
Abstract
The in vitro scratch assay is an easy, low-cost and well-developed method to measure cell migration in vitro. The basic steps involve creating a “scratch” in a cell monolayer, capturing the images at the beginning and at regular intervals during cell migration to close the scratch, and comparing the images to quantify the migration rate of the cells. Compared to other methods, the in vitro scratch assay is particularly suitable for studies on the effects of cell–matrix and cell–cell interactions on cell migration, mimic cell migration during wound healing in vivo and are compatible with imaging of live cells during migration to monitor intracellular events if desired. Besides monitoring migration of homogenous cell populations, this method has also been adopted to measure migration of individual cells in the leading edge of the scratch. Not taking into account the time for transfection of cells, in vitro scratch assay per se usually takes from several hours to overnight.
Similar content being viewed by others


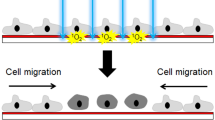
Introduction
The in vitro scratch assay is a straightforward and economical method to study cell migration in vitro1. This method is based on the observation that, upon creation of a new artificial gap, so called “scratch”, on a confluent cell monolayer, the cells on the edge of the newly created gap will move toward the opening to close the “scratch” until new cell–cell contacts are established again. The basic steps involve creation of a “scratch” on monolayer cells, capture of images at the beginning and regular intervals during cell migration to close the scratch, and comparison of the images to determine the rate of cell migration.
One of the major advantages of this simple method is that it mimics to some extent migration of cells in vivo. For example, removal of part of the endothelium in the blood vessels will induce migration of endothelial cells (ECs) into the denuded area to close the wound2. Furthermore, the patterns of migration either as loosely connected population (e.g., fibroblasts) or as sheets of cells (e.g., epithelial and ECs) also mimic the behavior of these cells during migration in vivo. Another advantage of the in vitro scratch assay is its particular suitability to study the regulation of cell migration by cell interaction with extracellular matrix (ECM) and cell–cell interactions. In other popular methods such as Boyden chamber assays, preparation of cells in suspension before the assays disrupts cell–cell and cell–ECM interactions. In addition, the in vitro scratch assay is also compatible with microscopy including live cell imaging, allowing analysis of intracellular signaling events (e.g., by visualization of green fluorescent protein (GFP)-tagged proteins for subcellular localization or fluorescent resonance energy transfer for protein–protein interactions) during cell migration. On the other hand, it is also probably the simplest method to study cell migration in vitro and only uses the common and inexpensive supplies found in most laboratories capable of cell culturing.
Although it is developed and more suitable for measuring migration of population of cells, the in vitro scratch assay has also been combined with other techniques, such as microinjection or gene transfection, to assess the effects of expression of exogenous genes on migration of individual cells3,4,5. The migration path of individual cells in the leading edge of the scratch is tracked with the aid of time-lapse microscopy and image analysis software. Capturing of an image in the beginning of the experiment with fluorescence microscopy can mark the cells with expression of exogenous gene or downregulation of endogenous genes by RNA interference (e.g., using a GFP marker). By comparing the tracks of these cells with surrounding control cells under the same experimental conditions allows determination of the role of a particular gene in the regulation of directional cell migration using the assay.
There are a number of disadvantages and limitations of the in vitro scratch assay compared to other available methods. It does not replace other well-established methods for chemotaxis such as the Boyden chamber assay, as no chemical gradient is established. It takes a relatively longer time to perform than some other methods. One to two days are needed for the formation of cell monolayer and then 8–18 h for cell migration to close the scratch. Last, relatively large amount of cells and chemicals will be required for the assay as it is usually performed in a tissue culture dish. Therefore, it is not a method of choice if the availability of cells (e.g., specialized primary cells that are hard to get in sufficient amount) or chemicals (e.g., expensive reagents) is limiting. The following table summarizes comparisons of the in vitro scratch assay with several other methods. Despite these limitations of the method, overall, in vitro scratch assay is still often the method of choice to analyze cell migration in a laboratory because it is easy to set up, does not require any specialized equipment and all materials required for the assay are available in any laboratory that performs cell culture.
In this protocol, Steps 1–9 describe the basic method of the in vitro scratch assay for measuring migration of cell populations. In Steps 10–13, the method is adopted to track migration of individual cells at the leading edge of the scratch. The latter is suitable to study the effect of particular gene products on cell migration when it is difficult to achieve high efficiency of transfection for exogenous gene expression or siRNA-mediated gene knockdown.
Materials
Reagents
-
Dulbecco's modified Eagle's medium with supplements (serum, antibiotics)
-
Versene (EDTA)with trypsin
-
Phosphate-buffered saline (PBS)
-
2 mg ml−1 bovine serum albumin (BSA)
-
1 mg ml−1 poly-L-lysine stock
-
LipofectAmine and PLUS transfection reagents (Life Technologies) (optional)
-
Plasmid-encoding GFP or other markers (optional)
-
CO2-independent medium (Optional)
Equipment
-
Tissue culture dishes (60 mm or of other size)
-
Razor or extra fine Sharpie marker
-
p200 Pipet tips
-
Hemocytometer
-
Phase-contrast microscope
-
Camera
-
Fluorescence microscope
-
Stage incubator
-
CO2 supply
-
Video camera
-
Image analysis software
Procedure
Coating of cell culture dishes
-
1
Coat 60-mm dishes with proper ECM substrates for the cell type to be studied (e.g., for fibroblasts, use 10 μg ml−1 fibronectin or 50 μg ml−1 poly-L-lysine as a control) by incubating the dishes overnight at 4 °C or for 2 h at 37 °C without rotation or shaking.
-
2
Remove the unbound ECM substrate and block the coated dishes with 3 ml of 2 mg ml−1 bovine serum albumin for 1 h at 37 °C. Then, wash the dishes once with PBS and refill the dishes with 3–5 ml of media before plating the cells. For the particular cell type used, the appropriate amount of serum in the medium during the in vitro scratch assay is required to be determined. It is recommended to use a lower percentage of serum than that used in the growth media to minimize cell proliferation, but just sufficient to prevent apoptosis and/or cell detachment.
Critical Step
Apart from the serum, if the assay is to study the effects of growth factors or other compounds, these soluble factors should be included in the media before addition of cells.
Passaging the cells in culture
-
3
Resuspend subconfluent growing cells in a tissue culture dish by washing cells twice with PBS, adding versene containing trypsin, and then mixing cells with medium containing serum. Gently pipette the solution and rock the dish to disperse the cells equally. Take an aliquot from the cell suspension and determine the cell counts using a hemocytometer.
-
4
Plate cells onto the prepared 60-mm dish to create a confluent monolayer. Incubate the dishes properly for approximately 6 h at 37 °C, allowing cells to adhere and spread on the substrate completely. The required number of cells for a confluent monolayer depends on both the particular cell type and the size of dishes and need to be adjusted appropriately.
Scratch or wound assay
-
5
The scratch assay can be performed on either native cells (A) or transfected cells (B) to study the effect of specific proteins overexpression (or knockdown) on cell migration.
-
A
Scratch assay on non-transfected cells
-
i
Scrape the cell monolayer in a straight line to create a “scratch” with a p200 pipet tip. Remove the debris and smooth the edge of the scratch by washing the cells once with 1 ml of the growth medium and then replace with 5 ml of medium specific for the in vitro scratch assay.
Critical Step
It is important to create scratches of approximately similar size in the assessed cells and control cells to minimize any possible variation caused by the difference in the width of the scratches.
-
ii
To obtain the same field during the image acquisition, create markings to be used as reference points close to the scratch. The reference points can be made by etching the dish lightly with a razor blade on the outer bottom of the dish or with an ultrafine tip marker. After the reference points are made, place the dish under a phase-contrast microscope, and leave the reference mark outside the capture image field but within the eye-piece field of view. Acquire the first image of the scratch.
-
iii
Place the dish in a tissue culture incubator at 37 °C for 8–18 h. The time frame for incubation should be determined empirically for the particular cell type used. The dishes can be taken out of the incubator to be examined periodically and then returned to resume incubation.
Critical Step
Choose a time frame of incubation that allows the cells under the fastest migrating condition to just achieve the complete closure of the scratch.
-
iv
After the incubation, place the dish under a phase-contrast microscope, match the reference point, align the photographed region acquired in Step 6 and acquire a second image.
-
v
The images acquired for each sample can be further analyzed quantitatively by using computing software of choice. For each image, distances between one side of scratch and the other can be measured at certain intervals (μm) using Image Pro-Plus software (Media Cybernetics) or a freeware (http://rsb.info.nih.gov/ij/). By comparing the images from time 0 (Step 6) to the last time point (Step 8), (see Fig. 1), obtain the distance of each scratch closure on the basis of the distances that are measured by software.
Figure 1: Analysis of primary EC migration by in vitro scratch assay. 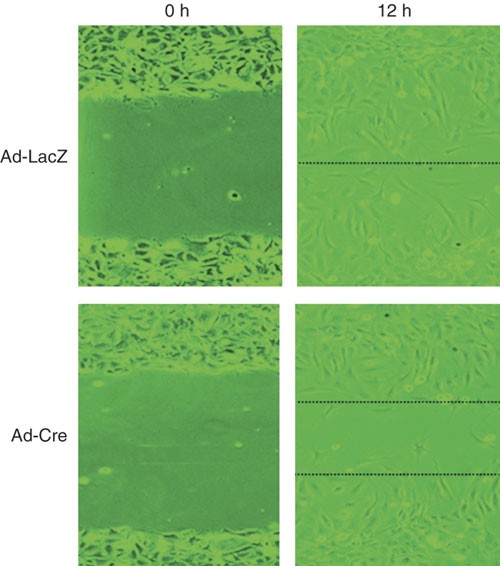
Primary ECs from floxed FAK mice were infected with Ad-LacZ or Ad-Cre, as indicated. Images were acquired at 0 and 12 h in in vitro scratch assay. The dotted lines define the areas lacking cells.
Critical Step
Measure at least 100 readings of distance for each sample and repeat each experiment at least three times. Alternatively, counting of the cells that cross into the scratch area from their reference point at time 0 can determine migration of cells. This method will provide large sample sizes that are easily quantified statistically. Cell counting can be processed by Image Pro-Plus software.
-
i
-
B
Scratch assay on transfected cells
-
i
Plate growing cells at 50–60% confluency for 12–18 h before transfection. Transfect cells with the plasmid encoding the gene of interest (or siRNA) along with a marker plasmid (i.e., GFP) in a 7:1 ratio, or with a vector-encoding GFP fusion protein containing the gene of interest by LipofectAmine and PLUS transfection reagents (Invitrogen). Incubate the dishes at 37 °C until cells reach 100% confluence to form a monolayer.
-
ii
Use a p200 pipet tip to create a scratch of the cell monolayer. Wash the plate once and replace with the desired medium. If time-lapse microscopy is used, CO2-independent media (e.g., HEPES-buffered media) may be required. The time-lapse microscope is used for acquiring the images from the same field automatically. It can be equipped with a stage incubator either with only temperature control or with both temperature and CO2 control. For the chamber with only temperature control, it is necessary to use CO2-independent medium during the assay. Nonetheless, it is not very practical to examine cell migration over a period of over 18 h by using the CO2-independent medium.
-
iii
Observe the cells under a fluorescence microscope to ensure that enough cells in the leading edge of the scratch are positively transfected (i.e., as marked by GFP). Create reference markings as described in Step 6 and acquire both phase-contrast and fluorescence images every 2 h for the same scratched region until the scratch completely close or within a desired time frame.
-
iv
Determine the rate of cell migration for the transfected cells by the available computing software that measures the distance traveled during the desired time frame (see Step A(v)). It should be noted that the neighboring untransfected cells could be used as controls for the positively transfected cells. It is also useful to draw an imaginary line in the middle of the scratch in the images captured (see Fig. 2).
Figure 2: Measurement of individual cell migration in in vitro scratch assay. 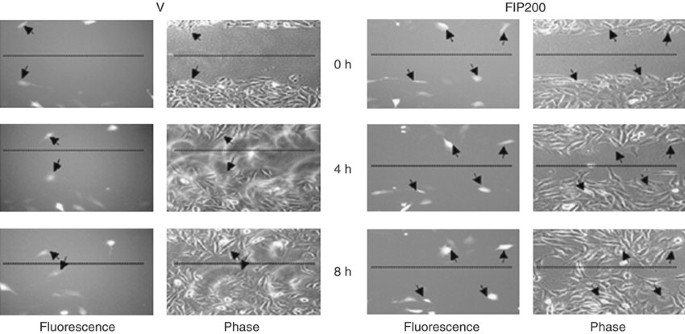
NIH3T3 cells grown on FN (10 μg ml−1) were co-transfected with the plasmid encoding GFP and expression vector encoding FIP200 or vector alone as control. They were then subjected to in vitro scratch assay with images captured at 0, 4 and 8 h after incubation using fluorescence or phase-contrast microscope. The rate of migration was measured by quantifying the total distance that the GFP+ cells (as indicated by arrows) moved from the edge of the scratch toward the center of the scratch (marked by imaginary dotted lines). This figure is reprinted from Mol. Biol. Cell 13, 3178–3191 (2002) with permission from the American Society for Cell Biology.
-
i
-
A



Timing
Step 1: 2 h/overnight (as needed)
Step 2: 1.5 h
Step 3: 30 min
Step 4: 6.5 h
Step 5A(i): 5 min
Step 5A(ii): 5 min
Step 5A(iii): 8–18 h (as needed)
Step 5A(iv): 2–5 min
Step 5A(v): 10–30 min (as needed)
Step 5B(i): 2–3 days (as needed)
Step 5B(ii): 5 min
Step 5B(iii): 12 h or as needed
Step 5B(iv): 10–30 min (as needed)
Anticipated results
An example of the in vitro scratch assay is shown in Figure 1, comparing migration of primary ECs upon deletion of focal adhesion kinase (FAK). Primary ECs from floxed FAK mice were infected with recombinant adenoviruses encoding Cre recombinase (Ad-Cre) to delete endogenous FAK or a control recombinant adenovirus encoding lacZ (Ad-lacZ) for 72 h before the in vitro scratch assay. The migration of population of cells is analyzed because of the high efficiency of adenovirus-mediated infection. FBS at 2% (vol/vol) was found to be optimal for the assay, although primary ECs are normally cultured in complete growth medium containing 20% (vol/vol) FBS. VEGF (50 ng ml−1) was included in the assay medium to induce migration of the cells. The images at the beginning and the end of a 12 h incubation period were captured, which showed significant migration of the cells toward the scratch. The incubation time was determined at 12 h when the faster moving cells (control, Ad-LacZ-infected cells) were just about to close the scratch.
In Figure 2, an example is shown for using the in vitro scratch assay to track migration of individual cells in the leading edge of the scratch. This method is used because only a fraction of the NIH3T3 cells were positively transfected as marked by the plasmids encoding GFP. On the left, the cells were transfected with a control vector that did not affect cell migration. As expected, the positively transfected cells (marked by arrows) migrated at the same rate as the surrounding untransfected cells in the leading edge of the scratch. On the right, the cells were transfected with a plasmid encoding FIP200. Analysis of the images showed that the positively transfected cells (marked by arrows) migrated at a slower rate compared with the untransfected control cells in the leading edge of the scratch in the same dish. Furthermore, comparison of the cells transfected with FIP200 (right panels) and those transfected with the vector alone (left panels) also showed inhibition of cell migration by FIP200.
References
Todaro, G.j. et al. The initiation of cell division in a contact-inhibited mammalian cell line. J. Cell Physiol. 66, 325–333 (1965).
Haudenschild, C.C. et al. Endothelial regeneration. II. Restitution of endothelial continuity. Lab. Invest. 41, 407–418 (1979).
Etienne-Manneville, S. et al. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 106, 489–498 (2001).
Fukata, Y. et al. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J. Cell Biol. 145, 347–361 (1999).
Abbi, S. et al. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol. Biol. Cell. 13, 3178–3191 (2002).
Acknowledgements
This work was supported by NIH grants GM48050 and HL73394 to J.-L. Guan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Liang, CC., Park, A. & Guan, JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2, 329–333 (2007). https://doi.org/10.1038/nprot.2007.30
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.30
This article is cited by
-
A new physiological medium uncovers biochemical and cellular alterations in Lesch-Nyhan disease fibroblasts
Molecular Medicine (2024)
-
Melatonin modulates L-arginine metabolism in tumor-associated macrophages by targeting arginase 1 in lymphoma
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Biopolymer-based Sustainable Membrane for Skin Regeneration
Materials Circular Economy (2024)
-
Synergistic effects of dendrosomal nanocurcumin and oxaliplatin on oncogenic properties of ovarian cancer cell lines by down-expression of MMPs
Biological Research (2023)
-
Nano-calcium incorporated piscean collagen scaffolds: potential wound dressing material
Future Journal of Pharmaceutical Sciences (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
