
Abstract
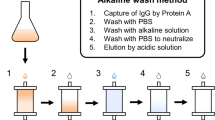
A protocol for the purification of polyclonal antibodies from ovine serum using the synthetic protein A absorbent MAbsorbent A2P is described. Clarified serum is loaded directly onto the affinity column without prior adjustment and albumin and unwanted serum components are washed from the column using a sodium octanoate buffer before elution of bound antibodies. MAbsorbent A2P was shown to bind ∼27 mg ml−1 of polyclonal immunoglobulin under overloading conditions, with eluted IgG purities of >90% and minor levels of albumin (∼1%). The anticipated time required to complete the purification protocol is 6–7 h. Although the protocol is similar to methods utilized for antibody purification using chromatography with protein A derived from the cell wall of the microorganism Staphylococcus aureus or protein G from Streptococcus as the affinity ligands, affinity absorbents based on synthetic ligands offer a number of advantages to compounds derived from biological sources, in particular robustness, relatively low cost, ease of sanitization and, in principle, lack of biological contamination.
Similar content being viewed by others

Introduction
Purified polyclonal antibodies are invaluable tools for biochemical research1 and for the production of antibody-derived biotherapeutics2,3. Common techniques used for the purification of antibodies often involve affinity absorbents utilizing protein A derived from the cell wall of the microorganism S. aureus (widely used for the purification of monoclonal antibodies)4 and protein G from Streptococcus as the affinity ligands5. However, the relatively high cost of these affinity absorbents for the purification of polyclonal antibodies6,7 combined with the additional cost of expensive cleaning solutions has resulted in the recent development of a number of commercial alternatives to protein A or G chromatography absorbents8,9. Many of these chemical alternatives are applicable for the direct capture of antibodies from crude feedstocks.
Although recombinant protein A is a robust molecule that may be cleaned using relatively harsh cleaning conditions, the use of protein A or G for a primary capture step of antibodies directly from a feedstock such as serum may be problematic. This is due to the viscosity of the serum and the relatively high concentration of antibodies, other proteins (albumin, transferrin and other serum proteins) and non-protein impurities (lipids and often cryoprecipitates) that may remain nonspecifically adsorbed to the column and thus adversely impact the capacity and reuse of the column matrix. Such problems are often diminished when using absorbents with bound synthetic chemical ligands due to the chemical stability of the ligand and the opportunity to evaluate a combination of harsh cleaning protocols. This may help to ensure that nonspecifically adsorbed impurities are removed before subsequent chromatographic runs.
The MAbsorbent A2P ligand (Prometic BioSciences) is composed of a di-substituted phenolic derivative of trichlorotriazine and is commercially available coupled to a 6% crosslinked agarose (Purabead) chromatography bead (Table 1).
Here we present a detailed protocol for the direct capture and purification of polyclonal antibodies from ovine serum (see Table 2 for an overview of the purification steps). The purification protocol was developed after identifying critical parameters and optimizing process conditions using factorial experimental design10. Although similar protocols describing the purification of polyclonal antibodies from serum using alternative synthetic affinity absorbents have recently been described11, a major technical challenge to overcome when isolating antibodies from serum is the removal of nonspecifically bound serum proteins (particularly serum albumin) from the absorbent before IgG elution. The protocol described outlines the critical process steps, technical challenges and techniques to avoid potential purification issues that may arise when purifying polyclonal antibodies from clarified serum.
In our experience, the major limitation of the present method is the high back pressures that may be observed when purifying antibodies from serum using MAbsorbent A2P. This appears to be largely independent of serum viscosity and occurs during pH transitions. Increased back pressure may be due to absorbent compression resulting from ionization of the bound ligand affecting the rigidity of the agarose base matrix. This may in turn impact the performance of the absorbent for the purification of antibodies from serum using large-diameter commercial-scale chromatography columns12,13.
Although the protocol described herein has previously been evaluated for the purification of antibodies for the production of antibody-derived biotherapeutics10, this approach may be equally applicable for the purification of monoclonal antibodies for diagnostic or analytical techniques and may therefore be adapted for use in a wider range of applications. The protocol may also be applied for the purification of normal (non-hyperimmune) plasma antibodies and use with other chromatography absorbents utilizing small molecule ligands, such as hydrophobic charge induction chromatography14,15.
Materials
Reagents
-
Ovine serum. Hyperimmunized ovine serum was obtained from Protherics UK Ltd for method development (serum may not be available owing to commercial limitations without a materials transfer agreement). However, ovine serum from other sources may be suitable (e.g., Sigma, cat. no. S 2263). Note that aliquoted serum should be kept frozen at −20 °C for long-term storage.
Caution
Serum may contain indigenous low-risk agents present in the community that may be associated with human disease of varying severity. Handle serum according to appropriate health and safety requirements.
-
MAbsorbent A2P (25 ml; ProMetic Biosciences Ltd, cat. no. 3901-00025). Note that the absorbent (supplied in 20% ethanol) is dark red in color owing to the absorptive characteristics of the bound aromatic triazine derivative. The absorbent should be stored at 2–8 °C
-
Sodium hydroxide (Sigma-Aldrich, cat. no. S8045-1KG)
-
Sodium phosphate dibasic, anhydrous (Sigma-Aldrich, cat. no. S-7907-500G)
-
Sodium phosphate monobasic, anhydrous (Sigma-Aldrich, cat. no. S-0751-500G)
-
NaCl (Sigma-Aldrich, cat. no. S-9625)
-
Sodium octanoate (Sigma-Aldrich, cat. no. C5038-100G)
-
Citric acid (Sigma-Aldrich, cat. no. C-0759-500G)
-
Sodium citrate dihydrate (Sigma-Aldrich, cat. no. S-4641)
-
Concentrated HCl (Sigma-Aldrich, cat. no. H-1758).
Caution
Hazardous and highly corrosive. Store and handle according to appropriate health and safety requirements.
-
Ethanol, 96% (VWR International Ltd, cat. no. 287196P).
Caution
Highly flammable. Store and handle according to appropriate health and safety requirements.
-
Analytical-grade H2O purified using an Elix 3 (Millipore (UK) Ltd, cat. no. ZLXS5003Y) water purification system (or equivalent)
-
Tris-glycine SDS running buffer (10×) (Invitrogen, cat. no. LC 2675)
-
Novex pre-cast 4–20% (1 mm, ten well) Tris-glycine gels (Invitrogen, cat. no. EC6025BOX)
-
Mark 12 unstained protein markers (Invitrogen, cat. no. LC5677)
-
Coomassie Brilliant Blue R250 (Sigma, cat. no. B0149-25G)
-
Jacksons ChromPure ovine polyclonal IgG standard (Jackson ImmunoResearch Europe Ltd, cat. no. 013-000-003, http://www.jireurope.com/home.asp)
-
Methanol (for preparation of gel destain; Sigma, cat. no. 154903-2L)
-
Glacial acetic acid (for preparation of gel destain; Merck, part no. 100063.1000)
-
Gel-Dry drying solution (1×) (Invitrogen, cat. no. LC4025)
Equipment
-
Sartolab P20-plus 0.2 μm + GF prefilter syringe filter units (Sartorius Ltd, cat. no. 18058)
-
Syringes (30 ml, BD Plastipak, BD, cat. no. 301229, or similar)
-
ÄKTA Explorer 100 (GE Healthcare, cat. no. 18-1112-41). A minimum of six buffer inlets are required for the protocol described. However, the method would be suitable for most commercially available laboratory-scale chromatography systems with minor modification to the method
-
ÄKTA detectors (GE Healthcare) used: UV-900 flow cell, 2 mm (cat. no. 18-1111-10), pH/C-900 pH and conductivity monitors (cat. no. 18-1107-76). An operational pressure limit of ≤0.7 MPa (101 psi, 7 bar) is recommended. This is determined from the combined operating pressure limit of the column (0.5 MPa) plus the back pressure observed from the flow restrictor included as part of the ÄKTA system (0.2 MPa)
-
XK16/20 chromatography column (GE Healthcare, cat. no. 18-8773-01).
Critical
Low-pressure columns (C10 columns, GE Healthcare) may not be suitable for the protocol described owing to back pressure observed during pH transitions.
-
Online ÄKTA filter kit (GE Healthcare, cat. no. 18-1027-11).
Critical
If filtered serum is loaded onto the column via the system pump, a new online filter should be installed or an apparent increase in back pressure will be observed. Alternatively, the in-line filter unit can be removed from the system before starting the method.
-
Novex XCell SureLock mini electrophoresis unit (Invitrogen, cat. no. EI0001)
-
Hot plate and beaker of water for boiling samples
-
Benchtop centrifuge (Eppendorf 5415D or equivalent)
-
Gel knife (Invitrogen, cat. no. EI9010)
-
Gel loading tips (Invitrogen, cat. no. LC 1001)
-
Platform shaker (Stuart Scientific STR6 or equivalent)
-
Stericup filter unit (Millipore, 0.45 μm HV Durapore Membrane 500 ml, cat. no. SCHVU05RE, or equivalent for filtration of stain before use)
-
ImageMaster TotalLab v2.01 image analysis software (GE Healthcare) or similar image analysis software
-
DryEase mini cellophane (Invitrogen, cat. no. NC2380)
-
DryEase gel drying frame (Invitrogen, cat. no. NI2200)
-
DryEase gel drying base (Invitrogen, cat. no. NI2300)
-
Gel-Dryer clamps (Invitrogen, cat. no. NI2100)
-
Flatbed scanner (Epson stylus CX5400) or equivalent
Reagent setup
-
Equilibration buffer 100 mM sodium phosphate pH 7.5 (±0.3). Dissolve sodium phosphate monobasic and dibasic solids in the proportions described. The pH of the resulting solution should be checked using a pH meter after preparation. As the buffer has been formulated, pH adjustment is not required. However, minor adjustment with 1 M NaOH or concentrated HCl may be required if the final solution is not within this specification.
Table 3 Table 4 -
Wash buffer 25 mM sodium phosphate, 100 mM NaCl and 25 mM sodium octanoate pH 7.5 (±0.3).
Critical
≥25 mM sodium octanoate is required during column washing to remove residual albumin adsorbed to the chromatography matrix. The wash buffer is prepared by dissolving solids in the proportions described. The pH of the resulting solution should be checked using a pH meter after preparation. As the buffer has been formulated, pH adjustment is not required. However, minor adjustment with 1 M NaOH or concentrated HCl may be required if the final solution is not within this specification.
Table 4 Table 5 -
Elution buffer 50 mM sodium citrate pH 3.0 (±0.2). A formulated citrate buffer is prepared by dissolving the solids in the proportions described: the pH of the resulting solution should be checked using a pH meter after preparation. Subsequent pH adjustment is not required.
Table 5 Table 6 -
Low-pH cleaning buffer A low-pH wash buffer (50 mM citrate pH 2.0 ± 0.2) is prepared by dissolving 9.61 g liter−1 citric acid (Sigma-Aldrich, cat. no. C0759) and adjusting pH with approximately 1.0 ml of concentrated HCl (Sigma Aldrich, cat. no. H-1758).
-
Matrix storage solution 20% (v/v) ethanol.
Table 6 Table 7 -
0.1% (w/v) Coomassie Brilliant Blue R250 Add 1.0 g of Coomassie Brilliant Blue R250 to 40% (v/v) methanol and 10% (v/v) glacial acetic acid.
Caution
Methanol is flammable and may cause skin irritation. May be absorbed through the skin and may cause kidney damage, respiratory tract irritation and may be fatal or cause blindness if swallowed. Acetic acid is strongly corrosive and may cause severe burns. Ensure safety glasses or face mask and nitrile gloves are worn. Gently agitate the stain solution for a minimum of 1 h. Filter the stain solution using a stericup filter unit (or alternative filtration equipment) to remove particulates before use.
Table 7 Table 8 -
Gel destain solution 40% (v/v) methanol and 10% (v/v) glacial acetic acid.
Table 8 Table 9
Equipment setup
-
ÄKTA setup Turn on the ÄKTA system. Rinse all buffer lines with H2O (to remove storage solution, typically 20% (v/v) ethanol) and sanitize the system using a pump wash with 0.5 M NaOH. Soak the system (minimum contact time 30 min) and then flush the system with H2O before use.
Procedure
Column preparation
Timing Approximately 1 h
-
1
Remove chromatography absorbent (MAbsorbent A2P) from the refrigerator (2–8 °C) and allow to equilibrate to room temperature (16–22 °C for at least 30 min).
-
2
Connect the bottom end of an XK16 chromatography column to the ÄKTA system.
-
3
Gently agitate the absorbent, then slowly pour matrix into the column and permit settling of the matrix bed under gravity until a bed height of 22 cm is obtained.
-
4
Connect the top of the column and lower top adaptor to within 5 mm of the settled bed.
-
5
Pack the column using H2O at a flow rate of 13 ml min−1.
Critical Step
Owing to potential compression of this absorbent during polyclonal IgG elution, a high linear flow rate of 400 cm h−1 is recommended for packing. Pack the column using H2O for a minimum of 5 column volumes (CVs; final packed bed height of 20 cm ± 10%, 40 ml volume).
Serum preparation
Timing A few minutes up to 1 h
-
6
Remove serum from refrigerator (or thaw if required).
Caution
Serum may contain indigenous low-risk agents present in the community that may be associated with human disease of varying severity. Handle serum according to appropriate health and safety requirements. Store thawed serum at 2–8 °C and discard after 24 h.
-
7
Filter serum through a 0.2 μm syringe filter.
Critical Step
Owing to the presence of particulates or cryoprecipitates, serum clarification is essential before direct loading onto the column.
Pause point
Filtered serum may be left overnight at 2–8 °C. The packed column may be left overnight at room temperature, although a continuous flow of H2O through the column at 0.2 ml min−1 is recommended. A slow continuous flow of water maintains a low level of back pressure within the system, thus preventing dissolved gases forming small bubbles within the column during the overnight pause.
IgG purification
Timing 3 h
-
8
Equilibrate the column with equilibration buffer at 3.35 ml min−1 (100 cm h−1) until constant pH and conductivity levels are observed (approximately 3 CVs). Load 30 ml of serum onto the column at 3.35 ml min−1 (100 cm h−1). No dilution or pH adjustment is necessary. Loading serum at 100 cm h−1 permits a residence time of 12 min. Assume a dynamic binding capacity of the matrix of 22.5 g liter−1 and a total ovine serum IgG titer of 30 g liter−1 (ref. 16).
-
9
Wash the column at 3.35 ml min−1 with wash buffer until the absorbance at 280 nm drops below 0.4 AU.
-
10
Start the fraction collector; collect 10 ml fractions. Elute bound antibodies from the column using elution buffer at 3.35 ml min−1.
Critical Step
During elution, many of the polyclonal antibodies transition through their isoelectric points and some haziness within concentrated fractions may be observed. Back pressure may be observed corresponding to an increased peak during IgG elution.
-
11
Collect eluted fractions (approximately 60 ml) until baseline absorbance is observed (≤0.05 AU). IgG fractions collected at the start of the elution peak may be hazy (see Step 10). Once all eluted fractions are pooled, the final IgG solution will become clear. The final IgG concentration is 6–12 mg ml−1 (depending on serum IgG titer and elution volumes). A mass balance will confirm that all of the IgG has been eluted. Also trending column capacities and recoveries from subsequent runs will also indicate whether complete IgG elution has been observed.
Column cleaning and sanitization
Timing 1 h
-
12
Wash the column with 1 CV of low-pH cleaning buffer at 4.0 ml min−1 (120 cm h−1) and then continue to wash until the pH is <2.5 and the absorbance is less than 0.1 AU.
-
13
Santize the column with 0.5 M NaOH at 2.0 ml min−1 for 40 min.
Critical Step
Approximately 5–15% of polyclonal IgG remains adsorbed to the column. A low-pH wash and NaOH sanitization are required to remove adsorbed components. If the column is not cleaned sufficiently, then a drop in column capacity (and back pressure) may be observed when using the column for subsequent purification runs. An increase in pressure may be observed during pH transition into NaOH owing to the physical properties of the hydrophobic ligand and compression or swelling of the agarose base matrix. In this case, reduce flow rates for column sanitization.
Re-equilibration
Timing 0.5–1 h
-
14
Re-equilibrate the column with equilibration buffer at 2.0 ml min−1 until the pH is below 8.0.
-
15
If the column is not to be used for subsequent purification runs, flush the column with 5 CVs of H2O followed by ≥3 CVs of matrix storage solution. The packed column may be detached, both ends capped and the column stored at 2–8 °C. Alternatively, the matrix may be carefully rinsed as a slurry from the column (using storage solution) and stored at 2–8 °C in 20% ethanol (1:1 volume of ethanol to matrix).
pH adjustment of the IgG-containing solution
Timing 15 min
-
16
Adjust the pH of the purified IgG to neutral (pH 6–7) using 2 M Tris base or sodium hydroxide, syringe filter (0.2 μm) and store as required (at 2–8 °C for ≤3 days, at −20 °C for long term). Neutral pH adjustment of the eluted IgG and ≥−20 °C storage is recommended to minimize aggregation, denaturation and degradation of the purified antibodies.
Analysis of antibody purity using Novex Tris-glycine gels
Timing 24–60 h
-
17
Prepare a 1× Tris-glycine SDS running buffer solution by adding 100 ml of Novex Tris-glycine SDS running buffer (10×) to 900 ml of H2O.
-
18
Remove a pre-cast Novex 4–20% Tris-glycine gel from the packet; remove the tape from the surface of the gel. Mark the position of the wells using a permanent marker.
Caution
Acrylamide is a possible human carcinogen and possible teratogen. Acrylamide is readily absorbed through the skin and may cause heritable genetic damage. Although polymerized polyacrylamide gels may present a reduced risk, ensure safety glasses and gloves are worn and handle in an area with adequate ventilation.
-
19
Place the gel into the Novex SureLock electrophoresis unit and fill the upper and lower buffer chambers with 600 ml of 1× Tris-glycine SDS running buffer.
-
20
Gently remove the well comb and ensure the top of sample wells are filled and covered with running buffer.
-
21
Dilute the antibody solution to 1.0 mg ml−1 using H2O. Mix 10 μl (10 μg) with 10 μl of Tris-glycine SDS sample buffer in a 1.5 ml Eppendorf tube (final volume 20 μl) and immerse the samples in a boiling water bath for 2 min.
-
22
Remove the samples and allow to cool for 3–5 min. Centrifuge the samples for 5–10 s at room temperature at 16,000g within a benchtop centrifuge if condensation on the wall of the Eppendorf tube is observed.
-
23
Load 10 μl of sample per well (5.0 μg protein load) using gel loading pipette tips. Load also 10 μl of mark 12 molecular weight marker into one lane.
-
24
Run the samples by electrophoresis at a constant voltage of 125 V (expected current 30–40 mA per gel at the start, 8–12 mA per gel at the end of the run) for approximately 70 min or until the sample is approximately 1 cm from the bottom of the gel.
-
25
While the gel is running, pour approximately 50 ml of Coomassie Brilliant Blue R250 stain into a clean staining tray of appropriate size.
-
26
After electrophoresis is complete, turn off the power, disconnect electrodes and remove the gel from the XCell SureLock Mini-Cell. Separate each of the three bonded sides of the cassette by inserting the gel knife into the gap between the cassette's two plates.
-
27
Gently push down the knife to separate the plates. Repeat on each side of the gel cassette until the plates are completely separated. Carefully remove and discard the top plate, allowing the gel to remain on the bottom plate.
-
28
Use the sharp edge of the gel knife to remove the bottom lip of the gel. Hold the plate and gel over a container with the gel facing downward and use the knife to carefully loosen one lower corner of the gel and allow the gel to peel away from the plate into the suitable staining tray containing 50 ml of Coomassie stain prepared in Step 25.
-
29
Stain the gel at room temperature with gentle agitation. After a minimum of 1 h, pour off the stain and destain the gel with 40% (v/v) methanol and 10% (v/v) glacial acetic acid until the background of the gel is clear upon visual inspection.
-
30
Destained gels may be scanned using a flatbed scanner and analyzed using densitometry analysis using ImageMaster TotalLab v2.01 image analysis software (or equivalent image capture and analysis software).
-
31
Gels may be dried using the Invitrogen DryEase mini-gel drying system (Invitrogen instruction manual 052002, IM-2380) (Steps 17–22).
-
32
After staining and destaining steps are completed, wash the destained gel three times for 2 min each with H2O (50 ml per mini gel) on a rotary shaker.
-
33
Decant the water and add Gel-Dry drying solution (35 ml per mini gel). Equilibrate the gel in Gel-Dry solution for 15–20 min with gentle agitation.
-
34
Remove two sheets of DryEase mini cellophane. Immerse one sheet in gel drying solution for 15–20 s. Place a DryEase gel drying frame on the DryEase gel drying base. Center a piece of pre-wetted cellophane over the base/frame combination so that the cellophane lays over the inner edge of the frame. Lay the gel on the center of the cellophane sheet ensuring no bubbles are trapped between the gel and the cellophane.
-
35
Carefully lay a second pre-wetted cellophane sheet over the gel. Gently smooth out any bubbles and wrinkles in the assembly with a gloved hand.
-
36
Align the remaining frame so that the corner pins fit into the appropriate holes on the bottom frame. Push four plastic clamps onto the edges of the frames. Lift the frame assembly from the DryEase gel drying base and pour off any excess solution.
-
37
Stand the gel dryer assembly upright.
Pause point
Dry at room temperature for 12–36 h.
-
38
When the cellophane is dry to the touch, remove the gel/cellophane sandwich and trim the edges. Press the dried gel between the pages of a notebook for 2 days. Gels will remain flat for scanning or photography.
Troubleshooting
Troubleshooting advice can be found in Table 3.
Timing
Steps 1–7, serum filtration and column preparation and packing: 1–2 h
Steps 8–11, antibody purification: 3 h
Steps 12 and 13, column cleaning and sanitization: 1 h
Step 14, column re-equilibration: 0.5–1 h
Step 15, matrix storage: 1.5–2 h
Step 16, pH adjustment of purified antibodies: 15 min
Steps 17–38, analysis of IgG purity by SDS-PAGE: 24–60 h (time will be dependent on whether gels are destained overnight and whether gels are dried before densitometry analysis)
Anticipated results
Typical isolated yields of purified polyclonal IgG will be ∼80%, although this may be dependent on polyclonal IgG titers within the serum. Although an approximate IgG serum titer of 30 mg ml−1 has been presented, an accurate serum IgG titer may be determined using protein G chromatography or an alternative analytical technique16. A representative chromatogram with absorbance (280 nm), pH and conductivity traces is shown in Figure 1. IgG purities (as determined by Coomassie-stained SDS-PAGE and scanning densitometry)10 will be 90% (Fig. 2).

A chromatogram showing A280 nm, pH and conductivity (mS cm−1) traces. IgG elution starts when the absorbance drops below 0.4 AU (see Step 9). Continued washing to baseline levels does not improve eluted IgG purity (as determined by SDS-PAGE, Steps 17–29) and extends the purification step.

Lane 1: Mark 12 molecular weight standards (Invitrogen, cat. no. LC5677); lane 2: hyper-immunized ovine serum (0.2 μl load); lanes 3–5: purified IgG from three consecutive purification cycles; lane 6: ovine polyclonal IgG standard (Jacksons ChromPure, representative sample lane from a separate gel shown). Minor cropping at the bottom of the gel has been undertaken to remove the unresolved insulin (3 kDa) standard.
References
Harlow, E. & Lane, D. Antibodies: A Laboratory Manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1988).
Newcombe, C. & Newcombe, A.R. Antibody production: polyclonal-derived biotherapeutics. J. Chromatogr. B 848, 2–7 (2007).
Thillaivinayagalingam, P., O'Donovan, K., Newcombe, A.R. & Keshavarz-Moore, E. Characterisation of an industrial affinity process used in the manufacturing of digoxin-specific polyclonal Fab fragments. J. Chromatogr. B 848, 88–96 (2007).
Racher, A.J., Tong, J.M. & Bonnerjea, J. Biotechnology. Recombinant Proteins Vol. 5a 247 (Wiley-VCH, Weinheim, Germany, 2003).
Lian, L.Y., Barsukov, I.L., Derrick, J.P. & Roberts, G.C. Mapping the interactions between streptococcal protein G and the Fab fragment of IgG in solution. Nat. Struct. Biol. 1, 355–357 (1994).
Chhatre, S. et al. The integrated simulation and assessment of the impacts of process change in biotherapeutic antibody production. Biotechnol. Prog. 22, 1612–1620 (2006).
Chhatre, S. et al. A decision-support model for evaluating changes in biopharmaceutical manufacturing processes. Bioprocess Biosyst. Eng. 30, 1–11 (2007).
Li, R., Dowd, V., Stewart, D.J., Burton, S.J. & Lowe, C.R. Design, synthesis, and application of a protein A mimetic. Nat. Biotechnol. 16, 190–195 (1998).
Kabir, S. Immunoglobulin purification by affinity chromatography using protein A mimetic ligands prepared by combinatorial chemical synthesis. Immunol. Invest. 31, 263–278 (2002).
Newcombe, AR. et al. Optimised affinity purification of polyclonal antibodies from hyper immunised ovine serum using a synthetic Protein A adsorbent, MAbsorbent A2P. J. Chromatogr. B 814, 209–215 (2005).
Bak, H. & Thomas, OR. Evaluation of commercial chromatographic adsorbents for the direct capture of polyclonal rabbit antibodies from clarified antiserum. J. Chromatogr. B 848, 116–130 (2007).
Birch, J.R. & Racher, A.J. Antibody production. Adv. Drug Deliv. Rev. 58, 671–685 (2006).
Bonnerjea, J. Purification of therapeutic proteins. Methods Mol. Biol. 244, 455–462 (2004).
Boschetti, E. Antibody separation by hydrophobic charge induction chromatography. Trends Biotechnol. 20, 333–337 (2002).
Ghose, S., Hubbard, B. & Cramer, S.M. Evaluation and comparison of alternatives to Protein A chromatography mimetic and hydrophobic charge induction chromatographic stationary phases. J. Chromatogr. A 1122, 144–152 (2006).
Newcombe, A.R. et al. Evaluation of a biosensor assay to quantify polyclonal IgG in ovine serum used for the production of biotherapeutic antibody fragments. Process Biochem. 41, 842–847 (2006).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Chhatre, S., Titchener-Hooker, N., Newcombe, A. et al. Purification of antibodies using the synthetic affinity ligand absorbent MAbsorbent A2P. Nat Protoc 2, 1763–1769 (2007). https://doi.org/10.1038/nprot.2007.253
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.253
This article is cited by
-
Performance evaluation of Mimetic Ligand™ B14-triazole-FractoAIMs adsorbents for the capture of human monoclonal immunoglobulin G from cell culture feed
Analytical and Bioanalytical Chemistry (2011)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
