
Abstract
We describe a protocol for mutating genes in the nematode Caenorhabditis elegans using the Mos1 transposon of Drosophila mauritiana. Mutated genes containing a Mos1 insertion are molecularly tagged by this heterologous transposable element. Mos1 insertions can therefore be identified in as little as 3 weeks using only basic molecular biology techniques. Mutagenic efficiency of Mos1 is tenfold lower than classical chemical mutagens. However, the ease and speed with which mutagenic insertions can be mapped compares favorably with the vast amount of work involved in classical genetic mapping. Therefore, Mos1 could be the tool of choice when screening procedures are efficient. In addition, Mos1 mutagenesis can greatly simplify the mapping of mutations that exhibit low penetrance, subtle or synthetic phenotypes. The recent development of targeted engineering of C. elegans loci carrying Mos1 insertions further increases the attractiveness of Mos1-mediated mutagenesis.
Similar content being viewed by others

Introduction
Forward genetic screens have been used with great success in the nematode C. elegans to identify the genes involved in a variety of biological processes such as programmed cell death1,2 or RNA interference (RNAi)3,4 (for review, see ref. 5). Chemical mutagens such as ethyl methanesulfonate (EMS) are most commonly used to generate random mutations in the C. elegans genome because they are easy to use, efficient and create a wide range of genetic lesions. For example, a loss-of-function mutation can be obtained for a given gene by screening on average 2,000–4,000 chromosomes mutated by EMS under standard conditions5. However, the relative ease with which mutants can be isolated often contrasts with the difficulty of identifying the mutated gene when no further information is available. Genetic mapping, although simplified and rendered more systematic by the advent of single-nucleotide polymorphism (SNP) mapping6, is still a tedious and time-consuming task. This is especially true when performing modifier screens (e.g., enhancers, suppressors or synthetic screens), which require manipulation of both the initial mutation and the modifier mutation over the course of the mapping experiments. When phenotypes are subtle or of low penetrance, following the relevant mutation reliably can be difficult, especially if the SNP-containing strain modifies the phenotypic expression of that mutation7.
One way to circumvent the need for genetic mapping is to use genome-wide RNAi screening strategies8. In C. elegans, systemic downregulation of a specific gene can be achieved by feeding animals with bacteria that express double-stranded RNA corresponding to that gene. Libraries that contain most open reading frames of the C. elegans genome are now available. Such screens have been spectacularly successful in the identification of the genes with roles during early embryogenesis9, for example. However, RNAi screens also have intrinsic limits. First, 15% (ref. 8) to 40% (ref. 10) of the predicted C. elegans genes are still absent from RNAi-feeding libraries. Second, RNAi does not downregulate all genes with the same efficiency. Some genes are not sensitive to RNAi or show “hypomorphic” behaviors when compared to actual genetic mutants. Third, RNAi by feeding is very inefficient at targetting genes expressed in neurons11. Mutant strains that are more sensitive to RNAi than wild type have been isolated and have been used successfully to RNAi genes in neurons12,13,14. However, these strains display intrinsic mutant phenotypes that can interfere with the process of interest. Therefore, there exists the need for random forward genetic screens.
We describe here a protocol for implementing a mutagenesis technique based on the mobilization of the Drosophila mauritiana transposon Mos1 (see ref. 15) in the germ line of C. elegans16,17.This technique has been used successfully in forward genetic screens to identify genes of interest18,19,20,21. This system has three main advantages.
-
Mutated genes are molecularly tagged by the insertion of a Mos1 transposon. By identifying genomic sequences flanking these transposons, they can be mapped with single-nucleotide resolution in as little as 3 weeks, hence circumventing the need for genetic mapping.
-
Mos1 mutagenesis is safe for the experimenter, in contrast to EMS, which is highly mutagenic in all metazoans, including Homo sapiens.
-
We recently developed a technique called MosTIC to engineer custom C. elegans alleles of genes of interest by homologous recombination22. This technique relies on the remobilization of a Mos1 transposon from the targeted locus. The remobilization of the transposon induces a double-strand break, which can be repaired by copying engineered sequences from a transgene into the genome. Therefore, Mos1-tagged alleles recovered in a screen represent valuable entry points to further manipulate genes of interest.
Mos1 is a member of the Tc1/mariner family of transposons. The Mos1 sequence is composed of a single open reading frame encoding a transposase, flanked by short terminal inverted repeats. All Tc1/mariner elements transpose via a conserved “cut-and-paste” mechanism23,24. The transposase binds to the inverted repeats and catalyzes the excision and subsequent insertion of the element from one genomic locus into the other. The general strategy of Mos1-mediated mutagenesis is depicted in Figure 1. It uses a two-component system to mobilize Mos1 in the C. elegans germ line. The first component, called the “transposon array”, is an extrachromosomal transgene carrying multiple copies of Mos1. The second is another transgene, the “transposase array”, in which the expression of the Mos1 transposase gene is under the regulation of a heat shock-inducible promoter. When worms carrying both transgenes are subjected to heat shock, the transposase is expressed and catalyzes the transposition of Mos1 from the “transposon array” into chromosomal loci. The F1 or F2 progeny of these heat-shocked worms can then be screened for phenotypes of interest. As Mos1 transposons are normally absent from C. elegans, Mos1 insertions represent molecular tags that are easy to identify using inverse PCR25 (Fig. 2), a DNA amplification method that is often used to identify flanking sequences to a known stretch of DNA. We have adapted this technique to identify Mos1 insertions in the C. elegans genome (see Fig. 2). The average number of insertions per line is 2.5 but can be up to 10 (ref. 17 and unpublished results). When multiple insertions are present, the mutagenic insertion can be identified either by serial outcrossing of the mutant strain in order to eliminate non-mutagenic insertions or by testing for linkage between a genetic marker (genetic mutant or SNP) and the mutation (Fig. 3). In ideal cases, it can take as little as 3 weeks to molecularly identify the mutated gene in a given mutant.

Set up crosses between oxEx166 hermaphrodites and N2 males at 25 °C (Steps 1–4). Three days later, select male cross-progeny carrying the “transposon array” oxEx166 and cross them to hermaphrodites carrying the “transposase array” oxEx229. Three days later, select doubly transgenic oxEx166; oxEx229 hermaphrodites. Pick pools of three doubly transgenic worms. Record the number of generations when both arrays are maintained together in a strain, starting with TT1 for the doubly transgenic worms picked from the second cross. At the TT2 generation, test the transposition rate (Steps 5–22). Amplify the population of doubly transgenic worms for another two to four generations until you are ready to begin a screen (Steps 23–26). As a control, test the transposition rate during the screen by setting aside 30 F1s (Step 26). While screening, maintain the doubly transgenic population to perform successive heat shocks and screens. This figure has been adapted with permission from Bessereau29.

(1) genomic DNA is first digested, then (2) DNA fragments are ligated onto themselves. (3) Using primers 1 and 2 in Mos1, a first PCR serves to amplify genomic sequences flanking the transposon insertion. (4) A nested PCR with primers 3 and 4 enriches for specific products. (5) Fragments are gel-purified and TA-cloned. Individual transformant colonies are screened by PCR. (6) The precise insertion point can be determined by sequencing PCR products using primer 3. This figure has been adapted with permission from Bessereau29.

Confirm that a candidate mutant carries Mos1 insertions by PCR (Steps 19 and 20). Outcross the candidate mutant and either (i, Step 28B) map the mutation to a chromosome and proceed with inverse PCR (Steps 29–43), or (ii, Step 28A) verify that all outcrossed lines carry Mos1 insertions (Steps 19 and 20), perform inverse PCR (Steps 29–43) and test for genetic linkage between candidate Mos1 insertions and the phenotype (Step 44A). Validate candidate mutants by additional means (Steps 44B–E). This figure has been adapted with permission from Bessereau29.
However, there are a number of points to consider before choosing Mos1-mediated mutagenesis for a specific screen.
-
Mos1 mutagenesis is not as straightforward as exposing worms to chemical mutagens such as EMS and requires careful monitoring of the different steps to ensure that mutagenesis will be successful.
-
Mos1 mutagenesis is about ten times less efficient than EMS17 and generates mostly strong loss-of-function or null alleles as mutations are caused by the insertion of the 1.3 kb Mos1 sequence into the target gene. It is therefore highly recommended to perform a small-scale pilot screen using EMS to verify that mutants of interest are generated at a frequency compatible with standard loss-of-function mutations before performing a Mos1-based screen. The Mos1 screen can then be scaled appropriately to maximize the chances of success.
-
Like for any mutagen, it is unlikely that all genes are equally mutable by Mos1. The only absolute requirement for Mos1 insertion is the presence of a TA dinucleotide in the target sequence. The analysis of 914 independent Mos1 insertions did not reveal significant hotspots for Mos1 insertions except for a region of chromosome I (see ref. 26). Conversely, one could imagine cold-spots, that is, regions of the genome from which Mos1 would be excluded. However, such cold-spots cannot be defined conclusively before more insertions are identified.
-
Transposons can cause mutations by “hit-and-run” mechanisms27,28. In this case, the transposon is re-excised from the genome, leaving a molecular scar behind and the resulting mutation is no longer tagged by the transposon. Out of 72 mutants, which we isolated using Mos1 mutagenesis screens, five mutations were not attributable to Mos1 insertions. However, we cannot be certain that these mutants have been caused by hit-and-run events because they were recovered at frequencies compatible with spontaneous mutations (unpublished results).
In conclusion, choosing Mos1-mediated mutagenesis will depend on the trade-off between time spent screening for mutants and time spent mapping and rescuing a mutation caused by a chemical mutagen. The recent development of MosTIC22, a genome-engineering technique based on the re-excision of Mos1 from the locus to manipulate, is an additional parameter to support the choice of Mos1 as a mutagen. Theoretically, all Mos1 alleles can serve as reagents for the MosTIC technique, opening new avenues to analyze gene function in a near-physiological context.
Materials
Reagents
-
C. elegans strains (see Table 1 and REAGENT SETUP) can be obtained, at no expense, from the Caenorhabditis Genetics Center (http://www.cbs.umn.edu/CGC/) or from the Bessereau Lab (http://www.biologie.ens.fr/bcsgnce/)
Table 1 C. elegans strains used for Mos1-mediated mutagenesis. -
Oligonucleotides, stored at −20 °C as 25 μM stocks (see Table 2). The position of each oligonucleotide on the Mos1 sequence is shown in Figure 4
Table 2 Oligonucleotides used in this protocol. Figure 4: Mos1 sequence features. 
Sequence of the Mos1 transposon (accession number X78906). When Mos1 is inserted in the genome, TA dinucleotides flank this sequence. Primers and restriction sites used in this protocol are indicated. Cleavage sites for class A enzymes are shown in red. Cleavage sites for class B enzymes are shown in orange. This figure has been adapted with permission from Bessereau29.
-
Mos1 sequence: NCBI accession number X78906 (http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nucleotide&val=1335619)
-
dNTPs 10 mM (Fermentas, #R0182)
-
Taq DNA polymerase (Invitrogen, 10342-020)
-
T4 DNA ligase (Fermentas, #EL0015)
-
TA-cloning kit pGEM-T Easy Vector System I (Promega, A1360)
-
Transformation-competent Escherichia coli bacteria
-
QIAquick gel extraction kit (QIAgen, 28706)
-
QIAquick PCR purification kit (Qiagen, 28704)
-
DNEasy DNA extraction kit (Qiagen, 69504)
-
1 kb Plus DNA Ladder (Invitrogen, 10787-026)
-
Worm lysis solution (see REAGENT SETUP)
-
Restriction enzymes: Class A: HaeIII (R0108S), HpaII (R0171S) and Sau3AI (R0169S); class B: AluI (R0137S), HhaI (R0139S) and MseI (R0525S); NEB
-
Agarose gels for molecular biology
-
LB agar plates with appropriate antibiotic (according to TA-cloning kit instructions)
-
Nystatin solution (Sigma, N3503, http://www.sigmaaldrich.com; see REAGENT SETUP)
-
Nematode growth media (NGM) plates (see REAGENT SETUP)

Equipment
-
Dedicated Pipetman to load PCR samples onto gels (Gilson)
Critical
This minimizes stock reagent contamination by Mos1 PCR products.
-
Parafilm M* Laboratory Wrapping Film (Fisher, 13-374-10, http://www.fishersci.com)
-
Dissecting scope equipped with epifluorescence to visualize GFP in C. elegans
-
Heating waterbath
-
Thermal cycler (MJ Research PTC-200)
-
15, 20 and 25 °C incubators
Reagent setup
-
C. elegans care and handling Instructions for maintaining C. elegans strains can be found at http://www.wormbook.org/chapters/ www_strainmaintain/strainmaintain.html. Strains are grown at 20 °C on NGM plates unless otherwise noted. Propagate the “transposase array” strain (EG2762 oxEx116) and the “transposon array” (EG1470 oxEx229) strain at 25 °C in order to diminish the likelihood of transgene silencing in the germ line. Freeze multiple vials of these two strains when you receive them to have multiple backup stocks, which can be thawed if the transgenes become silenced. Instructions on how to freeze C. elegans strains can be found at http://www.wormbook.org/chapters/ www_strainmaintain/strainmaintain.html. The strain carrying the “transposase array” can sometimes give rise to “abnormal” progeny (e.g., worms showing uncoordinated, sterile or protruding vulva phenotypes). These phenotypes are likely caused by leaky expression of the transposase in somatic cells and never inherited in the progeny. Strains containing the “transposon array” and the “transposase array” are maintained separately to prevent spontaneous insertion of Mos1 transposons and to avoid a decrease of transposition efficacy when the two transgenes are maintained together over many generations. Therefore, the first step of Mos1-mediated mutagenesis is to combine both arrays into a single strain (Figs. 1 and 5 and Steps 1–3).
Figure 5: Representative pictures of transgenic strains used in this protocol. 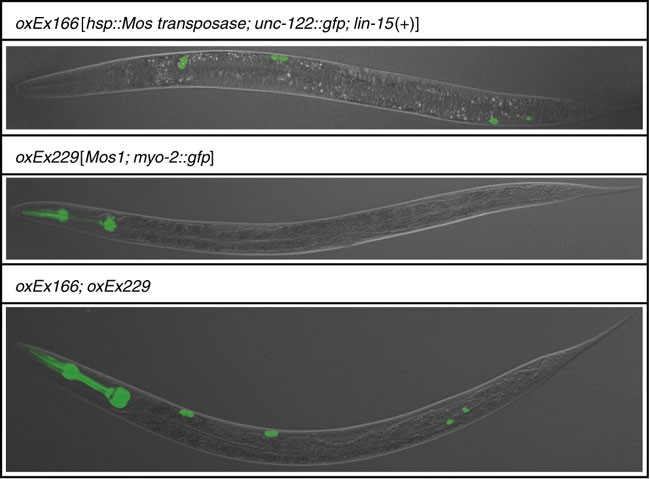
See Table 1 for detailed information about these strains.
-
Worm lysis buffer 50 mM KCl, 10 mM Tris-HCl (pH 8.3), 2.5 mM MgCl2, 0.45% (v/v) Tween 20, 0.45% (v/v) NP-40 and 0.01% (w/v) gelatin. Worm lysis buffer can be stored at 4 or −20 °C.
-
Worm lysis solution Add 1 mg ml−1 proteinase K to worm lysis buffer. Worm lysis solution is used to prepare crude worm lysates (Step 15) and should be prepared fresh.
-
Nystatin solution Dissolve 4 g of nystatin in 200 ml ethanol and 200 ml ammonium acetate. Heat to 50 °C until the solution is homogeneous and filter-sterilize using a 0.22 μm filter.
-
NGM plates 3 g NaCl, 16 g agar, 2.5 g peptone, 975 ml H2O, 1 ml cholesterol (5 mg ml−1 in ethanol), 2.5 ml nystatin, 1 ml of 1 M CaCl2, 1 ml of 1 M MgSO4 and 25 ml KH2PO4 pH 6; the last five ingredients should be filter-sterilized and added after autoclaving using sterile techniques.

Procedure
Generating doubly transgenic worms
Timing 12 days
-
1
Cross 12 N2 males with four hermaphrodites carrying the “transposase array” (oxEx166—labeled by gfp expression in coelomocytes) (see Figs. 1 and 5 and REAGENTS). Grow the worms at 25 °C for 3 days. Only a fraction of the male progeny produced by these crosses will carry the “transposase array” since the transgene is not integrated into the genome. To obtain a sufficient number of transgenic males for Step 2, set up at least five crosses.
-
2
From the crosses in Step 1, select male cross progeny carrying the “transposase array” and set up at least 5 crosses between 12 transgenic males and 4 L4 hermaphrodites carrying the “transposon array” (oxEx229—labeled by gfp expression in the pharynx) (see Figs. 1 and 5 and REAGENTS). Grow the worms at 25 °C for 3 days.
-
3
From the crosses in Step 2, select doubly transgenic worms carrying both the “transposon array” and the “transposase array” (see Figs. 1 and 5 and REAGENTS). Label these worms TT1 to indicate that this is the first generation in which the “transposase array” and the “transposon array” are combined. To amplify the population of doubly transgenic worms, pick pools of three doubly transgenic hermaphrodites to fresh plates and let them self-fertilize at 25 °C; pick five or more pools.
Critical Step
If brood size of doubly transgenic worms is very low at 25 °C (less than 50 progeny per animal), temperature can be decreased down to 20 °C without significant loss of transposition efficiency in the later steps.
Critical Step
It is important to track the number of generations where the transposon array and the transposase arrays are combined as transposition efficiency tends to decrease over time. In addition, it is possible that leaky expression of the transposase causes random insertions in the background of doubly transgenic worms. However rare, this could make it more difficult to identify mutagenic insertions in later mapping stages. Therefore, generate new doubly transgenic strains after 15 generations or sooner if transposition frequency decreases.
-
4
Maintain and amplify the population of doubly transgenic worms by picking pools of three doubly transgenic hermaphrodites to fresh plates and growing them at 25 °C. Repeat until a sufficiently large population is obtained.
Critical Step
The plates of each successive generation should be labeled TT2, TT3 and so on.
Critical Step
Set apart ten young TT2 adults to test the transposition efficiency (Steps 5–22).
Measuring transposition rate: heat-shock-induced Mos1 transposition
Timing ∼4 h
-
5
While the population of doubly transgenic worms grows, perform a test heat shock on ten doubly transgenic young adults from the TT2 generation to verify the transposition rate of the strain. Equilibrate a water bath to 33 °C ahead of time.
Critical Step
Heat shocking is deleterious for worms that contain both the transposase and the transposon arrays, most probably because the heat-shock promoter is driving high levels of transposase expression in somatic tissues and causes extremely high rates of transposition in somatic cells. We estimate that, on average, ten copies of Mos1 insert into each haploid genome in somatic tissues. Thus, heat-shock conditions have been optimized to achieve efficient transposition without overly affecting brood size. On average, heat-shocked worms will produce around 50 F1 progeny, which is significantly lower than wild type.
-
6
Use two layers of parafilm to seal the plate containing the ten doubly transgenic young adult TT2 worms set apart in Step 4.
Critical Step
Heat shock only young adult stage doubly transgenic worms. Do not heat shock L4 larvae because they are more likely to die or be sterile after a heat shock.
-
7
Submerge the plate in the water bath at 33 °C for 1 h.
-
8
Remove the plate from the water bath and place it at 20 °C for 1 h.
-
9
Put the plate back at 33 °C for 1 h.
-
10
Take the plate out of the water bath, remove the parafilm and let the worms recover and lay their first eggs for 12 h at 20 °C.
-
11
After 12 h, transfer pairs of worms onto five fresh plates and let them grow at 20 °C. The heat-shock regime outlined in Steps 7–11 is summarized in Table 3.
Table 3 Summary of heat-shock treatment. -
12
At 24–30 h after heat shock, randomly pick, under visible light, six individual F1 progeny from each of the five plates of heat-shocked worms from Step 11. Move each worm to individual plates. Grow these F1 worms for 3 days at 25 °C.
-
13
After 3 days, pick ten F2 worms that do not carry the “transposon array” oxEx229 (i.e., have no GFP in the pharynx) from each clonal F1 plate from Step 12 together onto a new plate. Grow these worms at 25 °C for 3 days.
Critical Step
As the PCR assay to detect Mos1 insertions in the genome also detects Mos1 in the “transposon array”, it is essential to pick only those worms that do not contain the array (i.e., show no gfp expression in the pharynx; see Fig. 5). Note that presence of the “transposase array” oxEx166 (labeled by gfp expression in coelomocytes; see Fig. 5) is inconsequential, as the PCR assay used to detect Mos1 transposons does not amplify the transposase array.
Measuring transposition rate: preparation of worm lysates for PCR
Timing ∼2 h
-
14
In the F3 generation, prepare worm lysates from each clone by lysing 20 worms that do not carry the “transposon array”.
Critical Step
Contamination of these worm lysates with worms that contain the “transposon array” will lead to false-positive PCR and erroneous values of transposition rates. If some of the F3 worms still carry the oxEx229 “transposon array”, either (a) pick ten worms that do not carry the “transposon array” to a fresh plate, wait for 4 days and proceed to Step 15 if none of the F4 worms carries the oxEx229 array; or (b) prepare worm lysates as described in Step 15 and verify that none of the lysed F3 worms is mosaic for the “transposon array” by performing an oJL103/oJL104 PCR, which detects the presence of the “transposon array” (see Step 20). Use worms carrying the oxEx229 “transposon array” as a positive control for this experiment.
-
15
For each clone, place 20 worms in 10 μl of worm lysis solution and freeze them for 15 min at −80 °C. Freezing is a crucial step that disrupts the envelope of the worm and promotes efficient lysis.
Critical Step
Remember to include positive controls (worms carrying the “transposon array”) and negative controls (N2 worms).
-
16
Incubate for 1 h at 65 °C to lyse the worms.
-
17
Incubate for 15 min at 95 °C to inactivate proteinase K in the lysis solution.
Pause point
Keep the lysates on ice or freeze them at −20 °C.
Measuring transposition rate: PCR for Mos1 transposon detection
Timing ∼3 h
-
18
Set up a PCR for each worm lysate as detailed below, using oligonucleotides oJL102 and oJL103 to detect Mos1 insertions (see REAGENTS and Fig. 4).
Table 4 Table 5 -
19
Run the PCR program detailed below.
Table 5 Table 6 -
20
If necessary (see Step 14), check for the absence of contaminant oxEx229 DNA by performing a PCR using oJL103 and oJL104 as outlined below. Presence of oxEx229 contaminant DNA will result in a PCR product of 726 bp. Use the PCR program detailed in Step 19.
Table 6 Table 7 -
21
Analyze PCR products on a 1% agarose gel. Presence of a Mos1 transposon will result in a PCR product of 355 bp.
Critical Step
Use a dedicated pipetman to load these PCR products to avoid contamination of future PCR experiments by Mos1 PCR products.
-
22
Calculate the transposition rate by dividing the number of Mos1-positive clones by the total number of clones that were analyzed. It should be 50 ± 15%. If the transposition rate is lower, refer to the TROUBLESHOOTING section.
Screening with Mos1
Timing variable
-
23
To start a screen, select doubly transgenic early adult stage worms using an epifluorescence dissecting microscope from the population that has been amplified in Step 4. Pool 50 doubly transgenic worms onto one plate. Make as many pools as necessary. These animals are the P0 population.
Critical Step
Remember that each heat-shocked P0 worm will produce only ∼50 F1 progeny. As Mos1 mutagenesis is about ten times less efficient than chemical mutagenesis with EMS, scale your experiment to screen the appropriate number of haploid genomes. Results from an EMS pilot screen are valuable to estimate this number. In addition, note that some of the P0 worms might die because of the heat shock. Therefore, more worms should be heat-shocked than theoretically necessary.
-
24
Heat shock the worms immediately, as outlined in Steps 7–11 and Table 3.
-
25
Once the worms have recovered for 12 h and depending on the screening strategy, move individual or groups of P0 worms onto small or large plates. Alternatively, let multiple heat-shocked worms lay eggs for a given period of time on a fresh plate (e.g., 12 h) and retransfer them to fresh plates at regular intervals. This will synchronize the F1 population and make it easier to manage large numbers of worms. Stop harvesting F1 progeny 36 h after heat shock as these worms will not carry Mos1 insertions17.
-
26
When the population of F1 animals is grown, set apart 30 F1s on individual plates to measure the transposition rate that was obtained in the heat-shocked P0s from Step 24. Proceed with these F1s as described in Steps 13–22.
Critical Step
A test of transposition rate must be performed during the screen to control the efficacy of the mutagenesis step.
-
27
Screen the F1 or the F2 population for the phenotype you are interested in. Clone individual mutants to establish mutant lines. Unless two worms with clearly different phenotypes are found on a given plate, assume that mutants on a plate are all siblings and represent a single mutant line.
Identifying Mos1 mutants: testing for mendelian segregation of the mutation
-
28
Once a candidate mutant has been isolated from a screen, confirm that its phenotype breeds true to the next generation and test whether worms that have lost the “transposon array” oxEx229 are positive for Mos1 by PCR (Steps 15–19). After the presence of Mos1 in the mutant strain is confirmed, use either option A or B. Option A involves outcrossing the mutant against N2 or another relevant genetic background. This is often the simplest approach. Option B involves rough genetic mapping using SNPs or genetic markers. Mapping is more informative but often requires more work and reagents.
-
A
Outcrossing against N2 or another relevant genetic background
-
i
Outcross the mutant strain against N2 (or another appropriate genetic background) and generate 24 independent outcrossed clones that have been reselected for the phenotype of interest.
-
ii
Test whether all of these independent clones carry at least one Mos1 insertion by PCR, as detailed in Steps 15–19.
-
iii
If all independent clones are positive for Mos1, select one clone on which to perform inverse PCR (Steps 29–43).
Pause point
The remaining clones can be kept at 15 °C and used later to validate a mutagenic insertion (see Step 44A).
Critical Step
If one or multiple mutant clones are found to contain no Mos1 transposon, then the mutation is unlikely to be tagged with a Mos1 insertion and will have to be identified by classical genetic mapping.
Timing variable
-
i
-
B
Mapping using SNPs or genetic markers
-
i
Map the mutation to a chromosome by using SNPs or visible genetic markers as outlined in the WormBook (http://www.wormbook.org/chapters/ www_introandbasics/introandbasics.html). This will provide genetic mapping information, which can be used to confirm or exclude Mos1 insertions identified by inverse PCR.
-
ii
Analyze independent mutant lines generated during the mapping process for the presence of Mos1 insertions by PCR, as detailed in Steps 15–19.
-
iii
If all independent clones are positive for Mos1, select one clone on which to perform inverse PCR (Steps 29–43).
Timing variable
-
i
Critical Step
Once a Mos1-carrying mutant line is established, you could theoretically perform inverse PCR (Steps 29–43) immediately. However, we recommend applying inverse PCR only after outcrossing the mutant line once to increase the chances of identifying relevant Mos1 insertions. This additional step also serves to obtain preliminary mapping or genetic linkage information, which will narrow down the list of candidate insertions when multiple insertions are localized in a strain.
-
A
Identifying Mos1 mutants: localizing Mos1 insertions by inverse PCR
Timing variable
-
29
Prepare genomic DNA.
Critical Step
Inverse PCR can be performed on purified genomic DNA (DNEasy DNA extraction kit; Qiagen) or on a worm lysate. Worm lysates work most of the time; a 20 ml lysate can be prepared from 20 worms as detailed in Steps 15–17, half of which should be used for digestion. However, we prefer to use purified genomic DNA because it gives more reliable results.
-
30
Digest approximately 150 ng of genomic DNA in a final volume of 30 ml, as detailed in the table below. Incubate the digestion mix for 3 h to overnight at 37 °C. Inactivate the restriction enzyme by incubating at 75 °C for 15 min. We recommend that each DNA sample be analyzed using at least two different enzymes.
Table 7 Table 8 Critical Step
Six different restriction enzymes can be used to digest the genomic DNA (see Fig. 4, REAGENTS and Step 37). Each enzyme will cut in a different position in Mos1 and the flanking genomic DNA sequence (see Fig. 4). Enzymes are grouped into two classes depending on where they cut in Mos1 and therefore which set of primers is used to perform the inverse PCRs: class A: HaeIII, HpaII and Sau3AI; class B: AluI, HhaI and MseI. When there are multiple insertions in a strain, often only a subset of insertions will be identified using one restriction enzyme. Indeed some insertions can be missed if the closest restriction site in the flanking genomic sequence is too far (resulting in no PCR product) or too close (resulting in too little sequence to be informative).
-
31
Set up ligations of digested genomic DNA as detailed below. Setting up the ligation in a large volume will ensure that intramolecular reactions are favored. Incubate overnight at 4 or 15 °C.
Table 8 Table 9 Pause point
The ligation reactions can be kept frozen at −20 °C indefinitely.
-
32
Set up inverse PCRs, as detailed below, using 3 μl of ligation mix (from Step 31) using the appropriate primer combination depending on whether a class A (primers oJL103 and oJL114) or class B (primers iPCR1a and iPCR1b) enzyme was used to digest the genomic DNA.
Table 9 Table 10 -
33
Perform PCR using the program outlined below. Choose an annealing temperature appropriate for the primer pair used in Step 32.
Table 10 Table 11 -
34
Set up a nested PCR using a 1:200 dilution of the PCR products from Step 33 and the appropriate primer pair: primers oJL115 and oJL116 for class A; primers oJL102 and IPCR2b for class B.
Table 11 Table 12 -
35
Perform PCR using the appropriate program (for class A or for class B) as outlined below.
Table 12 Table 13 -
36
Run PCR products from Step 35 on a 1.8% agarose gel. Each PCR can generate multiple products of different sizes, which can correspond to independent Mos1 insertions or to a single Mos1 insertion (owing to partial digests, religation of degraded DNA fragments, illegitimate PCR priming, etc.). Three examples of inverse PCR products are shown in Figure 6a.
Figure 6: Inverse PCR examples. 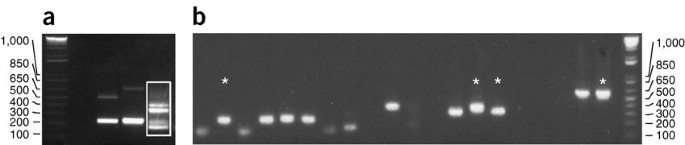
(a) Examples of inverse PCR on three different independent strains using the MseI restriction enzyme. The products in lane 3 (white box) were TA-cloned by cutting out the four most prominent bands. All visible bands were of sufficient size to be informative. (b) Colony PCRs derived from the TA cloning of the inverse PCR (white box in a). Stars indicate four different samples that were sequenced.
Critical Step
Use a dedicated pipetman to load these PCR products.
-
37
Gel-purify candidate PCR products using the QIAquick gel purification kit. Purify only those fragments that are longer than the minimal sizes listed in the table below. Shorter fragment would not be informative.
Table 13 Table 14 -
38
Gel-purified fragments could be sent for sequencing directly if individual bands can be clearly separated. However, we recommend TA-cloning the gel-purified PCR products in order to obtain individual clones that will guarantee successful sequencing. TA-clone gel-purified PCR products according to the manufacturer's protocol. Transform the TA-cloning reactions and select transformants using the appropriate antibiotic selection.
Pause point
TA-cloning ligations can be stored at −20 °C, or remain at 4 °C for 2 days before being transformed. Transformant colonies can be stored for some time at 4 °C.
-
39
Perform colony PCRs on individual transformant colonies from the TA cloning using the PCR protocol described in Steps 34 and 35, that is, pick a single colony using a sterile pipet tip and swirl the tip in the PCR tube containing the PCR mix. It is usually possible to recover clones representing all the principal PCR products obtained in Step 36.
-
40
Analyze PCR products on a 1.8% agarose gel (see Fig. 6b).
Critical Step
Use a dedicated pipetman to load these PCR products.
-
41
If PCR products of different sizes are obtained, select one representative sample for each size class and purify it using the QIAquick PCR purification kit (see Fig. 6b). Do not use samples containing multiple bands because sequencing will fail.
-
42
Sequence samples using either oJL115 (for class A enzymes) or oJL102 (for class B enzymes) as a primer.
-
43
Analyze sequencing results. Legitimate PCR products must contain the 5′ end of the transposon sequence directly followed by the C. elegans genomic sequence starting at a TA dinucleotide (Fig. 2). Mos1 insertions can be mapped to the genome by using BLAST or by using the Genome Browser function of wormbase (http://www.wormbase.org/db/seq/gbrowse/wormbase/).

Identifying Mos1 mutants: validating Mos1 alleles
-
44
Once a candidate insertion has been identified by inverse PCR, use one or more of the five basic strategies (options A–E) described below to validate the mutant.
-
A
Test for genetic linkage
-
i
This approach tests whether a mutant phenotype cosegregates with a given Mos1 insertion. First, design PCR primers located on each side of the Mos1 insertion. In combination with a primer inside Mos1, this allows one to differentiate between loci that do or do not contain a Mos1 insertion.
-
ii
Next, in the case of a recessive mutation, outcross the mutant against N2 (or another appropriate genetic background) and generate 24 independent mutant lines. If a candidate Mos1 insertion is homozygous in all 24 independent lines, the mutation is less than two genetic map units away from the Mos1 insertion. As Mos1 transposons are usually scattered throughout the genome, the likelihood that there will be no other Mos1 insertion in the vicinity is high. It is possible to further narrow down the genetic interval by increasing the number of independent clones, for example, 49 independent clones will place the mutation within less than one genetic map unit of the Mos1 insertion. Conversely, if a mutant clone that lacks the Mos1 insertion is found, this strain should be used to start a new mapping cycle.
-
i
-
B
Map the mutant phenotype to a chromosomal region
-
i
Use genetic mapping data to confirm or reject a candidate Mos1 insertion, as discussed in Step 28B.
-
i
-
C
Independent alleles
-
i
If a screen produces independent Mos1 mutants in the same gene, they validate each other. In addition, alleles previously generated in other screens (e.g., using chemical mutagens) or by reverse genetic approaches (http://www.wormbook.org/chapters/ www_introreversegenetics/introreversegenetics.html#d0e928) can be used to validate a Mos1 mutant candidate using genetic complementation tests (http://www.wormbook.org/chapters/ www_complementation/complementation.html).
-
i
-
D
Phenocopy by RNAi
-
i
If no other alleles are available, RNAi can be used to phenocopy the candidate mutant (http://www.wormbook.org/chapters/ www_introreversegenetics/introreversegenetics.html).
-
i
-
E
Rescue experiments
-
i
A definitive way to validate a candidate mutant consists of complementing the defect by providing a wild-type copy of the gene via a transgene (http://www.wormbook.org/chapters/ www_transformationmicroinjection/ transformationmicroinjection.html).
-
i
-
A
Troubleshooting
Troubleshooting advice can be found in Table 4.
Timing
Steps 1–4: generating doubly transgenic worms, 12 days
Steps 5–13: measuring transposition rate, heat-shock-induced Mos1 transposition, ∼4 h
Steps 14–17: measuring transposition rate, preparation of worm lysates for PCR, ∼2 h
Steps 18–22: measuring transposition rate, PCR for Mos1 transposon detection, ∼3 h
Steps 23–27: screening with Mos1, variable
Step 28: identifying Mos1 mutants, testing for mendelian segregation of the mutation, variable
Steps 29–43: identifying Mos1 mutants, localizing Mos1 insertions by inverse PCR, variable
Step 44: identifying Mos1 mutants, validating Mos1 alleles, variable
Anticipated results
Transposition rate
The average transposition rate should be 50 ± 15%. If the transposition rate is consistently lower than average, thaw the “transposon array” and “transposase array” strains again and generate new doubly transgenic strains.
Number of Mos1 insertions per strain
The number of Mos1 insertion per strain can range from 1 to 10 and averages 2.5 (see ref. 17).
Mutant yield
We estimate that Mos1 mutagenesis is roughly ten times less efficient than chemical mutagenesis with EMS17. This number was calculated by measuring the relative mutagenicity of EMS and Mos1 in a screen for mutants resistant to the antihelmintic drug levamisole. The mutagenic frequency of EMS was found to be 2.34 × 10−3 versus 4.3 × 10−4 for Mos1. Taking into account that Mos1 only affects gametes of the maternal germline, this means that ten times more genomes must be analyzed to obtain the same number of mutants. This is a critical parameter to take into account when deciding to undertake a Mos1 genetic screen and for future scaling of the experiment.
Nature of mutations
Most Mos1 insertions will lead to strong loss-of-function or null alleles. However, some insertions can generate hypomorphic alleles too. Insertions in promoters, introns or in late exons of genes have been found to partially decrease gene activity. In rarer cases, semi-dominant alleles have also been isolated17.
References
Ellis, H.M. & Horvitz, H.R. Genetic control of programmed cell death in the nematode C. elegans . Cell 44, 817–829 (1986).
Hedgecock, E.M., Sulston, J.E. & Thomson, J.N. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans . Science 220, 1277–1279 (1983).
Ketting, R.F., Haverkamp, T.H., van Luenen, H.G. & Plasterk, R.H. Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell 99, 133–141 (1999).
Tabara, H. et al. The rde-1 gene, RNA interference, and transposon silencing in C. elegans . Cell 99, 123–132 (1999).
Jorgensen, E.M. & Mango, S.E. The art and design of genetic screens: Caenorhabditis elegans . Nat. Rev. Genet. 3, 356–369 (2002).
Wicks, S.R., Yeh, R.T., Gish, W.R., Waterston, R.H. & Plasterk, R.H. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 28, 160–164 (2001).
Hodgkin, J. & Doniach, T. Natural variation and copulatory plug formation in Caenorhabditis elegans . Genetics 146, 149–164 (1997).
Kamath, R.S. et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237 (2003).
Sonnichsen, B. et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans . Nature 434, 462–469 (2005).
Rual, J.F. et al. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 14, 2162–2168 (2004).
Timmons, L., Court, D.L. & Fire, A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans . Gene 263, 103–112 (2001).
Kennedy, S., Wang, D. & Ruvkun, G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans . Nature 427, 645–649 (2004).
Simmer, F. et al. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 1, E12 (2003).
Wang, D. et al. Somatic misexpression of germline P granules and enhanced RNA interference in retinoblastoma pathway mutants. Nature 436, 593–597 (2005).
Jacobson, J.W., Medhora, M.M. & Hartl, D.L. Molecular structure of a somatically unstable transposable element in Drosophila . Proc. Natl. Acad. Sci. USA 83, 8684–8688 (1986).
Bessereau, J.L. et al. Mobilization of a Drosophila transposon in the Caenorhabditis elegans germ line. Nature 413, 70–74 (2001).
Williams, D.C., Boulin, T., Ruaud, A.F., Jorgensen, E.M. & Bessereau, J.L. Characterization of Mos1-mediated mutagenesis in Caenorhabditis elegans: a method for the rapid identification of mutated genes. Genetics 169, 1779–1785 (2005).
Ruaud, A.F. & Bessereau, J.L . Activation of nicotinic receptors uncouples a developmental timer from the molting timer in C. elegans . Development 133, 2211–2222 (2006).
Ruaud, A.F. & Bessereau, J.L. The P-type ATPase CATP-1 is a novel regulator of C. elegans developmental timing that acts independently of its predicted pump function. Development 134, 867–879 (2007).
Gally, C., Eimer, S., Richmond, J.E. & Bessereau, J.L. A transmembrane protein required for acetylcholine receptor clustering in Caenorhabditis elegans . Nature 431, 578–582 (2004).
Yook, K. & Hodgkin, J. Mos1 mutagenesis reveals a diversity of mechanisms affecting response of Caenorhabditis elegans to the bacterial pathogen Microbacterium nematophilum . Genetics 175, 681–697 (2007).
Robert, V. & Bessereau, J.L. Targeted engineering of the Caenorhabditis elegans genome following Mos1-triggered chromosomal breaks. EMBO J. (2006).
van Luenen, H.G., Colloms, S.D. & Plasterk, R.H. The mechanism of transposition of Tc3 in C. elegans . Cell 79, 293–301 (1994).
Dawson, A. & Finnegan, D.J. Excision of the Drosophila mariner transposon Mos1. Comparison with bacterial transposition and V(D)J recombination. Mol. Cell 11, 225–235 (2003).
Ochman, H., Gerber, A.S. & Hartl, D.L. Genetic applications of an inverse polymerase chain reaction. Genetics 120, 621–623 (1988).
Granger, L., Martin, E. & Segalat, L. Mos as a tool for genome-wide insertional mutagenesis in Caenorhabditis elegans: results of a pilot study. Nucleic Acids Res. 32, e117 (2004).
Salzberg, A. et al. P-element insertion alleles of essential genes on the third chromosome of Drosophila melanogaster: mutations affecting embryonic PNS development. Genetics 147, 1723–1741 (1997).
Hiesinger, P.R. & Bellen, H.J. Flying in the face of total disruption. Nat. Genet. 36, 211–212 (2004).
Bessereau, J.L. Insertional mutagenesis in C. elegans using the Drosophila transposon Mos1: a method for the rapid identification of mutated genes. Methods Mol. Biol. 351, 59–73 (2006).
Acknowledgements
We thank Iskra Katic for critical reading of the manuscript and members of the Bessereau lab and the Jorgensen lab for optimizing technical aspects of the protocol. T.B. is an EMBO long-term fellow. This work was funded by a European Union Grant (6th Framework program, PL-503334, code NemageneTAG) and an AVENIR grant from the Institut National de la Santé et de la Recherche Médicale.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Boulin, T., Bessereau, JL. Mos1-mediated insertional mutagenesis in Caenorhabditis elegans. Nat Protoc 2, 1276–1287 (2007). https://doi.org/10.1038/nprot.2007.192
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.192
This article is cited by
-
A role for worm cutl-24 in background- and parent-of-origin-dependent ER stress resistance
BMC Genomics (2022)
-
Transgenesis in parasitic helminths: a brief history and prospects for the future
Parasites & Vectors (2022)
-
Target lines for recombinase-mediated gene stacking in soybean
Theoretical and Applied Genetics (2022)
-
Two active bamboo mariner-like transposable elements (Ppmar1 and Ppmar2) identified as the transposon-based genetic tools for mutagenesis
Molecular Breeding (2016)
-
The codon-optimized Δ6-desaturase gene of Pythium sp. as an empowering tool for engineering n3/n6 polyunsaturated fatty acid biosynthesis
BMC Biotechnology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
