
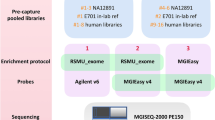
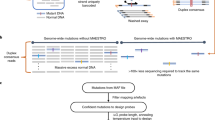
Abstract
The unprecedented increase in the throughput of DNA sequencing driven by next-generation technologies now allows efficient analysis of the complete protein-coding regions of genomes (exomes) for multiple samples in a single sequencing run. However, sample preparation and targeted enrichment of multiple samples has become a rate-limiting and costly step in high-throughput genetic analysis. Here we present an efficient protocol for parallel library preparation and targeted enrichment of pooled multiplexed bar-coded samples. The procedure is compatible with microarray-based and solution-based capture approaches. The high flexibility of this method allows multiplexing of 3–5 samples for whole-exome experiments, 20 samples for targeted footprints of 5 Mb and 96 samples for targeted footprints of 0.4 Mb. From library preparation to post-enrichment amplification, including hybridization time, the protocol takes 5–6 d for array-based enrichment and 3–4 d for solution-based enrichment. Our method provides a cost-effective approach for a broad range of applications, including targeted resequencing of large sample collections (e.g., follow-up genome-wide association studies), and whole-exome or custom mini-genome sequencing projects. This protocol gives details for a single-tube procedure, but scaling to a manual or automated 96-well plate format is possible and discussed.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

References
Wang, J. et al. The diploid genome sequence of an Asian individual. Nature 456, 60–65 (2008).
Li, Y. et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 42, 969–972 (2010).
Metzker, M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 11, 31–46 (2010).
Vissers, L.E. et al. A de novo paradigm for mental retardation. Nat. Genet. 42, 1109–1112 (2010).
Igartua, C. et al. Targeted enrichment of specific regions in the human genome by array hybridization. Curr. Protoc. Hum. Genet. 66, 18.3.1–18.3.14 (2010).
Durbin, R.M. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Mamanova, L. et al. Target-enrichment strategies for next-generation sequencing. Nat. Methods 7, 111–118 (2010).
Hodges, E. et al. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat. Protoc. 4, 960–974 (2009).
Nijman, I.J. et al. Mutation discovery by targeted genomic enrichment of multiplexed bar-coded samples. Nat. Methods 7, 913–915 (2010).
Harbour, J.W. et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330, 1410–1413 (2010).
Varela, I. et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469, 539–542 (2011).
Krawitz, P.M. et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 42, 827–829 (2010).
Gilissen, C. et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 87, 418–423 (2010).
Musunuru, K. et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl. J. Med. 363, 2220–2227 (2010).
Bilguvar, K. et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210 (2010).
Ng, S.B. et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42, 790–793 (2010).
Cummings, N. et al. Combining target enrichment with barcode multiplexing for high throughput SNP discovery. BMC Genomics 11, 641 (2010).
Teer, J.K. et al. Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 20, 1420–1431 (2010).
Harakalova, M. et al. Genomic DNA pooling strategy for next-generation sequencing-based rare variant discovery in abdominal aortic aneurysm regions of interest-challenges and limitations. J. Cardiovasc. Transl. Res. 4, 271–280 (2011).
Fisher, S. et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 12, R1 (2011).
Mokry, M. et al. Accurate SNP and mutation detection by targeted custom microarray-based genomic enrichment of short-fragment sequencing libraries. Nucleic Acids. Res. 38, e116 (2010).
Gnirke, A. et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189 (2009).
Bainbridge, M.N. et al. Whole exome capture in solution with 3 Gbp of data. Genome Biol. 11, R62 (2010).
Okou, D.T. et al. Microarray-based genomic selection for high-throughput resequencing. Nat. Methods 4, 907–909 (2007).
Johansson, H. et al. Targeted resequencing of candidate genes using selector probes. Nucleic Acids. Res. 39, e8 (2011).
Lee, H. et al. Improving the efficiency of genomic loci capture using oligonucleotide arrays for high throughput resequencing. BMC Genomics 10, 646 (2009).
Acknowledgements
We would like to thank I. Wortel and E. Slob for testing the protocol. B. Hrdlickova was supported by The Rector's grant MUNI/E0136/2009 provided by Masaryk University, Czech Republic.
Author information
Authors and Affiliations
Contributions
All authors contributed extensively to protocol development and to the preparation of the manuscript. M.H. and M.M. created the protocol and wrote the manuscript. M.H., M.M., B.H., I.R., K.D., H.V. and E.D. performed the experiments and optimized experimental steps. I.R., N.L. and E.D. performed the multiplexed sequencing runs. I.J.N. wrote custom scripts for the array-based probe design. M.V. and I.J.N. performed the bar code splitting, data mapping and distribution analysis. W.P.K. and E.C. supervised the experiments and the development of the protocol.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Figure 1
Graphical example of a manual for the level of multiplexing based on the size of design. (PDF 25 kb)
Supplementary Table 1
Oligonucleotide sequences (PDF 44 kb)
Rights and permissions
About this article
Cite this article
Harakalova, M., Mokry, M., Hrdlickova, B. et al. Multiplexed array-based and in-solution genomic enrichment for flexible and cost-effective targeted next-generation sequencing. Nat Protoc 6, 1870–1886 (2011). https://doi.org/10.1038/nprot.2011.396
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2011.396
This article is cited by
-
Few-shot learning creates predictive models of drug response that translate from high-throughput screens to individual patients
Nature Cancer (2021)
-
A systematic investigation of human DNA preservation in medieval skeletons
Scientific Reports (2020)
-
Inferring the evolution of the major histocompatibility complex of wild pigs and peccaries using hybridisation DNA capture-based sequencing
Immunogenetics (2018)
-
Joubert syndrome: genotyping a Northern European patient cohort
European Journal of Human Genetics (2016)
-
Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT
Kidney International (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
