Cell disruption. The ideal cell lysis protocol for LRC experiments breaks open cells but leaves nuclei intact such that the nuclear fraction can be removed by centrifugation before tryptic digest of the sample. Ideally, cells are disrupted through indirect sonication with an ultrasonic device. As this step is cell line dependent, it is highly recommended to optimize the disruption procedure before the initiation of an actual LRC experiment. When optimizing the procedure, short sonication cycles can be followed by centrifugation of the sample at 200g to pellet cells that are not yet disrupted. If intact cells are still present in the sample, additional sonication cycles can be performed. Other methods for cell disruption, such as detergent- or osmosis-based methods, can also be applied as long as they are compatible with the tryptic digestion of the resulting sample. When you are working with primary tissues, generally both detergent and mechanical lyses are required in order to effectively disrupt the tissue and lyse the cells.
Liquid chromatography-MS (LC-MS) analysis. LRC samples for MS analysis are of rather low complexity (300–800 total peptides are typically identified per sample) but need to be analyzed with a highly sensitive, high mass accuracy mass spectrometer such as a LTQ Orbitrap instrument (Thermo Scientific) or a comparable platform. Convincing LRC results have been obtained when peptides were identified using the SEQUEST algorithm40 and quantified using MS1-based label-free quantification using Progenesis LC-MS software (Nonlinear Dynamics)41. However, any other MS data analysis pipeline for the identification and label-free quantification of peptides should lead to comparable results. For comparisons of a single ligand and a single control sample, we recommend analyzing samples in technical triplicates (replicate LC-MS measurements of the same sample). This will allow for statistical evaluation of the experiment and will account for technical noise introduced during the acquisition of individual samples.
Statistical analysis. The preliminary output of an LRC experiment is not a list of quantified proteins but a list of quantified spectral features that represent glycopeptides. For the detection of proteins that are enriched in ligand samples compared with control samples, it is highly recommended to use the statistical model provided. This model views individual features as replicate measurements of a protein's abundance and explicitly accounts for this redundancy. Thereafter, it tests each protein for differential abundance in all pairwise comparisons of ligand and control samples and reports the P values of the tests. In the last step of the analysis, P values are adjusted for multiple comparisons to control the experiment-wide FDR at a desired level. As a result, this model adds confidence to receptor identifications in cases where several independent peptides representing the same receptor candidate are enriched in the ligand samples. This type of analysis considerably increases the sensitivity and specificity of testing and reports FDR-adjusted P values to rank receptor candidates in experiments that were carried out in biological or technical triplicate samples.
Data visualization. The processed LRC data of experiments that had been carried out in biological or technical triplicates can be presented in the form of a volcano plot, which plots significance versus fold-change on the y and x axes, respectively. Specifically, the adjusted P value obtained for every protein is plotted against the log2 of the magnitude of the fold enrichment. This enables quick visual identification in a single scatter plot of those proteins that show large-magnitude changes (fold-change >4) and are also significantly enriched (adjusted P value <0.01). This area in the scatter plot is considered the receptor space for positive control receptors and high-confidence receptor candidates. Notably, the number of identified glycopeptides for a given protein can influence the P value of the respective protein enrichment. Random glycoproteins that are only moderately enriched (fold change <4) but identified with many glycopeptides can easily get adjusted P values <0.01. In contrast, true positive receptor candidates that contain only one glycosite can be enriched substantially but will rarely get an adjusted P value <<0.01. For the final selection of receptor candidates for follow-up investigations, all proteins in the receptor space should thus be considered.
Data interpretation. The different enrichment ratios obtained for receptor glycopeptides can partly be caused by differences in the efficiency of the LRC reaction, i.e., because of the spatial arrangement of captured glycans in relation to the binding site of a ligand. Thus, it is advisable to additionally examine peptide-level volcano plots to detect specific glycopeptide enrichments. This can improve the sensitivity of testing in cases where not all of the identified glycosites of a receptor are sterically accessible by ligand-coupled TRICEPS. Glycosite-level volcano plots are also of special interest as they can contain ligand-binding site information for small ligands that bind to receptors with large extracellular domains. Once an LRC experiment has been carried out, glycoprotein-level, glycosite-level as well as glycopeptide-level volcano plots can be highly informative and should be inspected individually and thoroughly.
Limitations
The protein-directed ligand coupling reaction and the carbohydrate-directed receptor capture reaction of TRICEPS enable the efficient selective addressing of the two chemical functions in space and time. Another advantage of this strategy is that receptor capture is targeted away from receptor polypeptide domains that are important for interactions with the ligand. In contrast, only sterically accessible glycans are captured and only N-glycopeptides are subsequently released and processed for analysis by MS such that only N-glycoproteins can be identified as target receptors for ligands of interest.
The LRC protocol leads to a modification of ligands at primary amines. This treatment is likely to compromise a fraction of ligands in the coupling reaction (ligands in which the corresponding lysine residues are crucial for the interaction with a specific receptor). The fraction of ligands that remains functional after coupling (ligands that are coupled to TRICEPS via lysines that are not essential for the interaction with a specific receptor) is usually sufficient for successful receptor identification. However, coupling to TRICEPS can be problematic at high TRICEPS:ligand ratios and with ligands that only carry few primary amines. It is thus recommended to test whether a desired biological effect of a ligand is still observed after coupling of the ligand to TRICEPS at a given ratio. In addition, it is important to keep in mind that the gentle oxidation of cells leads to a modification of some components in the carbohydrate structures of cell surface receptors. Although this modification is not expected to interfere with most ligand-receptor interactions, a detrimental effect in some cases cannot be excluded.
The amine-reactive NHS ester in TRICEPS enables generic conjugation to amino group containing ligands of interest and LRC applications can directly be carried out with virtually any protein or peptide ligand with only minimal adaptations of the standard procedure. Small molecule–based LRC applications have not been carried out yet but are theoretically feasible through direct chemical derivatization of ligands or the attachment of chemical functionalities that enable coupling to TRICEPS. However, such strategies will typically lead to a uniform modification of coupled ligands at a particular site, and ligands have to be tested for functionality after derivatization. In addition, the number of receptor glycosylation sites that can be reached on average is likely to be reduced compared with LRC applications with larger protein ligands.
LRC technology can detect interactions of ligands with receptors that are embedded in their natural environment. Accordingly, LRC can lead to the identification of receptors that form and function as heterodimers or hetero-oligomers when present on living cells. During the incubation with ligands, the receptor capture reaction of TRICEPS reflects the respective equilibrium state between hydrazone formation and degradation, with the potential to capture receptor constellations over time. However, it remains to be shown whether the technology can also detect interactions where receptor oligomer formation is only induced upon ligand binding. Furthermore, LRC technology may fail to detect specific ligand-receptor interactions in cases where a ligand has an additional general affinity for molecular structures on the cell surface other than specific target glycoproteins.
LRC technology has the potential to decode complex virus-host cell interactions, in which multiple viral and cellular factors each contribute to successful and potentially cooperative binding and infection. Although the LRC technology provides a unique opportunity to identify defined sets of host receptor candidates, the enrichments of individual receptors are typically weaker than in experiments with comparatively simple ligands. This is expected owing to the comparatively large size of viruses and the many-to-many types of interactions involved between the multiple viral and host factors. Accordingly, such experiments are expected to reveal small lists of lower-confidence receptor candidates rather than single high-confidence receptors.
LRC technology may lead to the identification of false-positive receptor candidates, especially when applied under the nonstandard conditions described above. The LRC analysis pipeline has been optimized to lead to the enrichment of specific receptor candidates and the statistical model provided can be used for the identification and ranking of such candidates. However, the resulting data plots have to be analyzed carefully and more stringent receptor spaces can be defined on the basis of the scattering of the data and the identification of positive control receptors (e.g., insulin receptor). Eventually, the functional relevance of receptor candidates identified in LRC applications should be investigated with targeted and potentially combinatorial follow-up experiments, such as siRNA or antibody-based approaches. These approaches cannot be generalized and for every LRC application the type of follow-up experiment will depend on the type of ligand, the biological context and the tools available for the system under study.
Materials
REAGENTS
-
Acetonitrile LC-MS (Fisher Scientific, cat. no. A955-212)
-
Ammonium bicarbonate (Sigma-Aldrich, cat. no. 40867)
-
Benzonase nuclease HC (Merck Millipore, cat. no. 71205)
-
DMSO (Sigma-Aldrich, cat. no. D2438)
-
Formic acid, 98–100% (vol/vol) (Merck Millipore, cat. no. 100264)
-
Glycine (Sigma-Aldrich, cat. no. 50050)
-
Glycerol (Honeywell, cat. no. 10314830)
-
HEPES, 1 M buffer solution, 100 ml (Invitrogen, cat. no. 15630-056)
-
Insulin solution, human (Sigma-Aldrich, cat. no. I9278)
-
Iodoacetamide (IAA; Sigma-Aldrich, cat. no. I6125)
-
Methanol (Sigma-Aldrich, cat. no. 65543)
-
PBS, pH 7.4 (Invitrogen, cat. no. 10010-015)
-
Phosphoric acid (Sigma-Aldrich, cat. no. 04107)
-
PNGase F, glycerol free (New England Biolabs, cat. no. P0705)
-
RapiGest SF surfactant (Waters, cat. no. 186002123)
-
Sodium chloride (NaCl; Merck Millipore, cat. no. 106404)
-
Sodium bicarbonate (Sigma-Aldrich, cat. no. 13433)
-
Sodium hydroxide (NaOH; Sigma-Aldrich, cat. no. 221465)
-
Sodium (meta)periodate (Sigma-Aldrich, cat. no. S1878)
-
Streptavidin Plus UltraLink resin (Thermo Scientific, cat. no. 53117)
-
Tris(2-carboxyethyl)phosphine (TCEP; Thermo Scientific, cat. no. 20490)
-
Triton X-100 (Sigma-Aldrich, cat. no. X100)
-
Trypsin from bovine pancreas (Sigma-Aldrich, cat. no. T1426)
-
Water, high-purity, obtained from a Milli-Q integral water purification system (Merck Millipore)
-
Water, HPLC gradient grade (Fisher Scientific, cat. no. 10449380)
EQUIPMENT
-
C18 analytical column, PicoFrit (New Objective, cat. no. PF360-75-10-N-5)
-
C18 AQ Magic, 200 Å pore size, 3-μm particle size (Michrom, cat. no. PM3/61200/00)
-
C18 Ultra Micro SpinColumns (Harvard Apparatus, cat. no. 74-7206)
-
Centrifugal vacuum concentrator, acid-resistant CentriVap (Labconco)
-
Centrifuge 5415R (Eppendorf)
-
Eppendorf Safe-Lock micro test tubes (Sigma-Aldrich, EP0030120086)
-
Filter (large) 35-μm pore size for Mobicol (MoBiTec, cat. no. M523515)
-
HPLC system, EASY-nLC II (Thermo Scientific)
-
LTQ Orbitrap mass spectrometer (Thermo Scientific)
-
Milli-Q integral water purification system (Merck Millipore)
-
Mobicol 'Classic' with one closed screw cap (MoBiTec, cat. no. M1003)
-
NanoDrop 1000 spectrophotometer (Thermo Scientific)
-
AutoRep M repeater pipette (Rainin)
-
Thermomixer comfort (Eppendorf)
-
Tube rotator MACSmix (Miltenyi Biotec)
-
Ultrasonic bath, Sonorex Super (Bandelin)
-
Vac-Man laboratory vacuum manifold (Promega, cat. no. A7231)
-
Vac-Master manifold (Biotage, cat. no. 121-1016) with a PTFE stopcock/needle unit (Biotage, cat. no. 121-0001)
-
VialTweeter UIS250v (Hielscher)
-
Vortex mixer (VWR)
REAGENT SETUP
Critical
All of the steps of the LRC protocol such as incubations are usually carried out at room temperature or at 4 °C as specified for every step. A bucket of ice is required to cool cells and reagents when working at the bench.
TRICEPS reagent
-
The TRICEPS reagent is commercially available from Dualsystems Biotech (http://www.dualsystems.com) and from the corresponding authors for scientific collaborations.
Ligand of interest (day 1)
-
Ligands of interest are usually produced recombinantly or obtained from various commercial sources. At least 50 μg (ideally 100–200 μg) of ligand should be used per sample, and it is recommended to perform LRC experiments with biological triplicates (three ligand samples and three control samples). Ligands are ideally provided in lyophilized form or at a concentration of 100 μg of ligand per 50–100 μl of buffer in 25 mM HEPES (pH 8.2). For the coupling reaction with TRICEPS, ligand buffers must not contain primary amines and ligands have to be dialyzed if the composition of a buffer is unknown. It is highly recommended to include a control ligand (ideally insulin) in LRC experiments. Other commercially available control ligands with which successful LRC experiments have been carried out include epidermal growth factor, transferrin and a number of antibodies against cell surface proteins. Ligands should ideally be compatible with standard LRC buffers for TRICEPS coupling (pH 8.2) and incubation with oxidized cells (pH 6.5). For LRC applications with intact viruses, established virus preparation protocols may have to be scaled up to produce sufficient numbers of viruses (at least 5 × 108 per sample).
Caution
Make sure that the necessary laboratory safety requirements are met when working with intact viruses.
Target cells or tissue (day 1)
-
LRC experiments can be carried out with 107 cells per sample, but we highly recommend using up to 108 cells for every sample and performing experiments with biological triplicates (three ligand samples and three control samples). Different adherent cell lines show considerable variation in cell size and good results are usually obtained using four cell culture dishes (140 mm in diameter) of almost confluent cells per sample. The more cells are used per sample the better, as higher cell numbers generally lead to more robust MS peptide signals and to higher numbers of identified cell surface glycoproteins. Particularly good results have been obtained with Jurkat T, U2OS, U-251, HEK293, HepG2, Huh-7, A549 and BT-474 cells. When you are working with primary tissues, the methods for obtaining the target tissue may vary considerably depending on the species and type of tissue. Good results have been obtained with primary human hepatocytes obtained from a commercial source (Celsis, cat. no. M00995), frozen human breast carcinoma tissue that had been cut into 50-μm-thick slices38 and cultured mouse brain tissue slices39.
Caution
Make sure that all relevant safety and ethical requirements are met when working with primary tissue samples.
HEPES, 25 mM (pH 8.2; day 1)
-
Dilute 1 M HEPES with HPLC water and adjust the pH to 8.2 with NaOH. Store HEPES at 4 °C for up to 6 months.
PBS (pH 6.5; day 1)
-
Acidify 500 ml of PBS (pH 7.4–pH 6.5) by adding 105 μl of 85% (vol/vol) phosphoric acid. Store PBS at 4 °C for up to 6 months.
Ammonium bicarbonate, 50 mM (day 1)
-
Prepare 50 mM ammonium bicarbonate in high-purity Milli-Q water. Always freshly prepare the solution as pH (∼pH 7.8) increases over time. Store the solution at room temperature for no more than 2 d.
TCEP, 100 mM (day 1)
-
Prepare 100 mM TCEP in 500 mM ammonium bicarbonate in HPLC water and store it at −20 °C for up to 6 months.
IAA, 500 mM (day 1)
-
Prepare 500 mM IAA in HPLC water and store the solution at −20 °C in the dark for up to 6 months.
NaCl, 5 M (day 2)
-
Prepare 5 M NaCl in high-purity Milli-Q water, and then heat the solution under constant stirring until the NaCl is completely dissolved. Store the solution at room temperature for up to 6 months.
Detergent buffer (day 2)
-
Prepare the detergent buffer by mixing 150 mM NaCl, 1% (vol/vol) glycerol, 1% (vol/vol) Triton X-100 and 50 mM ammonium bicarbonate in high-purity Milli-Q water. Store the buffer at room temperature for up to 6 months.
Sodium bicarbonate 100 mM (pH 11) (day 2)
-
Dissolve sodium bicarbonate in high-purity Milli-Q water and adjust the pH to 11 with NaOH. Store the solution at room temperature for up to 6 months.
Formic acid, 10% (vol/vol) (day 3)
-
Prepare 10% (vol/vol) formic acid by pipetting formic acid into HPLC water using a glass pipette. Store the solution at room temperature for up to 6 months.
C18 wash buffer (day 3)
-
Combine 80% (vol/vol) acetonitrile and 0.1% (vol/vol) formic acid in HPLC water. Store the solution at room temperature for up to 6 months.
C18 loading buffer (day 3)
-
Combine 2% (vol/vol) acetonitrile and 0.1% (vol/vol) formic acid in HPLC water. Store the solution at room temperature for up to 6 months.
C18 elution buffer (day 3)
-
Combine 50% (vol/vol) acetonitrile and 0.1% (vol/vol) formic acid in HPLC water. Store the solution at room temperature for up to 6 months.
EQUIPMENT SETUP
LC-MS analysis
-
LRC samples are typically of low-to-moderate complexity and can be analyzed with any state-of-the-art LC-MS platform, provided that the system is sensitive enough to detect the low-abundance glycopeptides in the samples. Here we describe our recommended LC-MS configuration, but other methods and platforms that are optimized for the analysis of standard peptide samples should lead to comparable results. In the first step of the LC-MS analysis, peptide samples are separated by reversed phase chromatography on a high-performance LC column. For the analysis of LRC samples, we use analytical columns with a 75-μm inner diameter that are packed with a 100-mm stationary phase of C18 material (3-μm particles, 200-Å pore size). Peptides are loaded onto the column using a nano-flow HPLC system (EASY-nLC II). The HPLC system is directly coupled to a highly sensitive, high-mass-accuracy mass spectrometer (LTQ-Orbitrap XL) equipped with a nanoelectrospray ion source. Eight microliters of sample per analysis are loaded onto the column with buffer A (HPLC water, 0.1% (vol/vol) formic acid) and eluted with 300 nl min−1 over a 40-min linear gradient from 7–35% buffer B (acetonitrile, 0.1% (vol/vol) formic acid). After the gradient, the column is washed with 90% buffer B and re-equilibrated with buffer A. Mass spectra are acquired in a data-dependent manner, with an automatic switch between the MS and MS/MS scans during the acquisition. High-resolution MS scans are acquired in the Orbitrap (60,000 FWHM, target value 106) to monitor peptide ions in the mass range of 350–1,650 m/z, followed by collision-induced dissociation MS/MS scans in the ion trap (minimum signal threshold 150, target value 104, isolation width 2 m/z) of the five most intense precursor ions. To avoid multiple scans of dominant ions, the precursor ion masses of scanned ions are dynamically excluded from MS/MS analysis for 10 s. Single-charged ions and ions with unassigned charge states are also excluded from MS/MS fragmentation.
Software solution for the statistical analysis of LRC experiments
-
Regardless of the label-free quantification strategy chosen, the primary output of the analysis will be a list of quantified spectral features or glycopeptides. For the statistical analysis of such data sets on the peptide and protein level, we highly recommend using the specifically adapted R-based software solution provided (Supplementary Methods). To install this software on your local computer, go to http://www.stat.purdue.edu/~tclough/MSstats/MSstats.html and follow the instructions for the installation of the MSstats software. (i) Download and install R on your local computer. (ii) Install the following libraries in R using the Package Installer: Biobase (from BioConductor), lme4 (from CRAN), gmodels (from CRAN), lattice (from CRAN) and limma (from BioConductor). (iii) Download and install MSstats. After successful installation of the software, running the provided R script on the test data set will give you the output displayed in Figure 4. After successful analysis of the test data set, prepare your own data in the required format (.csv file) and run the script.
Figure 4: Anticipated results of LRC experiments. 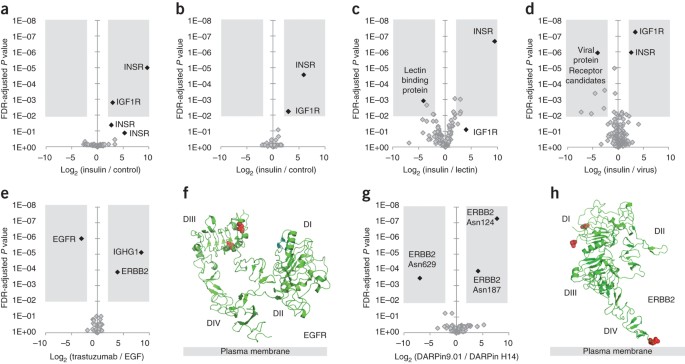
Volcano plots (FDR-adjusted P values are plotted against fold-changes between samples) can be used for the identification of receptor candidates in LRC experiments with biological or technical triplicates. Approximate ligand binding site information can be obtained for small ligands that bind to receptors with comparatively large extracellular domains. (a) LRC with insulin on BT-474 cells in biological triplicates. Data are shown on the peptide level. The receptor candidate space highlighted in gray is defined by an enrichment factor of greater than fourfold and an FDR-adjusted P value <0.01. (b) Same experiment as in a with data shown on the protein level. (c) LRC experiment with insulin and a lectin (carbohydrate-binding protein) ligand on HeLa Kyoto cells. Excessive scattering of the raw data makes the analysis less conclusive. Data are shown on the protein level. (d) LRC with insulin and intact viruses on A549 cells. Data are shown on the protein level. All of the identified binding factor candidates should be considered for follow-up investigations. (e) LRC with EGF and trastuzumab on U-251 cells (ref. 32). Data are shown on the protein level. (f) Tethered extracellular domain of EGFR with the glycosites that were enriched in the LRC experiment in e highlighted in red. (g) LRC with DARPin 9.01 and DARPin H14 on BT-474 cells (ref. 32). Data are shown on the glycosite level. (h) Extracellular domain of ERBB2 with the glycosites that were enriched in the LRC experiment in g highlighted in red. INSR, insulin receptor protein/peptide; IG1FR, insulin-like growth factor 1 receptor; ERBB2 ASN, glycosites of the human epidermal growth factor 2 receptor.

Procedure
Ligand coupling
Timing 0.5 h manual work plus 1–2 h incubation
Critical Step
This procedure describes the protocol for producing one ligand sample and one control sample. For discovery-driven LRC experiments, we highly recommend producing biological triplicates (three ligand samples and three control samples) in parallel to enable the identification of high-confidence receptor candidates. The quantities given in this protocol are for the generation of one ligand sample and one control sample and need to be multiplied by 3 for the generation of biological triplicates. Before initiating an actual LRC experiment, we recommend coupling a ligand of interest to TRICEPS at the recommended ratio (Steps 1–5) and testing the coupled ligand for receptor binding using streptavidin-coupled fluorophores and/or functional readouts.
-
1
Dissolve 1 mg of TRICEPS (MW 1,343) in 10 μl of DMSO to obtain a 75 mM working stock solution in a small glass vial.
Critical Step
Owing to the limited immediate water solubility and hydrolytic instability of TRICEPS, concentrated working stock solutions of the reagents are prepared in DMSO and immediately stored at −20 °C.
-
2
Dissolve 100 μg of your ligand of interest in 50 μl of 25 mM HEPES (pH 8.2) in an Eppendorf tube. This is your ligand sample.
Critical Step
Set up coupling reactions in buffers that do not contain primary amines or reducing agents (such as phosphate buffers or HEPES), and dialyze ligands if you are unsure about the composition of your buffer.
-
3
Dissolve 100 μg of your control ligand in 50 μl of 25 mM HEPES (pH 8.2) in an Eppendorf tube. This is your control sample.
Critical Step
Control ligands allow for quality control of the entire LRC protocol. We highly recommend using insulin as a control ligand: dilute 10 μl of insulin solution (100 μg) in 40 μl of 25 mM HEPES (pH 8.2). If no control ligand is available, quench TRICEPS with 100 μg of glycine in 50 μl of 25 mM HEPES (pH 8.2).
-
4
Add 0.5 μl of the TRICEPS working stock solution (50 μg of TRICEPS) to each of the ligand and control samples and mix them immediately.
Critical Step
Pipette in and out thoroughly with a 20-μl pipette to obtain a homogenous coupling reaction solution. Add 0.6 μl of the TRICEPS working stock solution (60 μg TRICEPS) if the ligand concentration in the coupling buffer is lower than 100 μg of ligand per 100 μl of buffer.
-
5
Incubate the mixture at room temperature under constant gentle agitation during the preparation of the target cells or tissues (60–120 min).
-
6
(Optional) Test the ligand for biological activity after coupling to TRICEPS at the recommended ratio. Reduce the TRICEPS:ligand ratio if considerable loss of activity is observed (i.e., the majority of coupled ligands are compromised by the attachment of TRICEPS) with the recommended ratio. In addition, the biotin group in TRICEPS can be used to monitor binding of coupled ligands to cells using streptavidin-conjugated fluorophores in flow cytometry or microscopy-based assays.
Receptor capture
Timing 2.5 h manual work plus 1.5 h incubation
-
7
Collect 2 × 108 cells. For adherent cells, remove the cell culture medium, add PBS, gently scrape the cells off the culture dishes and collect them in a single 50-ml centrifuge tube. This step is usually performed at room temperature for ease of handling and to avoid a cold shock effect. Use scrapers with a rubber-like blade and do not use trypsin to detach cells. For suspension cells, decant the cell suspension into 50-ml centrifuge tubes, centrifuge at 300g for 5 min and resuspend the cells in 50 ml of PBS in a single centrifuge tube. For primary tissues, dissociate tissues into smaller pieces by mechanical disruption and/or collagenase digestion and resuspend tissue pieces in 50 ml of PBS in a single 50-ml centrifuge tube.
Critical Step
LRC experiments can be carried out with 107 cells per sample, but it is highly recommended to use up to 108 cells per sample. When you are working with small numbers of cells or primary tissues, cells can be processed in 10 ml of buffer in a single 15-ml centrifuge tube to minimize cell losses during the oxidation and washing steps. Adherent cells are usually scraped off cell culture dishes first so all of the following steps can be done in centrifuge tubes, but cells can also be left attached to cell culture dishes for the oxidation and incubation with ligands.
-
8
Cool down the cell suspension on ice. We recommend performing all of the following steps up to Step 21 on ice (when working at the bench) or in a 4 °C refrigerator (for incubations under gentle agitation) in order to limit cellular processes such as endocytosis of oxidized cell surface proteins and/or ligand-receptor complexes. When a higher temperature is essential for the interaction of a given ligand with corresponding receptors, the incubation of TRICEPS-coupled ligands with target cells can also be carried out at room temperature or at 37 °C.
-
9
Centrifuge the cells at 300g for 5 min at 4 °C, discard the supernatant and resuspend the cells in 49 ml of capture buffer (PBS pH 6.5) in a single 50-ml centrifuge tube.
-
10
Add 16 mg of sodium (meta)periodate (MW 214) to 1 ml of capture buffer and vortex the solution until it is fully dissolved (75 mM). Always freshly prepare the buffer directly before the oxidation of target cells.
-
11
Add 1 ml of sodium (meta)periodate solution to 49 ml of cell suspension to obtain a final concentration of 1.5 mM, and then oxidize cells at 4 °C in the dark for 15 min under constant gentle agitation. Primary tissues are ideally oxidized for 30 min.
-
12
Centrifuge the cells at 300g for 5 min at 4 °C, discard the supernatant and resuspend the cells in 50 ml of capture buffer.
-
13
Centrifuge the cells at 300g for 5 min at 4 °C, discard the supernatant and resuspend the cells in 20 ml of capture buffer.
-
14
Split the cell suspension into 2 × 10 ml and decant into two 15-ml centrifuge tubes. Add the TRICEPS-coupled ligand of interest to one of the centrifuge tubes and the coupled control ligand to the other tube.
Critical Step
We highly recommend incubating ligands with target cells in capture buffer (PBS pH 6.5). The LRC protocol is efficiently fine-tuned by specific pH conditions and a slightly acidic pH during the capture reaction is beneficial for the efficiency and specificity of the procedure. Incubate at higher pH only if ligand binding is known to be highly pH dependent.
-
15
Incubate ligands with cells for 90 min at 4 °C under constant gentle agitation for the receptor capture reaction.
-
16
Centrifuge the cells at 300g for 5 min at 4 °C, aspirate the supernatant and resuspend the cells in 0.8 ml of freshly prepared 50 mM ammonium bicarbonate (pH 8). Pipette the suspension thoroughly in order to obtain a single-cell suspension for cell lysis, and then transfer it to an Eppendorf tube and put it on ice.
Critical Step
Always prepare 50 mM ammonium bicarbonate freshly as the pH of the solution increases over time.
Cell lysis and tryptic digest
Timing 1 h plus incubation time
-
17
Place the Eppendorf tubes in a VialTweeter and sonicate the samples (100% amplitude, 0.8 cycle) for 10–180 s depending on the cell line used. Sonicate the samples for maximally 60 s at a time, vortex and cool down on ice before performing additional cycles of sonication. In addition, we recommend precooling the VialTweeter by placing it in a 4 °C refrigerator to prevent excessive heating of samples during sonication.
Critical Step
This step highly depends on the target cells used. When optimizing the procedure, short sonication cycles (10 s) can be followed by centrifugation of the sample at 200g for 3 min at 4 °C. If intact cells are still present in the sample, additional sonication cycles should be performed. Alternative methods to indirect sonication, such as detergent-based methods, can be used to lyse cells or tissues (Step 20), but they need to be compatible with subsequent tryptic digestion of the resulting samples. Ideally, all cells should be disrupted but cell nuclei should remain intact.
-
18
Centrifuge the cells at 800g for 10 min at 4 °C to pellet the nuclear fraction of the homogenates. Pipette the supernatants (∼1 ml) into new Eppendorf tubes and discard the nuclear fractions.
-
19
Estimate the protein concentration of your cell lysates using a NanoDrop.
Critical Step
An estimate of the total protein concentration is needed to adjust the tryptic digest of the samples later on (Step 25). A NanoDrop device can be used to estimate the protein concentration in the samples. Note that nonprotein components such as nucleic acids and insoluble cell lysate factors can also absorb UV light, and NanoDrop absorbance measurements at 280 nm (A280) can only be used to estimate the upper limit of protein concentrations in cell lysates. For more accurate measurements of protein concentrations, use a BCA or Bradford assay.
-
20
Add 120 μl of a 1% (wt/vol) RapiGest SF stock solution per sample to obtain a final surfactant concentration of ∼0.1%.
Critical Step
RapiGest is used to enhance solubilization and enzymatic digestion of membrane proteins. Depending on the target cells used, the surfactant can also be used to lyse cells: add RapiGest SF to a concentration of 0.1% (wt/vol) to intact cells and place the samples on ice for 10 min. Centrifuge the samples at 200g for 3 min at 4 °C to pellet cells that are still intact. If intact cells are still present in the sample, vortex and sonicate (Step 17) until all cells are disrupted. This is also the recommended procedure to dissociate tissues, but it is expected to lead to considerable disruption of nuclear envelopes.
-
21
Place the Eppendorf tubes in a VialTweeter and sonicate the samples (50% amplitude, 0.5 cycle) for 60 s. Vortex the samples, cool down on ice and sonicate the samples again for 60 s until they become fully translucent.
-
22
(Optional) add 1 μl of benzonase nuclease (25–29 U μl−1) per sample, and then incubate the samples at room temperature for 30 min to degrade DNA and RNA. This step is recommended in cases where most cell nuclei were disrupted during cell lysis (i.e., if a slimy or no nuclear pellet is observed after Step 18).
-
23
Add the TCEP stock solution (100 mM TCEP in 500 mM ammonium bicarbonate) to the samples to a final concentration of 5 mM (60 μl of TCEP stock solution for a 1.2 ml of protein sample). Vortex and incubate the samples at room temperature for 30 min.
-
24
Add IAA stock solution (500 mM in HPLC water) to the samples to a final concentration of 10 mM (24 μl of IAA stock solution for a 1.2 ml of protein sample). Vortex and incubate the samples at room temperature for 30 min in the dark.
-
25
Calculate the amount of trypsin needed to digest the samples with a protein:trypsin ratio of ∼50:1 on the basis of the protein measurements in cell lysates (Step 19). Dissolve the total amount of trypsin needed (typically 200–600 μg of trypsin per sample) in 200 μl of 50 mM ammonium bicarbonate and add 100 μl to each sample.
-
26
Incubate the samples overnight at 37 °C under constant gentle agitation.
Pause point
After incubation overnight, the digested peptide sample can be frozen and stored at −20 °C.
Glycopeptide affinity purification
-
27
Incubate peptide samples at 96 °C for 12 min to inactivate trypsin.
-
28
Centrifuge the peptide samples at maximum speed for 10 min at room temperature to pellet the undissolved and undigested particles. Pipette the supernatants into new Eppendorf tubes.
-
29
Place 35-μm pore size filters into two Mobicols and place the columns on a Vac-Man Laboratory Vacuum Manifold.
-
30
Vortex streptavidin UltraLink Resin and pipette 80 μl of slurry (corresponds to 40 μl of settled resin) onto each column. Aspirate the liquid and wash the streptavidin resin three times with 500 μl of 50 mM ammonium bicarbonate.
-
31
Add 100 μl of 50 mM ammonium bicarbonate to the streptavidin resin, and then transfer the resin to the peptide solutions in the Eppendorf tubes. Repeat the step once to transfer the resin that remained in the columns. Incubate the tubes for 2 h at 4 °C on a slow end-over-end rotator.
-
32
Transfer 500 μl of streptavidin resin in peptide solution back to columns and aspirate the liquid. Repeat until all streptavidin resin is transferred back to the columns.
-
33
Wash the streptavidin resin with 10 × 500 μl of 5 M NaCl.
-
34
Wash the streptavidin resin with 10 × 500 μl of detergent buffer (150 mM NaCl, 1% (vol/vol) glycerol, 1% (vol/vol) Triton X-100, 50 mM ammonium bicarbonate).
-
35
Wash the streptavidin resin with 10 × 500 μl of 100 mM sodium bicarbonate of pH 11.
-
36
Wash the streptavidin resin with 20 × 500 μl of 50 mM ammonium bicarbonate.
Critical Step
We recommend using a repeater pipette for the washing procedure. Wash the streptavidin resins extensively with 50 mM ammonium bicarbonate at the end of the washing procedure to avoid contamination of the MS samples with residual material from the other washing buffers or the other steps of the PROCEDURE. At the end of the washing procedure, fill the columns with 50 mM ammonium bicarbonate to the very top and aspirate the liquid entirely; repeat the step two times.
-
37
Take the columns off the vacuum manifold and close them tightly with an outlet plug.
-
38
Pipette 3 μl of PNGase F to 800 μl of 50 mM ammonium bicarbonate and add 400 μl to each column (1.5 μl of PNGase F per sample). Close the columns tightly with screw caps and incubate them overnight at 37 °C on an end-over-end rotator.
Critical Step
The columns must be closed tightly and should additionally be wrapped with Parafilm to avoid leakage through the outlet.
-
39
Put the columns in 2-ml tubes and centrifuge them at 2,000g for 1 min at room temperature to collect the released liquid-containing, formerly glycosylated peptides.
-
40
Add 500 μl of 50 mM ammonium bicarbonate to the streptavidin resin and resuspend the resin by pipetting. Spin the tubes again at 2,000g for 1 min at room temperature and collect the flow-through in the same 2-ml tube.
Pause point
Samples containing released peptides can be frozen and stored at −20 °C.
LC-MS sample preparation
Timing 1–2 h plus incubation time
-
41
Add 40 μl of a 10% (vol/vol) formic acid stock solution to each sample to acidify to pH 2–3.
-
42
Cut the outlet (4 mm) of two C18 Ultra Micro SpinColumns, and connect them to a Vac-Master vacuum manifold.
-
43
Wash the C18 material with 400 μl of methanol and with 2 × 400 μl of C18 wash buffer (80% (vol/vol) acetonitrile, 0.1% (vol/vol) formic acid in HPLC water).
Critical Step
Do not let the C18 columns run dry at any point during the following procedure.
-
44
Equilibrate the C18 material with 2 × 400 μl of loading buffer (2% (vol/vol) acetonitrile, 0.1% (vol/vol) formic acid in HPLC water).
-
45
Add the peptide samples to the columns and bind peptides to the C18 material by slowly passing samples through the columns.
-
46
Wash the samples with 2 × 400 μl of loading buffer (2% (vol/vol) acetonitrile, 0.1% (vol/vol) formic acid in HPLC water).
-
47
Place the Eppendorf tubes in the Vac-Master manifold to collect peptide samples. Slowly elute samples with 2 × 300 μl of elution buffer (50% (vol/vol) acetonitrile, 0.1% (vol/vol) formic acid in HPLC water).
-
48
Put the samples in a centrifugal vacuum concentrator and evaporate the solvents at 45 °C. Evaporation of the solvents is expected to take ∼2 h.
Pause point
Tubes containing the peptide samples can be stored at −20 °C.
-
49
Pipette 10–30 μl of loading buffer into the Eppendorf tubes and pipette in and out 20× to re-dissolve the peptides.
Critical Step
This step depends on the configuration of your autosampler/HPLC system for MS analysis of the samples (typically 1–8 μl of the sample can be injected per analysis) and the setup of your LRC experiment. We highly recommend injecting the entire LRC samples into the mass spectrometer to maximize the number of identified glycopeptides. If a sample is analyzed in technical triplicates (replicate LC-MS measurements of the same sample) and 8 μl can be injected, the sample should be re-dissolved in at least 26 μl (three injections + 2 μl of residual volume). If biological triplicates have been produced that are analyzed in technical duplicates (recommended), each sample should be re-dissolved in at least 18 μl.
-
50
Place the samples in a thermomixer and shake the samples at 1,000 r.p.m. for 15 min at room temperature.
-
51
Place the samples in an ultrasonic bath for 3 min.
-
52
Centrifuge the peptide samples at 12,000g for 10 min and pipette the samples into MS vials.
Pause point
MS samples can be frozen and stored at −20 °C if not analyzed by MS within a week.
LC-MS sample acquisition
Timing 3–12 h, variable
-
53
Analyze the samples by LC-MS. See Equipment Setup for details about LC-MS configuration and settings.
Data analysis
Timing 3–8 h manual work, variable
-
54
Parameter setting for MS data searching: search the acquired tandem mass spectra against an appropriate protein database (UniProtKB/SwissProt is recommended) using an appropriate search algorithm (e.g., SEQUEST, Mascot, OMSSA and Andromeda). Add streptavidin (Uniprot P22629), PNGase F (Uniprot P21163) and cationic trypsin (Uniprot P00760) to your protein database. Specify trypsin as the proteolytic enzyme used, search for fully as well as semitryptic peptides and allow for maximally two missed cleavage sites per peptide. Include carbamidomethyl as a fixed modification on cysteine (+57.0215), oxidation as a variable modification on methionine (+15.9949) and deamidation as a variable modification on asparagine (+0.9840).
-
55
Filter your data to an FDR below 1% on the protein level. Determine the number of total peptides identified in each sample (typically 300–800 peptides) and the percentage of peptides that contain the N[115]-X-S/T signature (typically 20–50% of total peptides). These numbers can vary considerably depending on the amount of TRICEPS and the number of cells used, as well as on the target cell line chosen for the experiment. Verify that numbers are in the same range for the ligand sample and the control sample as a first quality-control measure. A minimum of 50 glycopeptides should be identified in every LRC sample.
-
56
Perform MS1-based relative label-free quantification for the identified peptides to determine the relative raw abundances of quantified spectral features or peptides. Filter your peptides for the presence of the N[115]-X-S/T signature and only consider these peptides for the following steps. Verify that the summed-up raw abundance of all N[115]-X-S/T peptides is in the same range for the ligand sample and the control sample as a second quality control measure. Furthermore, manually inspect highly enriched peptide features to ensure correct feature picking and integration during the quantification process. At this step, data can be exported and visualized in the form of an enrichment plot or volcano plot on the peptide or protein level. If technical or biological triplicates have been produced, we highly recommend exporting the feature or peptide raw abundances and analyzing the data with the provided script to make protein-level conclusions.
-
57
Perform protein quantification and significance analysis to detect specific receptor enrichments in your ligand sample compared with the control sample. Display your spectral feature or peptide raw abundances in a comma-separated value (.csv file) format according to the example provided (Supplementary Methods). Name the resulting file 'ligandVScontrol.csv' and copy the file into a folder named 'MSstats' on your local computer. Execute the individual steps of the R script provided. This script does the following: sums up intensities for peptide sequences with more than one entry such as multiple charge states of the same peptide, performs constant normalization across the samples, investigates the effect of normalization, creates trellis and profile plots for each protein, fits a fixed effects ANOVA model35 to each protein and tests each protein for differential abundance in all pairwise sample comparisons, adjusts the reported P values using the Benjamini-Hochberg method, makes a volcano plot of the results, performs peptide level analysis using a LIMMA model, adjusts the reported P values using the Benjamini-Hochberg method and makes a volcano plot of the results for quick visual identification of receptor candidates. The applied fixed effects model is fairly robust to missing peptide feature intensities, and only few missing values are usually present in the highly similar samples of a LRC experiment. The MSstats user manual and corresponding publication42 contain further information regarding this topic, including options to address the case of an extreme degree of missing intensities.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Preparation of ligand and target cells or tissue: variable
Steps 1–6, ligand coupling: 0.5 h manual work plus 1–2 h incubation
Steps 7–16, receptor capture: 2.5 h manual work plus 1.5 h incubation
Steps 17–26, cell lysis and tryptic digest: 1 h manual work plus 1h and overnight incubation
Steps 27–40, glycopeptide affinity purification: 1–2 h manual work plus 2 h and overnight incubation
Steps 41–52, LC-MS sample preparation: 1–2 h manual work plus 2–3 h incubation
Step 53, LC-MS sample acquisition: 3–12 h, variable
Steps 54–57, data analysis: 3–8 h manual work, variable
Anticipated results
The LRC technology enables the identification of receptors through quantitative comparison of a ligand sample with a control sample generated with a control probe with known or no binding preferences on a given target cell line or tissue. Depending on the number of samples generated and the data analysis strategy chosen, different LRC results can be anticipated. In the simplest conceivable experimental setup, one ligand sample and one control sample are produced and the results are displayed in an enrichment plot as shown in Figure 3. In experiments in which technical or biological replicate samples have been produced, it is highly recommended to use the provided statistical model for the analysis of the resulting data sets. This type of analysis considerably increases the sensitivity and specificity of testing and reports FDR-adjusted P values in order to rank receptor candidates in the resulting volcano plots (Fig. 4a,b). In such analyses, different receptor glycopeptides are usually not enriched uniformly owing to their variable steric accessibility in relation to the ligand-binding site and different peptide-specific parameters that affect label-free quantification. However, consistent enrichment of the independent receptor peptides usually leads to a robust protein-level identification of receptor candidates in the receptor space of volcano plots, whereas random cell surface proteins are not enriched in either condition. Such analyses usually enable a quick identification of receptor candidates in standard LRC applications but can be complicated by a number of factors. Notably, LRC experiments are usually less conclusive in cases where a ligand has an additional general affinity for molecular structures on the cell surface other than specific target glycoproteins. In LRC experiments with lectins (carbohydrate-binding proteins), we observed higher ion signals for most identified glycopeptides compared with control samples. This suggested that general binding of TRICEPS-coupled lectins to glycans on human cells increased the local concentration of the reagents and improved the overall capture of N-glycoproteins. These experiments led to the successful identification of a lectin-binding protein as a specific true receptor for the lectin (Fig. 4c). However, data analysis was made considerably more difficult and less conclusive by excessive scattering of the raw data. Similar results can be expected in experiments with complex viruses, where multiple viral and cellular factors may contribute to cooperative binding. In such experiments, the enrichment of individual receptors is typically weaker than in experiments with comparatively simple ligands, and the resulting volcano plots are expected to reveal small lists of lower confidence receptor candidates rather than single high-confidence receptors (Fig. 4d). In the opposite scenario, in which rather small ligands bind to receptors with large extracellular domains, LRC experiments have the potential to reveal approximate ligand binding site information through the enrichment of proximal receptor glycosites. This has previously been demonstrated in an experiment with the small ligand epidermal growth factor and the comparatively large therapeutic antibody trastuzumab32. For both of these ligands, the corresponding receptors epidermal growth factor receptor (EGFR) and ERBB2 were identified in a single experiment. Although the ERBB2 glycosites that were enriched with trastuzumab did not contain further information, enrichment of glycopeptides from only domain III of EGFR enabled an approximate localization of the EGF-binding site on this particular domain of the receptor as the molecular size of the EGFR by far exceeds that of its ligand (Fig. 4e,f). This additional information becomes more evident when LRC experiments are evaluated on the glycosite level. In the resulting volcano plot of an experiment performed with two small designed ankyrin repeat proteins (DARPins) that bind to the ERBB2 protein, the specific enrichment of corresponding glycosylation sites immediately revealed that one DARPin bound to domain I in ERBB2, whereas the other DARPin bound to domain IV (Fig. 4g,h; ref. 32). In summary, these anticipated results highlight the potential of LRC to identify N-glycoprotein receptor candidates for various ligands under near-physiological conditions. Identified receptor candidates should always be validated using orthogonal technologies, and such immediate follow-up experiments are especially recommended in LRC applications that lead to excessive data scattering or to only moderate enrichments of receptor candidates. Less-conclusive results are generally expected with large and/or versatile ligands, such as intact viruses, whereas LRC is expected to reveal high-confidence receptors and sometimes even binding site information under standard conditions.
References
Hopkins, A.L. & Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 (2002).
Fang, Y. Ligand-receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discov. 7, 969–988 (2012).
Hubner, N.C. et al. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J. Cell Biol. 189, 739–754 (2010).
Gingras, A.-C., Gstaiger, M., Raught, B. & Aebersold, R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654 (2007).
Glatter, T., Wepf, A., Aebersold, R. & Gstaiger, M. An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol. Syst. Biol. 5, 237 (2009).
Miteva, Y.V., Budayeva, H.G. & Cristea, I.M. Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal. Chem. 85, 749–768 (2012).
Jäger, S. et al. Global landscape of HIV-human protein complexes. Nature 481, 365–370 (2012).10.1038/nature10719
Elschenbroich, S., Kim, Y., Medin, J.A. & Kislinger, T. Isolation of cell surface proteins for mass spectrometry-based proteomics. Expert Rev. Proteomics 7, 141–154 (2010).
Helbig, A.O., Heck, A.J.R. & Slijper, M. Exploring the membrane proteome—challenges and analytical strategies. J. Proteomics 73, 868–878 (2010).
Rual, J.-F. et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178 (2005).
Stelzl, U. et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968 (2005).
Petschnigg, J., Wong, V., Snider, J. & Stagljar, I. Investigation of membrane protein interactions using the split-ubiquitin membrane yeast two-hybrid system. Methods Mol. Biol. 812, 225–244 (2012).
Meier, M., Sit, R., Pan, W. & Quake, S.R. High-performance binary protein interaction screening in a microfluidic format. Anal. Chem. 84, 9572–9578 (2012).
Ramachandran, N. et al. Next-generation high-density self-assembling functional protein arrays. Nat. Methods 5, 535–538 (2008).
MacBeath, G. Protein microarrays and proteomics. Nat. Genet. 32 (suppl.), 526–532 (2002).10.1038/ng1037
Braun, P. Interactome mapping for analysis of complex phenotypes: insights from benchmarking binary interaction assays. Proteomics 12, 1499–1518 (2012).
Bushell, K.M., Söllner, C., Schuster-Boeckler, B., Bateman, A. & Wright, G.J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Res. 18, 622–630 (2008).
Crosnier, C. et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 480, 534–537 (2011).
Savas, J.N., Stein, B.D., Wu, C.C. & Yates, J.R. Mass spectrometry accelerates membrane protein analysis. Trends Biochem. Sci. 36, 388–396 (2011).
Lee, A. How lipids affect the activities of integral membrane proteins. Biochim. Biophys Acta-Biomembranes 1666, 62–87 (2004).
Engelman, D.M. Membranes are more mosaic than fluid. Nature 438, 578–580 (2005).
Phillips, R., Ursell, T., Wiggins, P. & Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature 459, 379–385 (2009).
Lingwood, D. & Simons, K. Lipid rafts as a membrane-organizing principle. Science 327, 46–50 (2010).
Oates, J. & Watts, A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol. 21, 802–807 (2011).
Simons, K. & Gerl, M.J. Revitalizing membrane rafts: new tools and insights. Nat. Rev. Mol. Cell Biol. 11, 688–699 (2010).
Wollscheid, B. et al. Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat. Biotech. 27, 378–386 (2009).
Zeng, Y., Ramya, T.N.C., Dirksen, A., Dawson, P.E. & Paulson, J.C. High-efficiency labeling of sialylated glycoproteins on living cells. Nat. Methods 6, 207–209 (2009).
Hofmann, A. et al. Proteomic cell surface phenotyping of differentiating acute myeloid leukemia cells. Blood 116, e26–e34 (2010).
Bock, T. et al. Proteomic analysis reveals drug accessible cell surface N-glycoproteins of primary and established glioblastoma cell lines. J. Proteome Res. 11, 4885–4893 (2012).
Ramya, T.N.C., Weerapana, E., Cravatt, B.F. & Paulson, J.C. Glycoproteomics enabled by tagging sialic acid– or galactose-terminated glycans. Glycobiology 23, 211–221 (2012).
Bock, T., Bausch-Fluck, D., Hofmann, A. & Wollscheid, B. CD proteome and beyond—technologies for targeting the immune cell surfaceome. Front. Biosci. 17, 1599–1612 (2012).
Frei, A.P. et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nat. Biotech. 30, 997–1001 (2012).
Mädler, S., Bich, C., Touboul, D. & Zenobi, R. Chemical cross-linking with NHS esters: a systematic study on amino acid reactivities. J. Mass Spectrom. 44, 694–706 (2009).
Bruus-Jensen, K. et al. Chemoselective hydrazone formation between HYNIC-functionalized peptides and F18-fluorinated aldehydes. Nucl. Med. Biol. 33, 173–183 (2006).
Clough, T. et al. Protein quantification in label-free LC-MS experiments. J. Proteome. Res. 8, 5275–5284 (2009).
Bich, C. et al. Reactivity and applications of new amine reactive cross-linkers for mass spectrometric detection of protein-protein complexes. Anal. Chem. 82, 172–179 (2009).
Brinkley, M. A brief survey of methods for preparing protein conjugates with dyes, haptens and crosslinking reagents. Bioconjug. Chem. 3, 2–13 (1992).
Steu, S. et al. A procedure for tissue freezing and processing applicable to both intra-operative frozen section diagnosis and tissue banking in surgical pathology. Virchows Arch. 452, 305–312 (2008).
Falsig, J. et al. A versatile prion replication assay in organotypic brain slices. Nat. Neurosci. 11, 109–117 (2008).
Eng, J.K., McCormack, A.L. & Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 (1994).
Patel, V.J. et al. A comparison of labeling and label-free mass spectrometry-based proteomics approaches. J. Proteome Res. 8, 3752–3759 (2009).
Clough, T., Thaminy, S., Ragg, S., Aebersold, R. & Vitek, O. Statistical protein quantification and significance analysis in label-free LC-MS experiments with complex designs. BMC Bioinformatics 13, S6 (2012).
Acknowledgements
We greatly acknowledge T. Clough and O. Vitek for help with the statistical data analysis pipeline. We are grateful to A. Hofmann, T. Bock, D. Bausch-Fluck, A. Jacobs and A. Leitner for suggestions and support at all stages of the project. This work was supported by funding from the National Center of Competence in Research Neural Plasticity and Repair (to B.W.), the Swiss National Science Foundation (to B.W.) and SystemsX.ch/ InfectX (to B.W.)
Author information
Authors and Affiliations
Contributions
A.P.F. and B.W. designed the project and wrote the paper. A.P.F. performed all the experiments and analyzed all the data. H.M. and K.N. contributed ideas. All authors discussed the results and implications and commented on the manuscript at all stages.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Methods
R script for the statistical analysis of LRC datasets using MSstats (ZIP 14 kb)
Rights and permissions
About this article
Cite this article
Frei, A., Moest, H., Novy, K. et al. Ligand-based receptor identification on living cells and tissues using TRICEPS. Nat Protoc 8, 1321–1336 (2013). https://doi.org/10.1038/nprot.2013.072
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2013.072
This article is cited by
-
Loading characteristics of streptavidin on polypropylene capillary channeled polymer fibers and capture performance towards biotinylated proteins
Analytical and Bioanalytical Chemistry (2023)
-
Latrophilin-2 is a novel receptor of LRG1 that rescues vascular and neurological abnormalities and restores diabetic erectile function
Experimental & Molecular Medicine (2022)
-
Cell surface thermal proteome profiling tracks perturbations and drug targets on the plasma membrane
Nature Methods (2021)
-
Akkermansia muciniphila secretes a glucagon-like peptide-1-inducing protein that improves glucose homeostasis and ameliorates metabolic disease in mice
Nature Microbiology (2021)
-
Advances in high-throughput methods for the identification of virus receptors
Medical Microbiology and Immunology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
