
Abstract
In the past two decades, high-affinity nucleic acid aptamers have been developed for a wide variety of pure molecules and complex systems such as live cells. Conceptually, aptamers are developed by an evolutionary process, whereby, as selection progresses, sequences with a certain conformation capable of binding to the target of interest emerge and dominate the pool. This protocol, cell-SELEX (systematic evolution of ligands by exponential enrichment), is a method that can generate DNA aptamers that can bind specifically to a cell type of interest. Commonly, a cancer cell line is used as the target to generate aptamers that can differentiate that cell type from other cancers or normal cells. A single-stranded DNA (ssDNA) library pool is incubated with the target cells. Nonbinding sequences are washed off and bound sequences are recovered from the cells by heating cell-DNA complexes at 95 °C, followed by centrifugation. The recovered pool is incubated with the control cell line to filter out the sequences that bind to common molecules on both the target and the control, leading to the enrichment of specific binders to the target. Binding sequences are amplified by PCR using fluorescein isothiocyanate–labeled sense and biotin-labeled antisense primers. This is followed by removal of antisense strands to generate an ssDNA pool for subsequent rounds of selection. The enrichment of the selected pools is monitored by flow cytometry binding assays, with selected pools having increased fluorescence compared with the unselected DNA library. The procedure, from design of oligonucleotides to enrichment of the selected pools, takes ∼3 months.
Similar content being viewed by others

Introduction
Aptamers are single-stranded oligonucleotide (DNA or RNA) molecules, typically <100-mer, which have the ability to bind to other molecules with high affinity and specificity. They are evolved from random oligonucleotide pools by a process called systematic evolution of ligands by exponential enrichment (SELEX)1,2. Conceptually, the SELEX process is controlled by the ability of these small oligonucleotides to fold into unique 3D structures that can interact with a specific target with high specificity and affinity. Aptamers have been generated against a wide variety of targets, including small molecules such as metal ions3,4,5, organic dyes and amino acids6,7,8,9, and antibiotics10,11 and peptides12,13, as well as proteins of various sizes and functions2,14,15,16,17,18,19,20,21,22,23,24, whole cells25,26,27,28,29,30,31,32,33,34, viruses and virus-infected cells35,36,37,38,39, and bacteria40,41. The selective interactions between aptamers and these diverse targets have been exploited for different applications, including biotechnology, medicine, pharmacology, cell biology, microbiology and chemistry. For example, aptamers have been effectively used for therapeutic applications such as targeted therapy42,43,44, detection and diagnostics21,26,45,46. In fact, the first aptamer-based drug, called mucagen, is now available, and others, such as aptamer AS1411, which is specific for nucleolin, are in clinical trials47. This shows that aptamers can also be used directly as drugs.
On the basis of their target-recognition capability, selective binding and affinity, aptamers have been likened to antibodies. However, aptamers, by their unique features, have more flexibility in their development and range of applications. Specifically, the time needed for the generation of aptamers by the SELEX process is comparatively short. In addition, aptamers are chemically synthesized, which permits the biochemical manipulation required to incorporate various functional groups and specific moieties such as biotin, carboxyl, amino and thiol48,49,50,51, most of which do not affect the recognition of the aptamer to the target. This has allowed aptamer conjugation with drug molecules and nanomaterials50,51,52 and modification of nuclease-resistant bases, such as locked nucleic acids and 2′-O-methyl nucleotide analogs, to further enhance affinity and nuclease resistance53 so that they can be adopted for in vivo study49,54. Further, initial SELEX library containing 2′-fluoro-modified nucleotides has been used to generate high-affinity aptamers55. Modified bases such as 2′-fluoro-, 2′-O-methyl and locked nucleic acids can improve nuclease resistance of nucleic acids. Even with improved nuclease, aptamers have low blood residence time compared with antibodies, as well as low toxicity or immunogenicity, and these are important features when used for in vivo applications such as imaging. Other qualities, such as long shelf life and controllable or cyclical denaturation and renaturation, have expanded the flexibility of aptamers in various experimental designs such as biosensors.
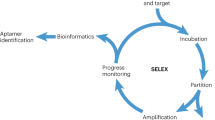
Aptamers are identified through an in vitro process. Briefly, this involves the incubation of the DNA library pool with the target of interest. After incubation, the binding complex (target and oligonucleotide sequences) is partitioned from the unbound sequences. Subsequently, the sequences binding to the target are eluted and then incubated with the control, and the remaining sequences are amplified by PCR. The process is repeated until the pool is enriched for sequences that specifically recognize the target. The enriched pool is cloned into bacteria, and positive clones are sequenced to obtain individual sequences. Representatives of these sequences, usually grouped into families (common in an enriched pool), are chemically synthesized, labeled with fluorescence dye and tested against the target to determine potential aptamer candidates. The mode of selection and the methodology used to generate these aptamers, as well as all the assays used in the identification of the potential aptamer candidates, depend to a large extent on the target of interest. In view of this, various selection modes have emerged, and each of these is rationally designed to suit the requirements of some specific purpose56,57,58,59,60,61,62. One such modality is the adoption of cell-based aptamer selection, termed cell-SELEX25,26,27,28,29,30,31,32,33,34 (Fig. 1).

DNA sequences that have specific recognition to target cells are evolved to enrich the selection pools. The enriched pools are cloned, and the positive clones are sequenced to identify individual aptamers.
Cell-SELEX is a term that describes the process whereby live cells are used to select aptamers for target recognition. Thus, cell-SELEX is a particularly promising selection strategy for various applications, including cancer research and therapy. Specifically, by adopting cell-SELEX, useful probes can be developed to potentially differentiate tumor cells from normal cells, as well as to differentiate between two different types of cancers. Thus far, live cells of different cancers have been used in this process, and as a result, a significant number of aptamers have been generated for most cancers studied26,27,28,29,30,31,32,33,34. Briefly, in the cell-SELEX method, oligonucleotides bind to molecules on the extracellular surface, a phenomenon that is important for the following reasons:
-
1
Prior knowledge of the target is unnecessary. The need to know the molecular composition of the cell surface is not important in cell-SELEX. Instead, the different types of cells used in the selection process are a critical parameter, as the selected aptamers will be useful for the specific recognition of these cells.
-
2
Aptamers can be generated for many targets. The cell membrane surface is a complex system that has a countless number of molecules, especially proteins. In cell-SELEX, each of these molecules is a potential target. At the end of a successful selection, aptamers are generated for many different targets. This feature is very important, as any one of these molecules may have one or more roles in the development of the cell or the disease they cause.
-
3
Aptamers can be generated for biomarker discovery. On the basis of the fact that cell-SELEX has the potential to develop aptamers for unknown molecules, as noted in (ii) above, these probes can be used through affinity precipitation to purify the target for aptamer recognition and then for disease biomarker discovery26,63,64.
-
4
Aptamers bind to the native state of target molecules. Aptamers are developed for molecules on the cell surface that are in their native state and would therefore represent their natural folding structures as well as their distribution; therefore, all post-translational modifications are left intact for proteins, and the aptamer will bind to the real folded conformation. It is known that the cell surface carries a net negative charge and thus one should expect repulsion to occur between the DNA polyanion and the cell; therefore, generating nucleic acid aptamers for cells will be difficult. However, we believe that the structure-related binding between the aptamer and the target will be strong enough to overcome the repulsion force. In addition, the presence of divalent cations could help to shield and reduce the effect of DNA-negative chargers.
The phosphodiester backbone of DNA can nonspecifically interact with mostly arginine or lysine and other side chain molecules that can potentially reduce the selectivity of the generated aptamers. However, as SELEX is an evolutionary process, the specific binding of the 3D structure of the DNA sequences dominates the random nonspecific binding. In addition, as selection progresses and the stringency of the procedure increases, the contribution of nonspecifically binding sequences, if any, is minimized to the barest minimum.
In this protocol, we present a detailed and proven working procedure for the development of DNA aptamers using the cell-SELEX model30,31,32,33,34 for different types of biological cells (Fig. 1). Although this protocol has been successfully implemented in many aptamer-development schemes using different cancer cell lines, it must be noted that the success of any particular selection will in part also depend on the experimenter's basic skills and efficiency in regular procedures such as PCR and cell culture techniques.
Experimental design
Primers and library. PCR is a major part of aptamer selection and therefore it is important to use primers and a library set that have high PCR amplification efficiency. The procedure to design primers and the library has been outlined below. The sense strand of the primer is labeled at the 5′ end with fluorescein isothiocyanate (FITC). The importance of FITC is to monitor the progress of selection by flow cytometry. The antisense strand is labeled at the 5′ end with biotin. The presence of biotin helps to separate the sense and antisense strands after PCR by streptavidin-biotin interaction, followed by alkaline denaturation. The initial library design is outlined under REAGENT SETUP. The length of the randomized region determines the diversity of the library. It is not necessary to label the initial library with FITC. All oligonucleotide sequences must be purified by high-performance liquid chromatography (HPLC). Two examples of good primer and library sets are shown below:
Set 1 (ref. 30) 5′-FITC-ATACCAGCTTATTCAATT-3′ 5′-biotin-AGATTGCACTTACTATCT-3′ 5′-ATACCAGCTTATTCAATT-52N-AGATAGTAAGTGCAATCT-3′ Set 2 (ref. 33) 5′′FITC-ATCCAGAGTGACGCAGCA-3′ 5′-biotin-ACTAAGCCACCGTGTCCA-3′ 5′-ATCCAGAGTGACGCAGCA-45N-TGGACACGGTGGCTTAGT-3′
Cell lines (positive and negative). The choice of cell lines depends on the purpose of the selection and what one seeks to achieve. Cell-SELEX has commonly been performed using cultured cancer cell lines, and aptamers have been generated that can differentiate between two different cancers or between cancer and normal cells. Table 1 shows examples of positive and negative cell lines that have been previously used to generate specific aptamers.
Maintenance of cell culture. Cell-SELEX uses live cells as target and therefore good cell culture maintenance is very important. Overgrown cell culture causes more cell death, and can probably cause an alteration in cell morphology and protein expression. Dead cells are detrimental to the success of selection, especially when using suspension-cultured cells. The reduction or elimination of dead cells can significantly enhance the enrichment of selected pools65. The viability of adherent monolayer cells is not a significant problem in selection because most dead cells normally float in the medium, and once the medium is removed, relatively healthy cells are obtained. That notwithstanding, cells must not be allowed to overgrow. There are two ways by which adherent monolayer cells can be used for selection: either directly in cultured dish or as cell-dissociated cells. The direct option may offer a better representation of the cells' natural environment. Dissociated cells either by short-term trypsin treatment or nonenzymatic dissociation buffer may be harsh on cells or may change the surface expression.
Elution of binding sequences. The recovery of the sequences binding to target cells is achieved by heating the cell-DNA complex at 95 °C. A twofold rationale underlies the necessity for heating the cell suspension at 95 °C to elute bound sequences: (i) Elevated temperature will cause denaturing of the cell surface proteins, and this will lead to the disruption of the interaction between DNA and protein. (ii) The folded structure of the DNA is disrupted, causing the release of DNA from the target protein. At 95 °C, any DNase that is released after cell disruption at the elevated temperature is inactivated and therefore cannot cause DNA digestion.
PCR procedures. PCR is a major part of this protocol and its success can enhance the success of the selection process. Before beginning the selection process, optimize the annealing temperature and the concentration of all PCR reagents including primers. After the first round of selection, three different PCR amplifications are performed: (i) amplification of the entire pool, (ii) determination of the optimum cycle for preparative PCR and (iii) preparation of PCR for the next round of selection. In the subsequent rounds of selection, however, only (ii) and (iii) are performed.
Preparation of single-stranded DNA. Symmetrical PCR for the preparative PCR generates double-stranded DNA that needs to be separated, and the FITC-labeled sequence needs to be recovered to continue the selection process. In this protocol, we have consistently and successfully separated sense single-stranded DNA (ssDNA) from biotinylated antisense ssDNA by alkaline denaturation and affinity purification with streptavidin-coated Sepharose beads. Recently, concerns have been raised about the problems associated with ssDNA preparation using streptavidin-coated magnetic beads66. The authors reported that ssDNA preparation using streptavidin-coated magnetic beads and subsequent alkaline separation is often contaminated by both streptavidin- and biotin-labeled sequences, which cause cell aggregation. They therefore could not enrich selected pools with the streptavidin-coated magnetic beads separation method. In our system, however, we have consistently used streptavidin-coated Sepharose beads without any difficulty. However, although we have not performed gel staining to assess the presence of streptavidin and biotin, we have never observed any cell aggregation as a result of this supposed contamination. We therefore believe that our system for ssDNA preparation is efficient for successful SELEX.
Negative selection. After the second round of selection, the selected DNA pool can be used to carry out negative selection to filter out sequences that may bind to the molecules existing on the surface of both the target and control cell lines. It is nearly impossible to remove all these sequences from the pool with negative selection, and the remaining sequences can increase in number after performing PCR. However, if negative selection is regularly carried out, most of these sequences can be eliminated, and they will be unable to populate the selection pool. All the selected DNA pools generated at the end of each round of selection are used for negative selection.
Monitor progress of selection. The enrichment of the selection pools, resulting in the evolution of potential aptamer candidates in the course of selection, is monitored by flow cytometry (suspension and adherent cells) or microscopy (adherent cells). To ensure consistency, it is desirable to perform binding assays to monitor the progress of selection after two or three continuous selections rather than after every round. Performing binding assays at the same time and under the same conditions makes it easier to compare results; therefore, any signal difference between two successive pools can be assessed well. In addition, this monitoring strategy enables one to evaluate the selection process in general.
End of selection. Selection is completed (i) when we observe a significant difference between the fluorescence signal intensity of the control background and the selected DNA pools and (ii) at a point at which no significant signal difference can be discerned between two or three successive selected pools. Once the selected pools are enriched with potential DNA aptamer sequences, they are cloned into bacteria, and the positive clones are sequenced to identify individual aptamer candidates.
Materials
REAGENTS
-
Target (positive) cell line (Table 1, CCRF-CEM)
-
Control (negative) cell line (Table 1, Ramos)
-
Appropriate cell culture media
-
Heat-inactivated fetal bovine serum (FBS; Invitrogen, cat. no. 10082-147)
-
Penicillin and streptomycin (Invitrogen, cat. no. 15140-122)
-
Trypsin-EDTA (Mediatech, cat. no. 25-052-CI)
-
Nonenzymatic cell dissociation solution (MP Biomedicals, cat. no. 1676949)
-
Trypan blue (Gibco, cat. no. 15250-061)
-
Hemocytometer (Hausser Scientific Partnership, cat. no. 3200)
-
Absolute ethanol (Decon Laboratories, cat. no. 2701)
-
Ethanol (70% (vol/vol); see REAGENT SETUP)
-
Culture dish (100 mm × 20 mm; Corning, cat. no. 430293)
-
Culture dish (60 mm × 15 mm; BD, cat. no. 430293)
-
5′ FITC-labeled sense primer (see REAGENT SETUP)
-
5′ Biotin-labeled antisense primer (see REAGENT SETUP)
-
5′ FITC-labeled random ssDNA library (see REAGENT SETUP)
-
D(+)-Glucose (Sigma, cat. no. G-8270)
-
Dulbecco's phosphate-buffered saline (DPBS; Sigma, cat. no. D8662 or MP Biomedicals, cat. no. 1960454)
-
Baker's yeast tRNA (Fluka Analytical, cat. no. 83853)
-
Magnesium chloride (Acros Organics, cat. no. 413415000)
-
Washing buffer (see REAGENT SETUP)
-
Binding buffer (see REAGENT SETUP)
-
Centrifuge tubes (15 ml; Corning, cat. no. 430052)
-
EZ-flip centrifuge tubes (15 ml; Nunc, cat. no. 362694)
-
Hot Start DNA Taq polymerase kit (HS Taq polymerase, TaKaRa, cat. no. TAKR007A)
-
dNTP mixture (2.5 mM each, TaKaRa, cat. no. 4030)
-
PCR buffer (10×; Sigma, cat. no. P2192-1VL)
-
Sterile water for RNA work (Fisher Scientific, cat. no. BP561-1)
-
PCR tubes (0.2 ml; Molecular BioProducts, cat. no. 3412A)
-
PCR tubes (0.5 ml; Molecular BioProducts, cat. no. 3430A)
-
Microfuge tubes (1.5 ml; Fisher Scientific, cat. no 05-408-129)
-
Microfuge tubes (2.0 ml; Fisher Scientific, cat. no 05-408-138)
-
Agarose (Fisher Scientific, cat. no. BP1356-500)
-
DNA step ladder molecular weight markers, including 6× blue/orange loading dye (25 bp; Promega, cat. no. PR-G4521)
-
Ethidium bromide (1% solution, Biotech grade, Fisher BioReagents, cat. no. BP-1302-10)
Caution
Ethidium bromide is a suspected mutagen and a possible carcinogen. Nitrile gloves and lab coats should be worn when handling this reagent and any gels or solutions carrying it. Ethidium bromide–containing material should be disposed in accordance with local institutional safety procedures.
-
Streptavidin Sepharose (GE, cat. no. 10024153)
-
Empty DNA synthesis column with filters (Glen Research, cat. no. 20-0021-01)
-
Syringe (10 ml; Becton Dickinson, cat. no. 309642)
-
Sodium hydroxide (200 mM; Fisher, cat. no. 5318-500)
-
Illustra NAP-5 Columns Sephadex G-25 DNA Grade (GE, cat. no. 17-0853-01)
-
Shealth fluid (BD, cat. no. 342003)
-
Flow cytometer tubes (Becton Dickinson, cat. no. 352008)
-
Bleach (Clorox Commercial Solutions, cat. no. 60234)
-
TOPO TA Cloning Kit for sequencing (Invitrogen, cat. no. K4500-01)
-
Agar granulated (Fisher, cat. no. BP1423-500)
-
X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside, 40 mg ml−1; Invitrogen, cat. no. 15520-018)
-
N,N-Dimethylformamide (Sigma-Aldrich, cat. no. 40255)
-
Luria-Bertani medium (LB medium; AccuGENE, cat. no. 51217)
-
Ampicillin (Fisher Scientific, cat. no. BP-1760-25)
-
Thermometer (Fisher Scientific, cat. no. 14983 10C)
-
Cell spreader (Fisher Scientific, cat. no. 08-100-13)
EQUIPMENT
-
Microscope (Olympus, cat. no. IX70)
-
Centrifuge (Eppendorf, cat. no. 5810R)
-
Microcentrifuge (Eppendorf, Minispin plus)
-
Heat block (Thermolyne, cat. no. DB17615)
-
Microwave (Kenmore)
-
Weight scale (Denver Instrument, cat. no. SI-602)
-
Analytical balance (Denver Instrument, cat. no. SI-64)
-
Flow cytometer (Becton Dickinson, FACScan)
-
Series II water-jacketed CO2 incubator (Thermo)
-
Class II biological safety cabinet, SterilGARD III advance (The Baker Company, Model SG603A)
-
Orbital shaker (Thermo, Model 420)
-
MyCycler thermal cycler from Bio-RAD
-
Purifier filtered PCR enclosure (Labconco)
-
Electrophoresis power supply (Bio-Rad, Power PAC 200)
-
UV transilluminator (Bio-Rad)
-
Spectrophotometer (Bio-Rad, SmartSpec plus)
-
Speedvac concentrator (Thermo, DNA120OP-115)
-
Water bath (Fisher Scientific, Isotemp 105)
-
Autoclave (Tuttnauer, model no. cat2007)
-
Isotemp incubator (Fisher Scientific)
REAGENT SETUP
Buffers and solutions
-
(i) Washing buffer: To 1 liter of DPBS, add 4.5 g of glucose and 5 ml of 1 M MgCl2. Store at 4 °C for up to 3 months. (ii) Binding buffer: To 1 liter of DPBS, and 4.5 g of glucose, 100 mg tRNA, 1 g BSA and 5 ml of 1 M MgCl2. Store at 4 °C for up to 1 month. (iii) 70% ethanol: To prepare 1 liter of 70% ethanol, add 700 ml of water to 300 ml of absolute ethanol.
Primers
-
Primers should be designed using appropriate software such as Integrated DNA Technologies (http://www.idtdna.com/analyzer/Applications/OligoAnalyzer/Default.aspx), according to standard primer design considerations. These include reasonable annealing temperature, >50% G-C content, avoidance of primer heterodimers and primer self-dimers; for example, 5′-FITC- ATCCAGAGTGACGCAGCA-3′ and 5′-biotin-ACTAAGCCACCGTGTCCA-3′.
Random library
-
Use the FITC-labeled primer at the 5′ end and the complementary strand of the biotin-labeled primer at the 3′ end flanking the random region between 25 and 60 nucleotides. Avoid a library with strong interaction between the constant regions. An example of a good library is shown at 5′-ATCCAGAGTGACGCAGCA-N40-TGGACACGGTGGCTTAGT-3′. The design of primers and library is very significant, as the efficiency of PCR amplification is the key to a successful selection. The value of 'N' signifies the potential number of sequences, as defined by the formula 4N, where 4 represents the four deoxyribonucleotides. Higher 'N' provides a more diverse library pool, generally ranging from 25 to 60.
Oligonucleotide synthesis and purification
-
The designed DNA library and primers can be synthesized and purified in the laboratory (if a facility is available) or purchased from commercial sources such as Integrated DNA Technologies. All synthesized oligonucleotides are purified by HPLC. For instance, in our laboratory we use reverse-phase HPLC with a C18 column (250 mm × 4.6 mm, 5-μm particle size) and a gradient mobile-phase mixture of acetonitrile and aqueous 0.1 M triethyl ammonium amine. Typically, a 1-μmol synthesis scale produces ∼50 nmol (DNA library) and 100–150 nmol of DNA primers each. Store the stock of pure oligonucleotides in higher concentrations (0.5–1.0 mM) in Tris-EDTA buffer.
EQUIPMENT SETUP
Software
-
Use the following software or their equivalents: (i) Integrated DNA Technologies, mfold (primers, library design and aptamer structure prediction), (ii) Winmdi, Cell Quest or FCS Express (analysis of flow cytometry data), (iii) ClustalX 1.83 (sequence alignment) and (iv) Systat Software, SigmaPlot for apparent dissociation constant determination.
Procedure
-
1
Prepare cells for the experiment. This procedure will present the detailed cell-SELEX protocol for both suspension and adherent cultured cell lines. For adherent cells, there are two options: the experiment can be performed either directly in a culture dish or using dissociated cells. If using dissociated cells, prepare them by following the steps given in Box 1 before following the procedure given below. If performing the experiment in a culture dish, refer to Box 2 and continue with the procedure from Step 61.
Initial DNA library pool preparation
Timing 20 min
-
1
Add 20 μl of 0.5 mM (10 nmol) DNA library to 350 μl of binding buffer, mix and heat the mixture at 95 °C for 5 min. Snap-cool on ice and keep on ice until ready to use (same day).
Pause point
If selection is not accomplished on the same day, store the DNA library pool at −20 °C. Thaw library on ice whenever ready to use. The denaturation step is not necessary once DNA has been thawed on ice.
Critical Step
Heating the DNA at 95 °C and subsequent fast cooling on ice are important to create folded ssDNA.


Preparation of target cells: cell viability
Timing 30 min
-
2
Determine cell viability using Trypan blue exclusion assay.
Critical Step
Cell viability assessment is very important, especially for suspension cells. Too many dead cells will seriously affect the efficiency of selection. DNA can nonspecifically adhere to and enter dead cells. This will cause the loss of important sequences, delay of enrichment or even the failure of selection. As the highest cell viability is ideal, it is preferable to use more than 95% viable cells.
Preparation of target cells: cell number
Timing 20 min
-
3
Determine the concentration of cells using a hemocytometer. On the basis of cell count, determine what volume will correspond to the number of cells needed for a specific round of selection.
Critical Step
For the first round of selection, use the highest number of cells possible for collecting specific sequences, preferably between 5 and 10 million cells.
Preparation of target cells: washing by centrifugation
Timing 30 min
-
4
Take the cell volume that yields the desired number of cells into a 15-ml centrifuge tube and centrifuge cells at 150g for 3 min at 4 °C. Remove the supernatant and add 3 ml of washing buffer. Resuspend cells by pipetting up and down, tapping the bottom of the centrifuge tube or mild vortexing. Pellet the cells at the same speed and duration. Repeat washing once more.
Critical Step
Avoid strong vortexing as it can cause cell breakage and may eventually affect selection.
Incubation of cells with DNA library pool
Timing 1 h
-
5
Resuspend cells in 330 μl of binding buffer. Add all of the 370 μl snap-cooled DNA library (Step 2) to the 330 μl cell suspension, mix thoroughly and incubate the mixture on ice for 1 h on a rotary shaker. Incubation temperature depends on the purpose of selection. In general, any temperature between 4 and 37 °C is applicable for this protocol; however, higher temperatures such as 37 °C can cause internalization. Once aptamer is selected at 4 °C, it is important to test the binding at 37 °C. This does not mean that all aptamers selected at 4 °C will not bind at 37 °C. From observation, most of the aptamers will bind very well at 37 °C, especially those with very high affinity. Some of the aptamers generated at 4 °C have been used in various applications at 37 °C (refs. 30,33). The most important point here is to generate aptamers with high affinity by gradually increasing the stringency of selection to generate aptamers that can bind at diverse binding conditions. For instance, aptamers that have been developed using DPBS as the binding buffer can still recognize the target in culture medium.
Critical Step
Because of the large number of cells relative to the small volume of incubation medium, it is sometimes common to see cells settling at the bottom of the tube, even while shaking, so occasionally check and resuspend the cells.
Washing step
Timing 40 min
-
6
After incubation, centrifuge cells at 150g for 3 min at 4 °C. Remove supernatant containing unbound sequences and resuspend cell pellets in 3 ml of washing buffer. Shake for about 30 s and centrifuge again using the same condition. Remove supernatant carefully with transfer pipette, avoiding cell loss. Quickly spin down (30 s) the residual buffer on the wall of the tube. To avoid cell loss, remove a small amount of residual buffer with folded kimwipe. Repeat the washing procedure two more times for a total of three washings.
Elution of bound sequences
Timing 15 min
-
7
For the first round of selection, elute the bound sequences in water. Add 500 μl of DNase-free water to the cell pellet. Resuspend cells and transfer cell suspension into a 1.5-ml microfuge tube.
Critical Step
Only in the first round is DNA eluted in water. In subsequent rounds, DNA is eluted in binding buffer. As the entire eluted pool is amplified by PCR, use only DNase-free water for the elution of the bound DNA sequences during the first round of selection. Otherwise, if the DNA is eluted in DPBS, the salts with high concentration will affect the efficiency of PCR.
-
8
Heat the cell mixture at 95 °C for 10 min, centrifuge at 13,100g for 5 min and collect supernatant containing eluted DNA.
Critical Step
Do not perform negative selection at this point. Minimize the loss of eluted sequences during the first selection round, as each sequence is theoretically represented only once, and when any sequence is lost, it can never be recovered.
Pause point
Store eluted DNA at −20 °C.
PCR procedures: PCR amplification of the entire first selected pool
Timing 1 h
-
9
This step is performed for pools generated from only the first selection. Set up a 1,000-μl PCR amplification reaction volume as shown below: adjust the volume of water to compensate for the total volume if the DNA selected pool volume differs.
Table 3 -
10
Mix thoroughly and pipet 100 μl into 10 individual tubes (using 96-well thermal cycler) for 100 μl per reaction, or pipet 200 μl into five individual tubes (60-well thermal cycler) for 200 μl per reaction.
Critical Step
Note that this step is only performed to step up the copies of individual sequences and is not necessarily preparative. Too many cycles may produce nonspecific amplicons, and this will affect the purity of the DNA library pool. Choose 8–10 cycles.
-
11
Perform PCR amplification. For example, in primer set 2 given above, use the following amplification conditions: denaturation at 95 °C for 30 s, annealing at 56.3 °C for 30 s and elongation at 72 °C for 30 s, followed by final extension for 3 min at 72 °C.
Critical Step
Always keep Taq polymerase at − 20 °C until ready to use. Thaw and keep all other PCR reagents on ice.
Table 4 -
12
After PCR amplification, pool all reaction mixtures together (total 1,000 μl). There is no need to perform agarose gel electrophoresis to assess template amplification. In most cases, the first-round PCR amplification product is insufficient for detection by ethidium bromide.
Critical Step
Note that the PCR-amplified pool can potentially contaminate PCR reagents and primers; therefore, keep them separated.
-
13
Label the PCR product as the first selected pool.
Pause point
Store the DNA pool at −20 °C.
PCR procedures: determine the optimum number of cycles for preparative PCR
Timing 1 h 30 min
-
14
The total PCR reaction mixture volume for each tube is 50 μl, with the amplified first selected pool at 10% serving as the template. Choose PCR samples at the following cycles: 4, 6, 8, 10 and 12, and one negative control at the twelfth cycle. Add the following reagents to PCR tubes as shown below:
Table 5 -
15
Mix thoroughly and pipet 50 μl of the positive reaction mixture into five individual tubes.
-
16
Perform PCR amplification using the PCR program described in Step 12 with the maximum number of cycles as 12.
-
17
Open the thermal cycler and take samples at the specified cycles as shown in Step 15.
PCR procedures: agarose gel electrophoresis
Timing 1 h 40 min
-
18
Prepare 3% agarose gel.
-
19
Prepare agarose gel samples as shown below:
Table 6 -
20
Load samples in lanes and perform electrophoresis at 100 V for 40 min.
-
21
Remove the gel, stain with ethidium bromide, and observe the bands under UV light and take image of the gel (Fig. 2).
Figure 2: Agarose gel electrophoresis image showing the products of the various cycles of selected DNA library amplification. 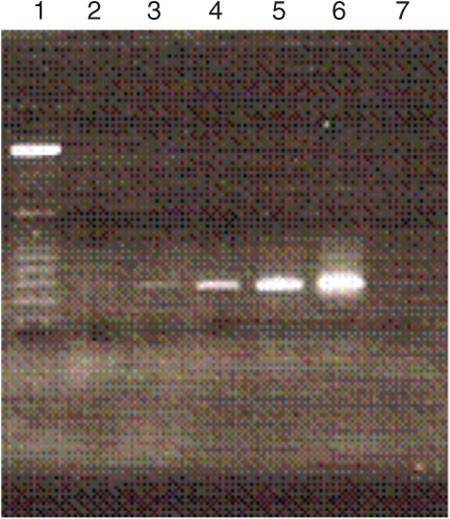
Lane 1 = 25-bp ladder; lane 2 = 4 cycles; lane 3 = 6 cycles; lane 4 = 8 cycles; lane 5 = 10 cycles; lane 6 = 12 cycles; and lane 7 = negative control.
-
22
Select a cycle number that yields a bright band without nonspecific amplicons. On the basis of Figure 2, a good example is the tenth cycle (lane 5). In preparative PCR, use this cycle of amplification to produce more PCR products for the preparation of ssDNA required for the second round of selection.
Pause point
One may store the library pool at −20 °C.

PCR procedures: preparative PCR
Timing 2 h
-
23
Use the same reagent concentrations as listed in Step 15, but in larger volumes. For the second round of selection, 1,000 μl of total PCR reaction mixture is enough to generate the required amount of ssDNA. Moreover, the entire ssDNA pool that is generated here is used for selection, whereas in the higher rounds, some are used for monitoring the selection progress. Prepare PCR reaction mixture as shown below:
Table 7 Critical Step
It is not necessary to perform control PCR for the preparative process once the cycle optimization control data are clean.
-
24
Mix thoroughly and pipet 100 μl or 200 μl of the mixture into ten PCR tubes (96-well thermal cycler) or five PCR tubes (60-well thermal cycler). Close the cap securely and perform PCR using the program described in Step 12, but with the chosen number of cycles.
-
25
After PCR, pool all PCR products together and perform gel electrophoresis (Step 20) to confirm the PCR product (Fig. 3).
Figure 3: Agarose electrophoresis gel image of preparative selected DNA library pool, confirming the efficiency of amplification. 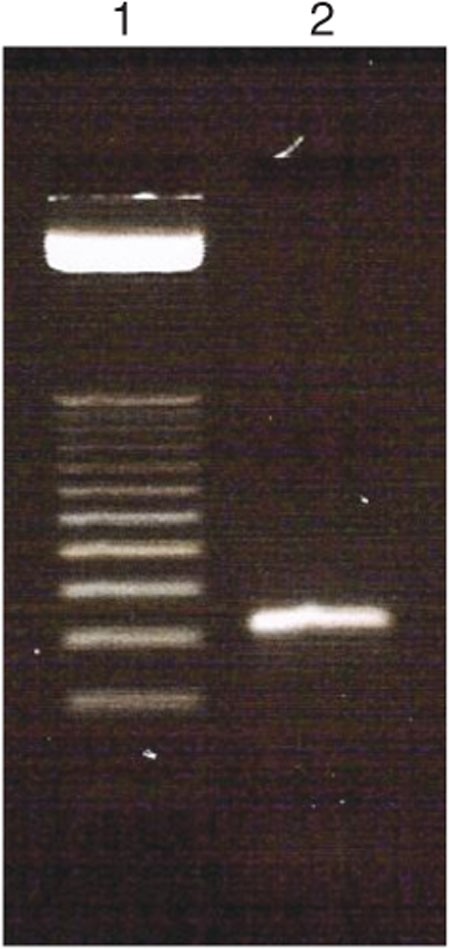
Lane 1 = 25-bp ladder and lane 2 = PCR product of 10 cycles.
Pause point
One may store the library pool at −20 °C.

Preparation of ssDNA from PCR product
Timing 40 min
-
26
Insert a filter and secure it firmly at one end of an empty DNA synthesis column. Remove the plunger from a 10-ml syringe without a needle and insert the empty syringe at the other end of the DNA synthesis column.
-
27
Mount the setup on a clamp. Add 200 μl of streptavidin Sepharose beads suspension. Insert the plunger and allow the storage buffer to drain.
-
28
Wash the beads with 2.5 ml of 1× DPBS.
-
29
Pass the PCR product three times through the column. Wash the beads again with 2.5 ml of DPBS.
-
30
Add 500 μl of 200 mM NaOH, insert the plunger and collect the eluate containing the FITC-labeled ssDNA.
Critical Step
Allow the PCR product and the NaOH solution to pass through the column gradually. Pushing the plunger hard will reduce the efficiency of the ssDNA yield. Furthermore, do not remove the plunger from the syringe while the syringe is connected to the column containing the beads. Always disconnect the syringe from the column before removing the plunger.
Desalting ssDNA
Timing 30 min
-
31
Wash the NAP5 column with at least 15 ml of deionized water. Add 500 μl of eluted ssDNA. Allow the entire sample volume to drain and then elute the ssDNA with 1,000 μl of DNase-free water.
Critical Step
Always keep the surface of the desalting column moist.
Quantifying eluted ssDNA
Timing 5 min
-
32
Determine the concentration of ssDNA by UV absorbance at 260 nm.
Drying ssDNA
Timing 3 h
-
33
Concentrate the ssDNA using a DNA Speedvac dryer. Technically, this step is the end of one round of DNA aptamer selection.
Pause point
One may store the dried ssDNA at −20 °C and resuspend it in binding buffer when needed for the next round of selection.
Second round of selection
Timing 2 h 30 min
-
34
Resuspend ssDNA in binding buffer to obtain a concentration of 1,000 nM.
-
35
Denature and renature the ssDNA pool as described in Step 2.
Preparation of cells for second round of selection
-
36
Follow the same procedure used in Steps 3 and 4 but with less than 5 million cells.
Washing
-
37
Repeat Step 5.
DNA incubation
-
38
Add 200 μl of ssDNA (Step 36) to 200 μl of cell suspension. Repeat Steps 7, 8 (use 600 μl binding buffer) and 9.
Pause point
One may store the selected DNA pool at −20 °C.
Negative selection procedure Cell viability and number of suspension cells
Timing 2 h 30 min
-
39
Check cell viability and count cells as described in Steps 3 and 4. Use at least 10 million control cells.
Critical Step
The viability of control cells is important in negative selection because sequences that nonspecifically adhere to and enter dead cells will cause the loss of specific sequences. To the extent possible, use more than 95% viable cells.
-
40
Wash cells as described in Step 5.
-
41
Incubate cell pellet with the entire eluted DNA pool from positive selection on ice with continuous shaking for 1 h.
-
42
After incubation, spin down the cells containing the binding sequences at 3,059g for 5 min.
-
43
Recover the supernatant containing sequences that do not bind to control cells and label this pool as the second selected DNA pool.
Pause point
One may store the selected DNA pool at −20 °C.
PCR amplifications Determination of the optimum number of cycles for preparative PCR of the second selected DNA pool
Timing 3 h 10 min
-
44
Estimate the minimum and maximum number of cycles to be assessed on the basis of the previous PCR cycle optimization procedures (Steps 15–23), but use the second selected DNA pool as the template. Reduce the number of cycles to be assessed to about four and maintain cycle intervals of two.
Preparative PCR
Timing 2 h
-
45
Follow the same procedure as described for Steps 24–26 (Fig. 4, panel b).
Figure 4: Agarose gel electrophoresis image showing products of cycle optimization of the second selected DNA library pool amplification. 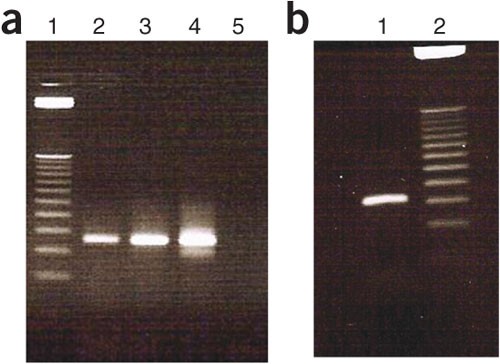
(a) Lane 1 = 25-bp ladder; lane 2 = 8 cycles; lane 3 = 10 cycles; lane 4 = 12 cycles; and lane 5 = negative control. (b) Lane 1 = 10 cycles and lane 2 = 25-bp DNA ladder.
Pause point
One may dry the selected DNA pool and store it at −20 °C.

ssDNA preparation
Timing 40 min
-
46
Follow Steps 27–31.
Desalting, quantifying and drying
Timing 3 h 35 min
-
47
Follows Steps 32–34.
Subsequent rounds of DNA aptamer selection
Timing 2 h–2 h 30 min per round
-
48
For subsequent rounds of selection (typically, between 12 and 16 rounds), follow the same steps as outlined for the second round of selection (Steps 35–48), but with the following modifications as selection progresses.
(1) Gradually reduce the cell number. For suspension cells, use from 10 million (first round) to 1 million (fifth round and beyond), and for adherent cells, use a 100-mm × 20-mm culture dish from the first to the fourth rounds and a 60-mm × 15-mm culture dish for the fifth round and beyond.
(2) Gradually reduce the incubation time from 1 h to 30 min.
(3) Increase the washing time and washing buffer volume.
(4) Add 10% of FBS from the fourth round of selection and gradually increase it to about 20%.
Monitor the progress of selection
-
49
Add 50 pmol of ssDNA pool in 100 μl binding buffer.
-
50
Increase the volume of the preparative PCR from the fourth round so that there will be enough ssDNA for both selection and monitoring.
Flow cytometry binding assays
Timing 2 h 30 min
-
51
Count the number of cells and check viability of both target and control cells (Steps 2 and 3). Use about 5 × 105 cells per sample.
-
52
Centrifuge target and control cells and wash each of them two times. Resuspend each cell line in binding buffer (1 × 106 cells per 100 μl).
-
53
Pipet 50 μl of either cell line into different flow cytometry tubes. The number of tubes depends on the number of DNA pools to be tested plus one control pool.
-
54
Add 50 μl of the ssDNA selected pool to each tube containing cells. Use unselected DNA library as a negative control pool. Add 10% of FBS.
-
55
Incubate the mixture on ice for 30 min.
-
56
Wash two times and then resuspend cells in 200 μl of binding buffer.
-
57
Determine the fluorescence signal intensity of FITC (FL-1) of each tube for target and control cells. Use unselected FITC library signal as the control DNA pool to set the fluorescence background.
-
58
Analyze flow cytometry data using Winmdi, Cellquest or FCS Express software (Fig. 5a and b).
Figure 5: Flow cytometry binding assay to assess the binding of initially selected DNA pools. 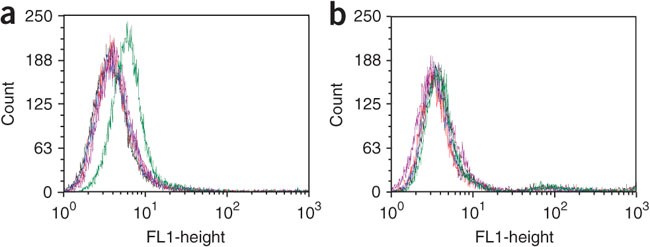
(a,b) Target and (a) control (b) cell lines. Red = unselected DNA library; green = round 4; blue = round 5; purple = round 6; and light blue = round 7. There is a clear fluorescence signal shift in the seventh round with positive cells, but not with negative cells. The transition of signal strength from round 6 to round 7 shows a detectable onset of enrichment.

End of selection
-
59
Selection is completed (i) when we observe a significant difference between the fluorescence signal intensity of the control background and the selected DNA pools and (ii) at a point at which no significant signal difference can be discerned between two or three successive selected pools (Fig. 6a and b).
Figure 6: Flow cytometry binding assay. 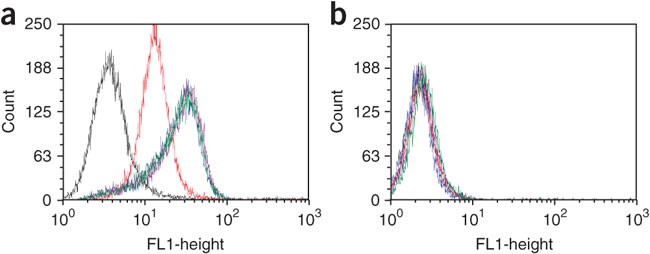
Shown: assessment of the binding of subsequent selected DNA pools to (a) target and (b) control cell lines. Red = unselected DNA library; yellow = round 8; brown = round 9; green = round 10; and dark green = round 11. No significant fluorescence shift from the 9th to the 11th round of selection (end of selection).

Cloning and sequencing of enriched pools
Timing 2 h
-
60
Select enriched pools on the basis of flow cytometry results.
-
61
Synthesize both primers without any label (FITC or biotin) and purify by HPLC.
-
62
Use these primers to optimize PCR amplification conditions for the pools to be cloned using the same amplification program, but extend the final extension time to 15 min to allow for complete adenylation.
-
63
Perform gel electrophoresis to assess the PCR amplification efficiency (Fig. 7), and select the PCR products with the cleanest band for cloning.
Figure 7: Agarose gel electrophoresis image to assess the PCR-amplified selected DNA pool for cloning. 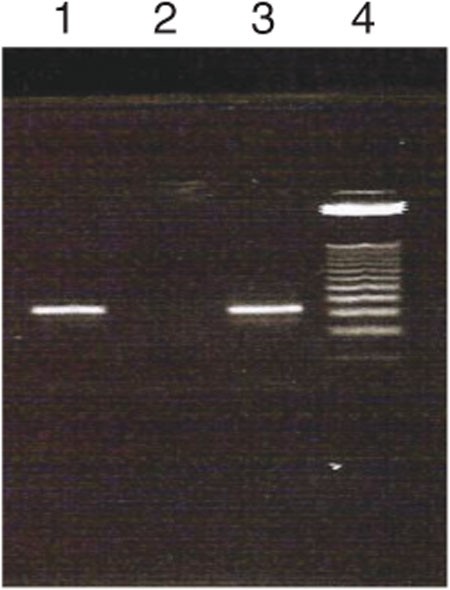
Lane 1 = selected pool; lane 2 = control; lane 3 = selected pool; and lane 4 = 25-bp DNA ladder.
Critical Step
Avoid nonspecific amplified sequences in PCR product for cloning and sequencing.

Cloning using the TOPO TA cloning kit
Timing 1 h 30 min
-
64
Prepare agar plates (Box 3).
-
65
Perform the TOPO TA cloning procedure as described by the manufacturer.
-
66
After 18 h, observe plates and choose the positive clones (white clones); perform any high-throughput sequencing.

Analyze sequencing data
Timing 5 h
-
67
Sequencing of the DNA pool produces both sense and antisense strands. To analyze the data, separate sense strands from antisense strands. To accomplish this, follow Steps 69–76, as detailed below.
-
68
Save the data using two file names, such as 'sense' and 'antisense'.
-
69
Using the sense file, search all sequences containing the forward primer and color code them.
-
70
Delete any sequences without the forward primer code.
-
71
Remove both the forward primer from the 5′ end of the sequences and the reverse primer at the 3′ end, leaving only the initial randomized region.
-
72
Repeat Steps 70–72 for the antisense.
-
73
Change all antisense sequences (Step 73) to the complementary strand using the Integrated DNA Technologies website.
-
74
Combine all these sequences together and determine the sequence alignment using sequence alignment programs such as ClustalX1.83.
-
75
Group sequences into families (Fig. 8).
Figure 8 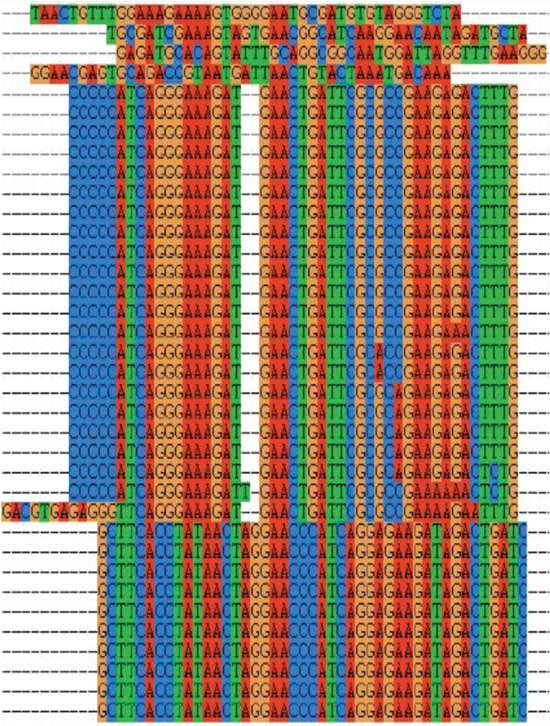
Sample data of sequence alignment using ClustalX1.83 showing, in two parts, large homologous families and nonhomologous sequences of potential aptamer sequences.

Synthesis and purification of potential aptamer candidates
Timing ∼24 h
-
76
Select representative sequences from the different families (Step 76).
-
77
Add the truncated primer regions to obtain the full length and predict structures.
Critical Step
Do not truncate sequences at this stage. Test full sequences to identify aptamers before truncation to obtain the minimal sequence length.
-
78
Chemically synthesize selected sequences and label them directly with (i) a fluorophore, such as FITC or cy5 or (ii) biotin. Detect biotin-labeled aptamers with streptavidin-conjugated fluorophore, such as AlexaFluor 488, phycoerythrin or phycoerythrin-cy5.5.
-
79
Purify sequences by HPLC and quantify.
Pause point
One may store DNA at −20 °C.
Screening of potential aptamer candidates
Timing 2 h
-
80
Prepare working dilutions of potential aptamer candidates at 500 nM using binding buffer.
-
81
Prepare target cells for flow cytometry as described in Steps 52–53 (suspension cells) and dissociated adherent monolayer cells (Box 1).
-
82
Add 100 μl of cell suspension to 100 μl of potential aptamer candidate (∼250 nM final concentration of aptamer) and 10% FBS.
-
83
For control, use unselected library.
-
84
Incubate the cells and DNA mixture for 30 min at 4 °C.
-
85
Wash the cells and determine fluorescence signal. If DNA is labeled with biotin, after washing, incubate the cell–aptamer complex with streptavidin-conjugated dye for 10 min at 4 °C. Wash and determine the fluorescence signal.
-
86
Analyze the flow cytometry data using Winmdi, Cellquest or FCS Express software (Fig. 9).
Figure 9: Binding assay of FITC-labeled potential aptamer candidates using flow cytometry. 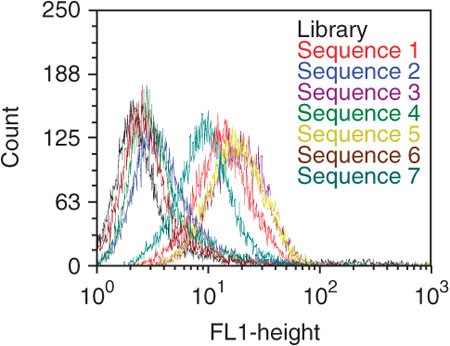
Aptamers were incubated with target cells at 250 nM final concentration. Four sequences (sequences 1, 3, 5 and 7) showed significantly higher fluorescence intensity than the control (black).

Specificity of aptamer candidates
Timing 3 h
-
87
Follow Steps 82–87 to test the selectivity of aptamers using the control cell line and other related cell lines available.
Critical Step
Some of the selected aptamers can also bind to the controls and other related cancer cells.
Determination of the apparent dissociation constant of aptamers
Timing 4 h
-
88
Prepare cells for flow cytometry (Steps 52 and 53).
Critical Step
A larger number of cells is needed for this procedure.
-
89
Prepare at least six different dilutions of the aptamer and control library in triplicate.
-
90
Incubate cells with DNA solution.
-
91
Use any flow cytometry analysis software to determine the mean fluorescence intensity of each concentration for both the aptamer and the control unselected library.
-
92
Subtract the mean fluorescence background of controls from the corresponding mean fluorescence intensity of the aptamer.
-
93
Using SigmaPlot, the apparent dissociation constants (Kd) of the aptamer-cell interaction are obtained by fitting the dependence of fluorescence intensity of specific binding on the concentration of aptamers to the one-site saturation equation Y=Bmax X/(Kd+X). Figure 10 shows the Kd determination curve using SigmaPlot.
Figure 10: Typical apparent dissociation curve of aptamer-cell interaction as analyzed by Sigma Plot. 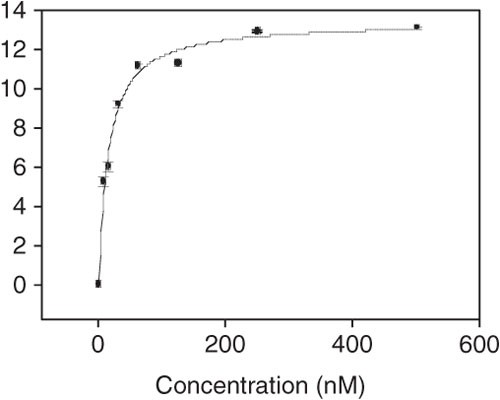
In this graph, Kd = 15.1 ± 1.3 nM and R = 0.9934.
Critical Step
Higher fluorescence signal intensity is not an indication of the aptamer's dissociation constant. It only shows the relative abundance of the target molecule.

Troubleshooting
Troubleshooting advice can be found in Table 2.
Timing
Steps 1 and 2, Initial DNA library pool preparation: 20 min
Step 3, Preparation of target cells: cell viability: 30 min
Step 4, Preparation of target cells: cell number: 20 min
Step 5, Preparation of target cells: washing by centrifugation: 30 min
Step 6, Incubation of cells with DNA library pool: 1 h
Step 7, Washing step: 40 min
Steps 8 and 9, Elution of bound sequences: 15 min
Steps 10–14, PCR procedures: PCR amplification of the entire first selected pool: 1 h
Steps 15–18, PCR procedures: determine the optimum number of cycles for preparative PCR: 1 h 30 min
Steps 19–23, PCR procedures: agarose gel electrophoresis: 1 h 40 min
Steps 24–26, PCR procedures: preparative PCR: 2 h
Steps 27–31, Preparation of ssDNA from PCR product: 40 min
Step 32, Desalting ssDNA: 30 min
Step 33, Quantifying eluted ssDNA: 5 min
Step 34, Drying ssDNA: 3 h
Steps 35–39, Second round of selection: 2 h 30 min
Steps 40–44, Cell viability and number of suspension cells: 2 h 30 min
Step 45, Determination of the optimum number of cycles for preparative PCR of the second selected DNA pool: 3 h 10 min
Step 46, Preparative PCR: 2 h
Step 47, ssDNA preparation: 40 min
Step 48, Desalting, quantifying and drying: 3 h 35 min
Steps 49–51, Subsequent rounds of DNA aptamer selection: 2 h–2 h 30 min per round
Steps 52–60, Flow cytometry binding assays and end of selection: 2 h 30 min
Steps 61–64, Cloning and sequencing of enriched pools: 2 h
Steps 65–67, Cloning using the TOPO TA cloning kit: 1 h 30 min
Steps 68–76, Analyze sequencing data: 5 h
Steps 77–80, Synthesis and purification of potential aptamer candidates: ∼24 h
Steps 81–87, Screening of potential aptamer candidates: 2 h
Step 88, Specificity of aptamer candidates: 3 h
Steps 89–94, Determination of the apparent dissociation constant of aptamers: 4 h
Anticipated results
All potential aptamer candidates that recognize any target on the cell surface will show a fluorescence signal intensity significantly higher than that of the unselected control library (Fig. 9). Repeat screening to confirm the results. For sequences that do not show any significant binding to target cells, repeat the screening process with higher concentrations (500 nM or 1 μM final concentration). The importance of this step is underscored by the wide range of binding affinities of selected aptamers. Drop any candidate that will not bind at these concentrations. Characterize the generated aptamers by determining their specificity and affinity, as described in the general procedure. In general, the affinities of selected aptamers are in the lower to mid-nanomolar range. It can also be expected that some selected aptamers will recognize both target and control cells29,32. In general, however, the procedure is effective in generating many aptamers, some of which are very specific to the target and possibly to closely related cell lines.
References
Tuerk, C. & Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 (1990).
Ellington, A.D. & Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 (1990).
Ciesiolka, J. & Yarus, M. Small RNA-divalent domains. RNA 2, 785–793 (1996).
Hofmann, H.P., Limmer, S., Hornung, V. & Sprinzl, M. Ni2+-binding RNA motifs with an asymmetric purine-rich internal loop and a G-A base pair. RNA 3, 1289–1300 (1997).
Rajendran, M. & Ellington, A.D. Selection of fluorescent aptamer beacons that light up in the presence of zinc. Anal. Bioanal. Chem. 390, 1067–1075 (2008).
Geiger, A., Burgstaller, P., von der Eltz, H., Roeder, A. & Famulok, M. RNA aptamers that bind l-arginine with sub-micromolar dissociation constants and high enantioselectivity. Nucleic Acids Res. 24, 1029–1036 (1996).
Connell, G.J., Illangesekare, M. & Yarus, M. Three small ribooligonucleotides with specific arginine sites. Biochemistry 32, 5497–5502 (1993).
Famulok, M. Molecular recognition of amino acids by RNA-aptamers: an l-citrulline binding RNA motif and its evolution into an l-arginine binder. J. Am. Chem. Soc. 116, 1698–1706 (1994).
Harada, K. & Frankel, A.D. Identification of two novel arginine binding DNAs. EMBO J. 14, 5798–5811 (1995).
Wallis, M.G. et al. In vitro selection of a viomycin-binding RNA pseudoknot. Chem. Biol. 4, 357–366 (1997).
Wallace, S.T. & Schroeder, R. In vitro selection and characterization of streptomycin-binding RNAs: recognition discrimination between antibiotics. RNA 4, 112–123 (1998).
Nieuwlandt, D., Wecker, M. & Gold, L. In vitro selection of RNA ligands to substance P. Biochemistry 34, 5651–5659 (1995).
Williams, K.P. et al. Bioactive and nuclease-resistant l-DNA ligand of vasopressin. Proc. Natl Acad. Sci. USA 94, 11285–11290 (1997).
Tuerk, C., MacDougal, S. & Gold, L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl Acad. Sci. USA 89, 6988–6992 (1992).
Chen, H., McBroom, D.G., Zhu, Y.-Q., Gold, L. & North, T.W. Inhibitory RNA ligand to reverse transcriptase from feline immunodeficiency virus. Biochemistry 35, 6923–6930 (1996).
Dang, C. & Jayasena, S.D. Oligonucleotide inhibitors of Taq DNA polymerase facilitate detection of low copy number targets by PCR. J. Mol. Biol. 264, 268–278 (1996).
Kubik, M.F., Stephens, A.W., Schneider, D., Marlar, R.A. & Tasset, D. High-affinity RNA ligands to human α-thrombin. Nucleic Acids Res. 22, 2619–2626 (1994).
Jellinek, D. et al. Potent 2′-amino-2′-deoxypyrimidine RNA inhibitors of basic fibroblast growth factor. Biochemistry 34, 11363–11372 (1995).
Green, L.S. et al. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry 35, 14413–14424 (1996).
Wiegand, T.W. et al. High-affinity oligonucleotide ligands to human IgE inhibit binding to Fc epsilon receptor I. J. Immunol. 157, 221–230 (1996).
Davis, K.A., Lin, Y., Abrams, B. & Jayasena, S.D. Staining of cell surface human CD4 with 2′-F-pyrimidine-containing RNA aptamers for flow cytometry. Nucleic Acids Res. 26, 3915–3924 (1998).
Smith, D., Kirschenheuter, G.P., Charlton, J., Guidot, D.M. & Repine, J.E. In vitro selection of RNA-based irreversible inhibitors of human neutrophil elastase. Chem. Biol. 2, 741–750 (1995).
Mallikaratchy, P., Stahelin, R.V., Cao, Z., Chob, W. & Tan, W. Selection of DNA ligands for protein kinase C-δ. Chem. Commun. 3229–3231 (2006).
Mendonsa, S.D. & Bowser, M.T. In Vitro selection of high-affinity DNA ligands for human IgE using capillary electrophoresis. Anal. Chem. 76, 5387–5392 (2004).
Morris, K.N., Jensen, K.B., Julin, C.M., Weil, M. & Gold, L. High affinity ligands from in vitro selection: complex targets. Proc. Natl Acad. Sci. USA 95, 2902–2907 (1998).
Blank, M., Weinschenk, T., Priemer, M. & Schluesener, H. Systematic evolution of a DNA aptamer binding to rat brain tumor microvessels. selective targeting of endothelial regulatory protein pigpen. J. Biol. Chem 276, 16464 (2001).
Hicke, B.J. et al. Tenascin-C aptamers are generated using tumor cells and purified protein. J. Biol. Chem. 276, 48644–48654 (2001).
Wang, C. et al. Single-stranded DNA aptamers that bind differentiated but not parental cells: subtractive systematic evolution of ligands by exponential enrichment. J. Biotechnol. 102, 15–22 (2003).
Cerchia, L. et al. Neutralizing aptamers from whole-cell SELEX inhibit the RET receptor tyrosine kinase. PLoS Biol. 3, 0697–0704 (2005).
Shangguan, D. et al. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl Acad. Sci. USA 103, 11838–11843 (2006).
Tang, Z. et al. Selection of aptamers for molecular recognition and characterization of cancer cells. Anal. Chem. 79, 4900–4907 (2007).
Chen, H.W. et al. Molecular recognition of small-cell lung cancer cells using aptamers. ChemMedChem 3, 991–1001 (2008).
Sefah, K. et al. Molecular recognition of acute myeloid leukemia using aptamers. Leukemia 23, 235–244 (2009).
Shangguan, D. et al. Identification of liver cancer-specific aptamers using whole live cells. Anal. Chem. 80, 721–728 (2008).
Pan, W. et al. Isolation of virus-neutralizing RNAs from a large pool of random sequences. Proc. Natl Acad. Sci. USA 92, 11509–11513 (1995).
Kumar, P.K.R. et al. Isolation of RNA aptamers specific to the NS3 protein of hepatitis C virus from a pool of completely random RNA. Virology 237, 270–282 (1997).
Misono, T.S. & Kumar, P.K.R. Selection of RNA aptamers against human influenza virus hemagglutinin using surface Plasmon resonance. Anal. Biochem. 342, 312–317 (2005).
Gopinath, S.C.B. et al. An RNA aptamer that distinguishes between closely related human influenza viruses and inhibits haemagglutinin-mediated membrane fusion. J. Gen. Virol. 87, 479–487 (2006).
Tang, Z., Parekh, P., Turner, P., Moyer, R.W. & Tan, W. Generating aptamers for recognition of virus-infected cells. Clin. Chem. 55, 813–822 (2009).
Bruno, J.G. & Kiel, J.L. In vitro selection of DNA aptamers to anthrax spores with electrochemiluminescence detection. Biosens. Bioelectron. 14, 457–464 (1999).
Hamula, C.L.A., Zhang, H., Guan, L.L., Li, X.-F. & Le, C. Selection of aptamers against live bacterial cells. Anal. Chem. 80, 7812–7819 (2008).
Floege, J. et al. Novel approach to specific growth factor inhibition in vivo: antagonism of platelet-derived growth factor in glomerulonephritis by aptamers. Am. J. Pathol. 154, 169–179 (1999).
Ostendorf, T. et al. The effects of platelet-derived growth factor antagonism in experimental glomerulonephritis are independent of the transforming growth factor–ß system. J. Am. Soc. Nephrol. 13, 658–667 (2002).
White, R.R. et al. Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for angiopoietin-2. Proc. Natl Acad. Sci. USA 100, 5028–5033 (2003).
Conrad, R. & Ellington, A.D. Detecting immobilized protein kinase C isozymes with RNA aptamers. Anal. Biochem. 242, 261–265 (1996).
Murphy, M.B., Fuller, S.T., Richardson, P.M. & Doyle, S.A. An improved method for the in vitro evolution of aptamers and applications in protein detection and purification. Nucleic Acids Res. 31, e110 (2003).
Ireson, C.R. & Kelland, L.R. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 5, 2957–2962 (2006).
Da Pieve, C., Williams, P., Haddleton, D.M., Palmer, R.M.J. & Missailidis, S. Modification of thiol functionalized aptamers by conjugation of synthetic polymers. Bioconjug. Chem. 21, 169–174 (2010).
Hicke, B.J. et al. DNA aptamers block L-selectin function in vivo. J. Clin. Invest. 98, 2688–2692 (1996).
Smith, J.E. et al. Aptamer-conjugated nanoparticles for the collection and detection of multiple cancer cells. Anal. Chem. 79, 3075–3082 (2007).
Kang, W.J., Chae, J.R., Cho, Y.L., Lee, J.D. & Kim, S. Multiplex imaging of single tumor cells using quantum-dot-conjugated aptamers. Small 5, 2519–2522 (2009).
Chu, T.C. et al. Aptamer:toxin conjugates that specifically target prostate tumor cells. Cancer Res. 66, 5989–5992 (2006).
Primo, C.D. et al. Systematic screening of LNA/2'-O-methyl chimeric derivatives of a TAR RNA aptamer. FEBS Lett. 581, 771–774 (2007).
Schmidt, K.S. et al. Application of locked nucleic acids to improve aptamer in vivo stability and targeting function. Nucleic Acids Res. 32, 5757–5765 (2004).
Gregory Biesecker, G., Dihel, L., Enney, K. & Bendele, R.A. Derivation of RNA aptamer inhibitors of human complement C5. Immunopharmacology 42, 219–230 (1999).
Golden, M.C, Collins, B.D., Willis, M.C. & Koch, T.H. Diagnostic potential of PhotoSELEX-evolved ssDNA aptamers. J. Biotechnol. 81, 167–178 (2000).
Cox, J.C. & Ellington, A.D. Automated selection of anti-protein aptamers. Bioorg. Med. Chem. 9, 2525–2531 (2001).
Kim, S.J., Kim, M.Y., Lee, J.H., You, J.C. & Jeong, S. Selection and stabilization of the RNA aptamers against the human immunodeficiency virus type-1 nucleocapsid protein. Biochem. Biophys. Res. Commun. 291, 925–931 (2002).
Bruno, J.G. & Kiel, J.L. Use of magnetic beads in selection and detection of biotoxin aptamers by electrochemiluminescence and enzymatic methods. Biotechniques 32, 178–183 (2002).
Bryant, K.F. et al. Binding of herpes simplex virus-1 US11 to specific RNA sequences. Nucleic Acids Res. 33, 6090–6100 (2005).
Noma, T. et al. A screening method for DNA aptamers that bind to specific, unidentified protein in tissue samples. Biotechnol. Lett. 28, 1377–1381 (2006).
Shamah, S.M., Healy, J.M. & Cload, S.T. Complex target SELEX. Acc. Chem. Res. 41, 130–138 (2008).
Shangguan, D. et al. Cell-specific aptamer probes for membrane protein elucidation in cancer cells. J. Proteome Res. 7, 2133–2139 (2008).
Mallikaratchy, P. et al. Aptamer directly evolved from live cells recognizes membrane bound immunoglobin heavy mu chain in Burkitt′s lymphoma cells. Mol. Cell Proteomics 6, 2230–2238 (2007).
Raddatz, M.-S.L. et al. Enrichment of cell-targeting and population-specific aptamers by fluorescence-activated cell sorting. Angew. Chem. Int. Ed. 47, 5190–5193 (2008).
Paul, A., Avci-Adali, M., Ziemer, G. & Wendel, H.P. Streptavidin-coated magnetic beads for DNA strand separation implicate a multitude of problems during cell-SELEX. Oligonucleotides 19, 243–254 (2009).
Acknowledgements
The authors would like to acknowledge many of our coworkers whose work is reported here. This work has been supported over the years by US National Institutes of Health and National Science Foundation and Florida State Biomedical Program grants. It is also supported by China 2009ZX10004-312 and a 985 project grant.
Author information
Authors and Affiliations
Contributions
D.S. and W.T. originated the concept of cell-SELEX; K.S., D.S., M.O., X.X. and W.T. were responsible for the experimental results and procedures of the project. K.S. wrote most of the manuscript, but all authors contributed to the writing and revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Sefah, K., Shangguan, D., Xiong, X. et al. Development of DNA aptamers using Cell-SELEX. Nat Protoc 5, 1169–1185 (2010). https://doi.org/10.1038/nprot.2010.66
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2010.66
This article is cited by
-
DNAzyme-based faithful probing and pulldown to identify candidate biomarkers of low abundance
Nature Chemistry (2024)
-
Generalizable anchor aptamer strategy for loading nucleic acid therapeutics on exosomes
EMBO Molecular Medicine (2024)
-
The Application of Aptamer and Research Progress in Liver Disease
Molecular Biotechnology (2024)
-
The application of Aptamer in biomarker discovery
Biomarker Research (2023)
-
A dynamic DNA tetrahedron framework for active targeting
Nature Protocols (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
