
Abstract
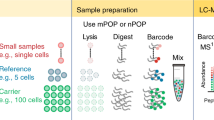
Mass spectrometry (MS)-based proteomics is increasingly applied in a quantitative format, often based on labeling of samples with stable isotopes that are introduced chemically or metabolically. In the stable isotope labeling by amino acids in cell culture (SILAC) method, two cell populations are cultured in the presence of heavy or light amino acids (typically lysine and/or arginine), one of them is subjected to a perturbation, and then both are combined and processed together. In this study, we describe a different approach—the use of SILAC as an internal or 'spike-in' standard—wherein SILAC is only used to produce heavy labeled reference proteins or proteomes. These are added to the proteomes under investigation after cell lysis and before protein digestion. The actual experiment is therefore completely decoupled from the labeling procedure. Spike-in SILAC is very economical, robust and in principle applicable to all cell- or tissue-based proteomic analyses. Applications range from absolute quantification of single proteins to the quantification of whole proteomes. Spike-in SILAC is especially advantageous when analyzing the proteomes of whole tissues or organisms. The protocol describes the quantitative analysis of a tissue sample relative to super-SILAC spike-in, a mixture of five SILAC-labeled cell lines that accurately represents the tissue. It includes the selection and preparation of the spike-in SILAC standard, the sample preparation procedure, and analysis and evaluation of the results.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Wolters, D.A., Washburn, M.P. & Yates, J.R. III. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 73, 5683–5690 (2001).
Aebersold, R. & Mann, M. Mass spectrometry-based proteomics. Nature 422, 198–207 (2003).
Steen, H. & Mann, M. The ABC's (and XYZ's) of peptide sequencing. Nat. Rev. Mol. Cell. Biol. 5, 699–711 (2004).
Ong, S.E. & Mann, M. Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 (2005).
Bachi, A. & Bonaldi, T. Quantitative proteomics as a new piece of the systems biology puzzle. J. Proteomics 71, 357–367 (2008).
Bantscheff, M., Schirle, M., Sweetman, G., Rick, J. & Kuster, B. Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 (2007).
Gruhler, A. et al. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell. Proteomics 4, 310–327 (2005).
Kohlbacher, O. et al. TOPP—the OpenMS proteomics pipeline. Bioinformatics 23, e191–e197 (2007).
Mueller, L.N. et al. SuperHirn—a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 7, 3470–3480 (2007).
Luber, C.A. et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 32, 279–289 (2010).
Leitner, A. & Lindner, W. Chemistry meets proteomics: the use of chemical tagging reactions for MS-based proteomics. Proteomics 6, 5418–5434 (2006).
Beynon, R.J. & Pratt, J.M. Metabolic labeling of proteins for proteomics. Mol. Cell. Proteomics 4, 857–872 (2005).
Gouw, J.W., Krijgsveld, J. & Heck, A.J. Quantitative proteomics by metabolic labeling of model organisms. Mol. Cell. Proteomics 9, 11–24 (2010).
Oda, Y., Huang, K., Cross, F.R., Cowburn, D. & Chait, B.T. Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. USA 96, 6591–6596 (1999).
Ong, S.E. et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 (2002).
Mann, M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell. Biol. 7, 952–958 (2006).
Hanke, S., Besir, H., Oesterhelt, D. & Mann, M. Absolute SILAC for accurate quantitation of proteins in complex mixtures down to the attomole level. J. Proteome Res. 7, 1118–1130 (2008).
Soufi, B. et al. Stable isotope labeling by amino acids in cell culture (SILAC) applied to quantitative proteomics of Bacillus subtilis. J. Proteome Res. 9, 3638–3646 (2010).
de Godoy, L.M. et al. Status of complete proteome analysis by mass spectrometry: SILAC labeled yeast as a model system. Genome Biol. 7, R50 (2006).
Sury, M.D., Chen, J.X. & Selbach, M. The SILAC fly allows for accurate protein quantification in vivo. Mol. Cell. Proteomics 9, 2173–2183 (2010).
Kruger, M. et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364 (2008).
Geiger, T., Cox, J., Ostasiewicz, P., Wisniewski, J.R. & Mann, M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385 (2010).
Ishihama, Y. et al. Quantitative mouse brain proteomics using culture-derived isotope tags as internal standards. Nat. Biotechnol. 23, 617–621 (2005).
Ong, S.E. & Mann, M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 1, 2650–2660 (2006).
Gerber, S.A., Rush, J., Stemman, O., Kirschner, M.W. & Gygi, S.P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. USA 100, 6940–6945 (2003).
Beynon, R.J., Doherty, M.K., Pratt, J.M. & Gaskell, S.J. Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods 2, 587–589 (2005).
Pratt, J.M. et al. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protoc. 1, 1029–1043 (2006).
Singh, S., Springer, M., Steen, J., Kirschner, M.W. & Steen, H. FLEXIQuant: a novel tool for the absolute quantification of proteins, and the simultaneous identification and quantification of potentially modified peptides. J. Proteome Res. 8, 2201–2210 (2009).
Brun, V. et al. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol. Cell. Proteomics 6, 2139–2149 (2007).
de Godoy, L.M. et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 (2008).
Bicho, C.C., de Lima Alves, F., Chen, Z.A., Rappsilber, J. & Sawin, K.E. A genetic engineering solution to the 'Arginine Conversion Problem' in stable isotope labeling by amino acids in cell culture (SILAC). Mol. Cell. Proteomics 9, 1567–1577 (2010).
Walther, D.M. & Mann, M. Accurate quantification of more than 4,000 mouse tissue proteins reveals minimal proteome changes during aging. Mol. Cell. Proteomics 9 published online, doi: 10.1074/mcp.M110.004523 (3 November 2010).
Scigelova, M. & Makarov, A. Orbitrap mass analyzer—overview and applications in proteomics. Proteomics 6 (Suppl 2): 16–21 (2006).
Makarov, A. et al. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 78, 2113–2120 (2006).
Olsen, J.V. et al. A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics 8, 2759–2769 (2009).
Shevchenko, A., Tomas, H., Havlis, J., Olsen, J.V. & Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2006).
Masuda, T., Saito, N., Tomita, M. & Ishihama, Y. Unbiased quantitation of Escherichia coli membrane proteome using phase transfer surfactants. Mol. Cell. Proteomics 8, 2770–2777 (2009).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705 (2009).
Cox, J. & Mann, M. Computational principles of determining and improving mass precision and accuracy for proteome measurements in an Orbitrap. J. Am. Soc. Mass. Spectrom. 20, 1477–1485 (2009).
Wisniewski, J.R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Wisniewski, J.R., Zougman, A. & Mann, M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8, 5674–5678 (2009).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007).
Bendall, S.C. et al. Prevention of amino acid conversion in SILAC experiments with embryonic stem cells. Mol. Cell. Proteomics 7, 1587–1597 (2008).
Acknowledgements
We thank P. Roerth for suggesting the term 'spike-in SILAC'. T.G. is supported by the Humboldt Foundation. This project was supported by the European Commission's 7th Framework Program PROteomics SPECification in Time and Space (PROSPECTS, HEALTH-F4-2008-021,648).
Author information
Authors and Affiliations
Contributions
T.G. developed the super-SILAC mix protocol. J.R.W. contributed the FASP and SAX protocols. J.C. developed MaxQuant and adapted it to spike-in SILAC. S.Z. contributed to the development, analysis and maintenance of the SILAC mice. M.K. developed and analyzed the SILAC mice. Y.I. contributed to the use of SILAC in a cell line as a spike-in standard. M.M. initiated and supervised all of these projects. T.G. and M.M. wrote the manuscript. All authors revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Geiger, T., Wisniewski, J., Cox, J. et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc 6, 147–157 (2011). https://doi.org/10.1038/nprot.2010.192
Issue Date:
DOI: https://doi.org/10.1038/nprot.2010.192
This article is cited by
-
A palmitate-rich metastatic niche enables metastasis growth via p65 acetylation resulting in pro-metastatic NF-κB signaling
Nature Cancer (2023)
-
Proteomic mapping of atrial and ventricular heart tissue in patients with aortic valve stenosis
Scientific Reports (2021)
-
Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker
Nature Cell Biology (2021)
-
Merging Fungal and Bacterial Community Profiles via an Internal Control
Microbial Ecology (2021)
-
Dual chemical probes enable quantitative system-wide analysis of protein prenylation and prenylation dynamics
Nature Chemistry (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
