
Abstract
This method is designed to maximize recovery of PCR-amplifiable DNA from ancient bone and teeth specimens and at the same time to minimize co-extraction of substances that inhibit PCR. This is achieved by a combination of DNA extraction from bone powder using a buffer consisting solely of EDTA and proteinase K, and purification of the DNA by binding to silica in the presence of high concentrations of guanidinium thiocyanate. All steps are performed at room temperature (20–23 °C), thereby reducing further degradation of the already damaged and fragile ancient DNA and providing an optimal trade-off between DNA release and degradation. Furthermore, the purification step removes most of the various types of PCR inhibitors present in ancient bone samples, thereby optimizing the amount of ancient DNA available for subsequent enzymatic manipulation, such as PCR amplification. The protocol presented here allows DNA extraction from ancient bone and teeth with a minimum of working steps and equipment and yields DNA extracts within 2 working days.
Similar content being viewed by others

Introduction
Rationale
Since its beginning, research on ancient DNA has suffered from the problem that, in almost all ancient specimens, any DNA that is preserved is present only in small amounts and in various states of degradation. Therefore, it is crucial to make this DNA available as much as possible for further enzymatic manipulation. This usually involves amplification by PCR, but more recent approaches also include ligation into bacterial vectors1,2 or addition of oligonucleotides for direct sequencing3,4,5. However, this is not a trivial task. First, most DNA extraction methods are designed to deal with fresh tissue containing intact cells and high molecular weight DNA. In ancient specimens, usually no cell structures are preserved (but see ref. 6 for rare exceptions) and, owing to as yet uncharacterized chemical modifications, it may even be difficult to get the DNA into aqueous solution7. Second, ancient DNA is damaged in various ways8,9,10,11, so extraction methods also have to avoid overly aggressive treatments, such as high temperatures or use of strong detergents12. Although these treatments might increase DNA release, they would decrease overall DNA yield by inflicting further damage to the ancient DNA molecules. Third, ancient bones and teeth often contain large amounts of PCR inhibitors13,14,15 that interfere with DNA amplification and are co-purified with ancient DNA. Thus, ancient DNA extraction methods have to deal with a number of problems that are sometimes difficult to reconcile. The method described here is the result of testing, on a number of Pleistocene samples, a wide range of conditions and ingredients from one published method16, followed by a final comparison of the optimized procedure to other published ancient DNA extraction methods12. It provides a trade-off between DNA release, DNA degradation during extraction and separation of DNA and inhibitors, thereby maximizing the DNA available for further applications. We have applied it successfully to extractions from bone and teeth samples originating from both cave12 and open sites17, from permafrost17 as well as non-permafrost environments12, and with sample sizes ranging from less than 50 mg (unpublished) to 40 g of bone powder if a concentration step is included1,17.
As is generally the case when working with ancient DNA, a number of precautions have to be taken during extraction of ancient specimens; these have been extensively described elsewhere18,19,20,21.
Advantages of silica extraction over other ancient DNA extraction methods
Several methods for ancient DNA extraction exist, including ethanol or isopropanol precipitation14,15, concentration of DNA using membranes22 and binding DNA to silica16. Compared to these extraction methods, the improved silica method described here has several advantages.
-
1
Quick and easy: this is a relatively fast method; it yields ancient DNA extracts within 2 working days and involves only a limited workload.
-
2
Scalable: the protocol can easily be adapted to different amounts of bone powder and extraction volumes. We have used it for extractions ranging from as little as 50 mg bone powder in 1 ml extraction solution (unpublished) to 2 g of bone powder in 10 ml extraction solution (unpublished). If a concentration step1,17 is included between extraction and purification of DNA, up to 40 g of bone powder in 1 liter of extraction solution can easily be processed.
-
3
Simple to implement: the method requires only standard laboratory equipment and a small number of chemicals and is processed at room temperature (20–23 °C). It can therefore be established rapidly.
-
4
Efficient removal of PCR inhibitors: we found this method to efficiently separate ancient DNA and PCR inhibitors12, a crucial problem in ancient DNA research13,14,15.
In fact, in a comparison with published methods, we found this protocol on average to yield the highest values of amplifiable DNA when tested on a number of Pleistocene cave bear samples12. This is not to claim that for certain samples, other protocols may not yield better results. However, given its superior performance on average and in the absence of prior knowledge of which method works best on a particular sample, this method can be seen as the most promising extraction method to start with for ancient samples. Moreover, its average high performance offers the greatest chance of success in analyses of large numbers of specimens23,24, where it is impractical to test various extraction methods on each sample, or when the value of a sample allows only one extraction, for example, in the case of Neanderthal samples25,26,27.
Other potential applications of this method
We have tested the method on a number of Pleistocene samples12,17 and on samples from various types of fossil sites (e.g., caves12, open sites (unpublished) and permafrost environments17). So far, our experience has been positive; results have even been obtained from samples that had failed previously. We have also been able to amplify nuclear genes17 from extracts obtained using this method. Thus, we would recommend this protocol for extracting DNA from bone and teeth samples of any age, including museum specimens, historical samples and even modern bone. However, it should be noted that other methods22,28,29 may yield better results with other types of samples, such as museum specimens, especially if they have been treated for storage30.
Optimization of the method
If unsatisfactory results are obtained for a certain type of sample, the extraction solution can be adjusted to the attributes of the sample type. Reducing agents (e.g., 50 mM dithiothreitol) and detergents (e.g., 1% (v/v) Triton X-100) can be added to the extraction solution to destroy proteins and intact cell membranes if modern or historical bones or teeth are extracted. N-phenacylthiazolium bromide (PTB) can be added to cleave Maillard products (sugar-derived protein crosslinks, e.g., in feces samples)31. Alternatively, other extraction methods14,15,22,29 may be evaluated.
For samples that do not contain PCR inhibitors, salts other than guanidinium thiocyanate (GuSCN) may be used, for example sodium chloride12. These non-chaotropic salts are much cheaper and perform equally well or even better in terms of DNA recovery. However, they tend to co-purify PCR inhibitors and therefore should not be used for samples that are likely to contain PCR inhibitors.
Limitations of this extraction method
A clear advantage of this method is its broad applicability to bone and teeth samples and the relatively short time necessary for obtaining DNA extracts. However, a limitation lies in the fact that it is specific to bone and teeth samples; this is due to the high amounts of EDTA in the extraction solution, which is necessary to dissolve part of the hydroxylapatite matrix specific to bone and teeth samples. The method may well be adapted to other types of samples by changing the extraction solution; for example, the addition of PTB has been shown to help DNA release from feces samples, possibly by releasing DNA from DNA–protein crosslinks31; the use of detergents and reducing agents is generally recommended when processing fresh samples where intact proteins and/or cells are present. Other ingredients may support DNA release from various sources (e.g., polyvinylpyrrolidone for plants32); however, this has not been tested comprehensively. Finally, reducing the concentration of EDTA for samples other than bones and teeth, or addition of Tris-HCl to buffer the extraction solution may help when other source materials are used. However, it should be noted that none of these chemicals improved DNA yields from ancient bones and teeth12.
All the above suggested modifications will need careful evaluation and optimization for each type of sample (such as feces, soft tissues, plants or sediments), to yield optimal results. However, as the vast majority of sequences used in ancient DNA research are obtained from bone or teeth specimens, this shortcoming is of limited importance.
Another drawback of this extraction method is that not only endogenous DNA is extracted from the sample, but also bacterial and fungal DNA that accumulated on the sample owing to microorganisms living in the surrounding sediment, on the sample itself or that have been introduced during collection or storage of the samples. As shown many times, the amount of exogenous DNA usually exceeds the amount of endogenous DNA several fold (refs. 1, 5 and 9, but see ref. 4 for an example of high amounts of endogenous DNA). However, no method that overcomes this problem is currently available.
General considerations
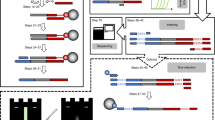
This protocol (for an overview, see Fig. 1) is written for a sample amount of around 500 mg. If the amount of sample is smaller or larger, adjust the volume of extraction solution proportionally (Step 6). For DNA binding to silica (Step 10), use four times the volume of binding buffer compared to the volume of extraction solution. Add 100 μl of silica suspension also for smaller amounts of sample/buffer, but do not add more silica if you exceed the recommended volumes mentioned here, as a proportion of the elution buffer will remain in the silica and the elution volume will fall below 50 μl. If you use more than 100 μl silica suspension, increase the elution volume. Note that the amount of HCl required to adjust the pH (Step 10) will also need to be adjusted. All steps described in this procedure are performed at room temperature (20–23 °C) if not stated otherwise.

Flow diagram for DNA extraction from ancient bones and teeth.
Materials
Reagents
-
Sodium hypochlorite solution (10–13%)—Bleach (Sigma, cat. no. 425044)
Caution
Alkaline, may cause skin irritation; wear protective clothes and gloves.
-
Water, HPLC–grade (Sigma, cat. no. 270733)
-
EDTA disodium salt dihydrate (Sigma, cat. no. E5134)
-
Proteinase K (Sigma, cat. no. P6556)
-
GuSCN (Sigma, cat. no. G9277)
Caution
Harmful; wear protective clothes and gloves.
-
Tris(hydroxymethyl)aminomethane (Tris base) (Sigma, cat. no. T1503)
-
Sodium chloride (Sigma, cat. no. S7653)
-
Silicon dioxide (Sigma, cat. no. S5631) (see REAGENT SETUP)
-
HCl (Fluka, cat. no. 17077) 30% w/v
Caution
Acidic, may cause skin irritation; wear protective clothes and gloves.
-
Absolute ethanol (Merck, cat. no. 1.00983.2500)
-
TE buffer, 10 mM Tris, 1 mM EDTA, pH 8.0
-
Extraction solution (see REAGENT SETUP)
-
Binding buffer (see REAGENT SETUP)
-
Washing buffer (see REAGENT SETUP)
Equipment
-
Two separate rooms with one hood in the first and two hoods (laminar flow hoods are recommended) in the second room; the minimum requirement is at least two hoods in one room to separate the dust-producing bone preparation from the buffer preparation, extraction procedure and setup of PCR, as these latter processes are very susceptible to contamination (see EQUIPMENT SETUP)
-
Drilling/cutting equipment with exchangeable cutting blades, discs and/or drilling bits (e.g., MICROMOT 40/E from Proxxon, cat. no. NO28515, and grinding and cutting discs from Proxxon, cat. nos. NO28812 and NO28830)
-
Mortar and pestle (one set per sample) or alternatively a freezer mill (e.g., Spex 6770 freezer mill from Spex SamplePrep, cat. no. 6770-230) with accessories (cat. no. 6751)
Critical
A sufficient number of grinding vials are required to prepare more than one sample per day. Grinding vials need to be thoroughly cleaned before reuse (see EQUIPMENT SETUP).
-
Liquid nitrogen, if using a freezer mill
Caution
Wear protective clothes and gloves to prevent freeze burning.
-
Rotary wheel, or similar equipment to keep the tubes agitated during incubation, with holders for different sized tubes (2-, 15- and 50-ml tubes)
-
Two balances, one for chemicals and one for samples
Critical
To prevent contamination of chemicals with sample material, the balance for the chemicals must not be used for weighing the samples and vice versa.
-
Table top centrifuges: one suitable for the use of 15- and 50-ml conical tubes with speed up to 5,000g (e.g., Multifuge 3 S from Heraeus), another for 1.5- and 2-ml tubes with speed up to 16,000g (e.g., Microcentrifuge 5415D from Eppendorf)
-
Filter tips are recommended to minimize the risk of cross-contamination owing to DNA aerosols.
-
pH indicator strips (e.g., pH-Fix 3.6-6.1, Macherey-Nagel, art. no. 921 30)
-
Standard laboratory equipment such as different sized tubes, freezer and refrigerator for storing extracts and chemicals.
Reagent setup
-
• TIMING 5 h for preparation of silica suspension, 30–60 min for all other buffers
General considerations Prepare all buffers, solutions and suspensions with HPLC-grade water and use only disposable equipment to weigh chemicals and prepare buffers. To minimize the risk of contamination, never put chemicals back into the storage container. It has been shown33 that UV irradiation of PCR reagents reduces the amplification success by causing blocking lesions in the DNA strands. This 'decontamination' procedure could also be applied to UV-insensitive reagents, buffers, reaction tubes and surfaces to reduce the risk of contamination. For further details (recommended wavelengths and distance), see ref. 33.
-
Extraction solution 0.45 M EDTA and 0.25 mg ml−1 proteinase K, pH 8.0. A volume of 10 ml is required per 500 mg of sample.
Critical
Always prepare fresh and do not forget to include sufficient buffer volume for at least one negative control during each extraction to check for cross-contamination and reagent contamination; we recommend a minimum of one extraction control. For more than seven samples, two or more extraction controls should be included.
-
Binding buffer 5 M GuSCN, 25 mM NaCl and 50 mM Tris. Per sample 40 ml are required plus 1 ml per sample for the first washing step (Step 13).
Critical
If possible, prepare fresh; however, it can be used for a maximum of 3 weeks if stored in the dark at room temperature (20–23 °C).
-
Washing buffer 50% v/v ethanol, 125 mM NaCl, 10 mM Tris and 1 mM EDTA, pH 8.0.
-
Preparation of silica suspension Suspend 4.8 g of silicon dioxide in water to a final volume of 40 ml and leave to settle for 1 h. Transfer 39 ml of the supernatant to a new tube and allow to sediment for an additional 4 h. Remove and discard 35 ml of the supernatant and add 48 μl of 30% w/v HCl to the pellet. Aliquots (we recommend amounts of 850 μl for extraction of seven samples and one blank control) should be stored at room temperature in the dark.
Critical
Silica suspension should be used within 1 month of preparation.
Equipment setup
-
Laboratory requirements Two rooms are necessary to safely separate the dust-producing working steps from the contamination-susceptible steps like buffer preparation and PCR setup. The first room is used for cutting and grinding the samples with appropriate equipment and disposables. The second room is used for preparation of buffers and setup of PCR reagent mix (first hood) and extraction of the samples and addition of DNA extract to the PCR (second hood). If only one room with two hoods is available, we would recommend separating the sample preparation from all other steps (buffer preparation, extraction procedure and PCR setup).
General considerations about handling
-
The samples used for extraction will usually contain only trace amounts of DNA. To avoid cross-contamination among samples, all steps that do not involve handling of bone samples (such as preparation of buffers, labeling of tubes, etc.) should, as far as possible, be performed before manipulation of the samples. Equipment where a different set is used for each sample (such as mortar and pestle or cutting discs) should be stored in a way that makes it easily accessible but prevents contamination of later samples with dust from the earlier samples. Manipulation of each bone sample must be completed to the point where the amount of powder necessary for extraction has been weighed and placed in the extraction tube before commencing with the next sample. Additional powder, as well as the remaining bone sample, must be stored away safely to avoid cross-contamination between samples. Single-use equipment must be removed from the working area. Finally, the working area and equipment such as the drill bit have to be cleaned appropriately (e.g., with sodium hypochlorite solution followed by water or ethanol) before work on the next sample is started. Various ways of cleaning/decontamination of equipment and work areas are possible. If no work is in progress, equipment and working area should be UV-irradiated33. Equipment should be first chemically cleaned (e.g., with sodium hypochlorite solution), followed by thorough removal of any potential DNA-damaging chemicals using water and/or ethanol. Finally, equipment should be UV-irradiated at least overnight. Care has to be taken with equipment that may be damaged by the treatment, such as metal equipment, which will be damaged by hypochlorite solution. In such cases, cleaning/decontamination can be done using water and UV irradiation.
Procedure
Preparation of the bone or tooth sample
Timing 15–30 min per sample
-
1
Remove dirt from the surface of the specimen with a tissue. If necessary, moisten the tissue with HPLC-grade water.
Critical Step
Dirt may introduce a variety of inhibitory substances to the extraction procedure, and therefore to the extract itself; these substances may interfere or even completely block subsequent enzymatic manipulations of the DNA extracts.
-
2
Remove the outer surface of part of the specimen with a single-use grinding tool.
Critical Step
This step removes possible contamination introduced during excavation, storage or collection, or other investigations of the specimen; although no 100% efficient procedure to remove contamination exists34, this step may improve the ratio between endogenous and contaminating DNA; moreover, it may further reduce the amount of inhibitors introduced into the extraction.
-
3
Cut off or cut out a piece of the specimen (you may have to reconcile with curatory requirements to minimize physical damage); if possible, sample from a compact part of the bone. When using teeth, cut off the root or use a part of the dentine, for example from inside the root, depending on curatory requirements. Most curators may not allow a piece to be cut from the (often unique) specimen. An alternative is to drill inside the specimen to obtain a fine powder without the need of further grinding. Drill with low speed to prevent overheating, which would damage the DNA.
Caution
Be careful with sharp cutting discs.
Critical Step
It is assumed that DNA is better preserved in compact parts of the bone than in more spongiose parts; therefore, we recommend using compact parts of bones, for example, the diaphyses of long bones. Use dentine rather than enamel from teeth, as dentine is assumed to contain more DNA.
Pause point
The sample can be stored at room temperature (e.g., together with the original specimen).
-
4
Grind the sample intended for DNA extraction with a mortar and pestle until a fine-grained powder is obtained. If necessary (e.g., if the sample is very hard), use a freezer mill.
Critical Step
Try to obtain as fine a powder as possible; the finer the powder, the more DNA released12. However, if using a freezer mill or similar equipment, do not overgrind, as this may fragment the DNA.
-
5
Weigh out no more than 500 mg of sample powder and transfer it to a 15 ml tube.
Pause point
The sample powder can be stored at room temperature, but should be subjected to the extraction as soon as possible.
DNA release
Timing 1 day (only 10 min work required)
-
6
Add 10 ml extraction solution to each 500-mg sample powder. Also include a blank extraction (10 ml extraction solution without a sample); this has to be treated identically to the experimental samples throughout the procedure. It monitors for contamination of the chemicals or cross-contamination during the procedure.
Critical Step
A positive control should be included only if testing the protocol the first time, testing if new chemicals interfere with the protocol or if a previous extraction failed. If a positive control (of the same kind of material) is included, it is recommended that a different species is used to control for cross-contamination by testing both the blank extraction and all extracts for DNA from the positive control.
Critical Step
Use separate pipette tip for each sample to avoid cross-contamination.
-
7
Seal the capped tubes with Parafilm and incubate with gentle agitation (e.g., slow rotation) overnight (∼16–24 h) in the dark.
-
8
Optional: Next day, to improve DNA yields, incubate with agitation for an additional 1–3 h at 56 °C. This step improves the digestion of the bone powder and thereby releases more DNA, especially in cases when the powder used is relatively coarse. If using a fine powder, this step can be omitted.
Critical Step
This step may cause further damage or degradation of the DNA owing to the high incubation temperature.
DNA purification by binding to silica: preparation of DNA binding
Timing 30 min (for seven samples plus a negative control)
-
9
Centrifuge the samples for 2 min at 5,000g.
Critical Step
Keep the remaining sample material; you may wish to retain it for a second round of extraction, especially if working with rare samples.
-
10
Transfer the supernatant into 40 ml binding buffer in a 50 ml conical tube, add 100 μl silica suspension and adjust the pH to ∼4.0 by adding ∼300 μl of 30% w/v HCl. First add only 200 μl of 30% w/v HCl, mix gently and measure the pH by pipetting (this minimizes the chance of introducing contamination) a few microliters to indicator paper. If the pH is higher than 4.0, add more HCl in 25 μl aliquots until pH 4.0 is reached.
Critical Step
Silica needs to be vortexed before pipetting, as the particles settle down quickly.
Critical Step
The amount of HCl you need to add may vary from sample to sample, as the pH of the extraction solution depends on the amount and type of sample and the extent of decalcification (EDTA complexes calcium ions, thereby releasing hydrogen ions, and therefore influences the pH).
Critical Step
Do not add too much HCl to the solution, as DNA will be destroyed at lower pH values. It is better to have a pH of 4.5 than 3.5.
Caution
HCl is acidic and may cause skin irritation; wear protective clothes and gloves.
DNA purification by binding to silica: incubation
Timing 3 h (no work required)
-
11
Close the tubes and seal with Parafilm. Incubate with agitation for 3 h in the dark.
DNA purification and elution
Timing 1–2 h (for seven samples plus a negative control)
-
12
Centrifuge the samples for 2 min at 5,000g. Pour the supernatant into a new tube.
Critical Step
Keep the supernatant in the refrigerator until you know the extraction worked, otherwise you can repeat the binding step with the same binding buffer by adding new silica suspension.
-
13
Add 1 ml binding buffer to the silica pellet and resuspend the silica by pipetting up and down.
-
14
Transfer the buffer–silica suspension into a fresh 2 ml tube. This transfer makes handling more convenient, as 2 ml tubes rather than 50 ml tubes can be used in all following steps.
-
15
Centrifuge for 15 s at 16,000g.
-
16
Discard the supernatant and remove the remaining solution with a pipette.
Critical Step
If the binding solution is not completely removed, the salt concentration in the elution buffer will be too high and all DNA will not be released from the silica during elution.
-
17
Add 1 ml washing buffer to the silica pellet and resuspend the silica by pipetting up and down.
-
18
Centrifuge for 15 s at 16,000g.
-
19
Discard the supernatant and remove the remaining liquid with a pipette.
-
20
Repeat Steps 17–19 once.
-
21
Centrifuge again for 15 s at 16,000g and remove the remaining liquid with a pipette.
-
22
Dry the silica at room temperature for ∼15 min with open lids.
-
23
Add 50 μl TE buffer to the dried silica and resuspend by stirring with the pipette tip and pipetting up and down.
-
24
Incubate with closed lids for ∼10 min; gently shake occasionally.
-
25
Centrifuge for 2 min at 16,000g.
-
26
Transfer the supernatant into a fresh tube. Optionally, the elution steps (Steps 23–26) can be repeated. Note that the second eluate will contain lower amounts of DNA compared to the first. Thus, combining both elutions will increase the total amount of DNA, but decrease the DNA concentration in the extract. It is also possible to store the first and second eluates separately, so that the first is not diluted by the second.
Critical Step
Try to avoid transferring large amounts of silica, as this may interfere with or even inhibit downstream applications.
-
27
Use this extract, or a dilution, for downstream applications. If necessary, determine the concentration of DNA in the extract as outlined in Box 1.
Pause point
Store the extract at −20 or −80 °C. It is better to aliquot the DNA before freezing to avoid DNA loss during freeze–thaw cycles.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Silica preparation: 5 h
Buffer preparation: 30–60 min
Steps 1–5, preparation of the bone or tooth sample: 15–30 min per sample
Steps 6–8, DNA release: 1 day (only 10 min work required)
Steps 9 and 10, DNA purification by binding to silica: preparation of DNA binding: 30 min (for seven samples plus a negative control)
Step 11, DNA purification by binding to silica: incubation: 3 h (no work required)
Steps 12–27, DNA purification and elution: 1–2 h (for seven samples plus a negative control)
Anticipated results
We extracted DNA from a number of cave bear and mammoth samples (Supplementary Table 1) using this protocol. Samples ranged in weight from 295 to 580 mg. We amplified mitochondrial products from the cave bear samples12 and nuclear products from the mammoth samples17 using regular16 or multiplex PCR21. Although not every PCR was successful in every attempt, 13 out of 17 cave bear samples gave positive mitochondrial PCR products in at least one trial using various dilutions of the extracts. Using mammoth samples and primers amplifying very short fragments of the nuclear MC1R gene, four out of six samples gave positive results in at least one trial using various dilutions (see Supplementary Table 1).
There seems to be no noticeable difference in the efficiency of this protocol between caves and open sites or between the different types of samples, as the majority of the PCR attempts were successful.
Note: Supplementary information is available via the HTML version of this article.
References
Noonan, J.P. et al. Genomic sequencing of Pleistocene cave bears. Science 309, 597–599 (2005).
Noonan, J.P. et al. Sequencing and analysis of Neanderthal genomic DNA. Science 314, 1113–1118 (2006).
Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).
Poinar, H.N. et al. Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA. Science 311, 392–394 (2006).
Green, R.E. et al. Analysis of one million base pairs of Neanderthal DNA. Nature 444, 330–336 (2006).
Rogaev, E.I. et al. Complete mitochondrial genome and phylogeny of Pleistocene mammoth Mammuthus primigenius. PLoS Biol. 4, e73 (2006).
Geigl, E.-M. On the circumstances surrounding the preservation and analysis of very old DNA. Archaeometry 44, 337–342 (2002).
Pääbo, S. Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc. Natl. Acad. Sci. USA 86, 1939–1943 (1989).
Höss, M., Dilling, A., Currant, A. & Pääblo, S. Molecular phylogeny of the extinct groundsloth Mylodon darwinii. Proc. Natl. Acad. Sci. USA 93, 181–185 (1996).
Hofreiter, M., Jaenicke, V., Serre, D., Haeseler Av, A. & Pääbo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 29, 4793–4799 (2001).
Hansen, A.J. et al. Crosslinks rather than strand breaks determine access to ancient DNA sequences from frozen sediments. Genetics 173, 1175–1179 (2006).
Rohland, N. & Hofreiter, M. Comparison and optimization of ancient DNA extraction. Biotechniques 42, 343–352 (2007).
Höss, M. & Pääbo, S. DNA extraction from Pleistocene bones by a silica-based purification method. Nucleic Acids Res. 21, 3913–3914 (1993).
Hänni, C., Brousseau, T., Laudet, V. & Stehelin, D. Isopropanol precipitation removes PCR inhibitors from ancient bone extracts. Nucleic Acids Res. 23, 881–882 (1995).
Kalmar, T., Bachrati, C.Z., Marcsik, A. & Rasko, I. A simple and efficient method for PCR amplifiable DNA extraction from ancient bones. Nucleic Acids Res. 28, E67 (2000).
Hofreiter, M. et al. Evidence for reproductive isolation between cave bear populations. Curr. Biol. 14, 40–43 (2004).
Rompler, H. et al. Nuclear gene indicates coat-color polymorphism in mammoths. Science 313, 62 (2006).
Hofreiter, M., Serre, D., Poinar, H.N., Kuch, M. & Pääbo, S. Ancient DNA. Nat. Rev. Genet. 2, 353–359 (2001).
Pääbo, S. et al. Genetic analyses from ancient DNA. Annu. Rev. Genet. 38, 645–679 (2004).
Willerslev, E. & Cooper, A. Ancient DNA. Proc. Biol. Sci. 272, 3–16 (2005).
Roempler, H. et al. Multiplex amplification of ancient DNA. Nat. Protoc. 1, 720–728 (2006).
Leonard, J.A., Wayne, R.K. & Cooper, A. Population genetics of ice age brown bears. Proc. Natl. Acad. Sci. USA 97, 1651–1654 (2000).
Haak, W. et al. Ancient DNA from the first European farmers in 7500-year-old Neolithic sites. Science 310, 1016–1018 (2005).
Shapiro, B. et al. Rise and fall of the Beringian steppe bison. Science 306, 1561–1565 (2004).
Serre, D. et al. No evidence of neandertal mtDNA contribution to early modern humans. Plos Biol. 2, 313–317 (2004).
Orlando, L. et al. Revisiting Neanderthal diversity with a 100,000 year old mtDNA sequence. Curr. Biol. 16, R400–R402 (2006).
Caramelli, D. et al. A highly divergent mtDNA sequence in a Neanderthal individual from Italy. Curr. Biol. 16, R630–R632 (2006).
Iudica, C.A., Whitten, W.M. & Williams, N.H. Small bones from dried mammal museum specimens as a reliable source of DNA. Biotechniques 30, 732–736 (2001).
Vigilant, L., Hofreiter, M., Siedel, H. & Boesch, C. Paternity and relatedness in wild chimpanzee communities. Proc. Natl. Acad. Sci. USA 98, 12890–12895 (2001).
Schander, C. & Halanych, K.M. DNA, PCR and formalinized animal tissue—a short review and protocols. Org. Divers. Evol. 3, 195–205 (2003).
Poinar, H.N. et al. Molecular coproscopy: dung and diet of the extinct ground sloth Nothrotheriops shastensis. Science 281, 402–406 (1998).
Reynolds, M.M. & Williams, C.G. Extracting DNA from submerged pine wood. Genome 47, 994–997 (2004).
Ou, C.Y., Moore, J.L. & Schochetman, G. Use of UV irradiation to reduce false positivity in polymerase chain reaction. Biotechniques 10, 442, 444, 446 (1991).
Gilbert, M.T.P., Hansen, A.J., Willerslev, E., Turner-Walker, G. & Collins, M. Insights into the processes behind the contamination of degraded human teeth and bone samples with exogenous sources of DNA. Int. J. Osteoarchaeol. 16, 156–164 (2006).
Sambrook, J., Fritsch, E.F. & Maniatis, T. Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989).
Tuma, R.S. et al. Characterization of SYBR Gold nucleic acid gel stain: a dye optimized for use with 300-nm ultraviolet transilluminators. Anal. Biochem. 268, 278–288 (1999).
Singer, V.L., Jones, L.J., Yue, S.T. & Haugland, R.P. Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal. Biochem. 249, 228–238 (1997).
Heid, C.A., Stevens, J., Livak, K.J. & Williams, P.M. Real time quantitative PCR. Genome Res. 6, 986–994 (1996).
Acknowledgements
We thank the members of the MPI EVA ancient DNA groups and Holger Römpler for discussion. This work was funded by the Max Planck Society.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Table 1
Anticipated results. (PDF 42 kb)
Rights and permissions
About this article
Cite this article
Rohland, N., Hofreiter, M. Ancient DNA extraction from bones and teeth. Nat Protoc 2, 1756–1762 (2007). https://doi.org/10.1038/nprot.2007.247
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.247
This article is cited by
-
Ancient human DNA recovered from a Palaeolithic pendant
Nature (2023)
-
Genetic sexing of subadult skeletal remains
Scientific Reports (2023)
-
Improving kinship probability in analysis of ancient skeletons using identity SNPs and MPS technology
International Journal of Legal Medicine (2023)
-
SNP analysis of challenging bone DNA samples using the HID-Ion AmpliSeq™ Identity Panel: facts and artefacts
International Journal of Legal Medicine (2023)
-
Hares from the Late Pleistocene of Ukraine: a phylogenetic analysis and the status of Lepus tanaiticus (Mammalia, Lagomorpha)
Biologia (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
