
Abstract
2-Alkyl-4-quinolones (AHQs) such as 2-heptyl-3-hydroxy-4-quinolone (PQS) and 2-heptyl-4-quinolone (HHQ) are quorum sensing signal molecules. Here, we describe methods for AHQ detection, tentative identification and quantification, which employ a lux-based Pseudomonas aeruginosa AHQ biosensor strain. The protocol describes both thin-layer chromatography (TLC) and microtiter plate assays, which use bioluminescence or the green color of pyocyanin as detection end points. Organic solvent extracts of bacterial cells or cell-free culture supernatants are chromatographed on TLC plates, which are dried and overlaid with the AHQ biosensor. AHQs appear as both luminescent and green spots. For the microtiter assay, either spent bacterial culture supernatants or extracts are added to a growth medium containing the AHQ biosensor. Light output is proportional to the AHQ content of the sample. The assays described take approximately 2 days to complete, are simple to perform, do not require sophisticated instrumentation and are highly amenable to screening large numbers of bacterial samples. However, apart from PQS and HHQ in P. aeruginosa, definitive AHQ identification will require additional MS and NMR analyses.
Similar content being viewed by others


Introduction
Bacteria release a wide variety of small molecules including secondary metabolites such as antibiotics and siderophores (iron chelators), metabolic end products and cell-to-cell signaling molecules. The latter facilitate the coordination of gene expression within a bacterial population1,2,3,4. Cell-to-cell communication in bacteria is referred to as “quorum sensing” (QS) and depends on the action of diffusible signal molecules or “autoinducers”. As a bacterial culture grows, signal molecules are released into the extracellular milieu and accumulate. Once a critical threshold concentration of the QS signal molecule (and consequently a specific population density) has been achieved, a coordinated change in bacterial behavior is initiated via a specific sensor kinase or response regulator, so facilitating the expression of QS-dependent target genes1,2,3,4. Secondary metabolite production, bioluminescence, competence, plasmid transfer, biofilm development and pathogenicity are all known to be controlled by QS in many different Gram-positive and Gram-negative bacteria.
QS signal molecules are chemically diverse, ranging from peptides and N-acylhomoserine lactones (AHLs) to furanones and AHQs1,2,3,4. Given the vast number of extracellular metabolites, the chemical diversity among known QS signal molecules is likely to represent only the “tip of the iceberg”. Indeed, it has been suggested that the majority of low-molecular-weight organic compounds made and secreted by microbes are likely to function as QS signal molecules5.
Studies of QS in diverse bacteria have greatly benefited from the availability of biosensor assays, which respond sensitively to specific QS signal molecule classes and do not require sophisticated instrumentation. Such assays can provide tentative information on the chemical identity, number and concentrations of the QS signal molecules present in bacterial culture supernatants. For example, AHLs can be readily detected and quantified using AHL biosensors based on lux6,7, gfp8 or lacZ9 reporter gene fusions or violacein pigment induction10 while autoinducer-2 is generally assayed using a bioluminescent Vibrio harveyi strain11.
In the 1940s and 1950s, a number of antibacterial AHQ compounds were isolated and purified from P. aeruginosa culture supernatants4. Subsequently, AHQs were discovered to inhibit the growth of Gram-positive bacteria, algae and phytoplankton, to modulate host immune defences, chelate iron and act as QS signal molecules12,13,14,15,16,17,18. Other pseudomonads, Burkholderia and Alteromonas species also produce AHQs and it is highly likely that other Gram-negative bacteria such as Ralstonia species synthesize these compounds as they possess homologs of the P. aeruginosa and B. pseudomallei AHQ biosynthetic genes19.
Although over 50 different AHQs have been identified in P. aeruginosa, most are present at low concentrations where they are unlikely to be physiologically active20,21. AHQs exist in another tautomeric form and can also be termed 4-hydroxy-2-alkylquinolines (HAQs)17,20. However, the neutral 4-quinolone rather than the 4-hydroxy-quinoline is the predominant species in the pH range 4–6 and hence under physiological conditions17; so here we use the AHQ rather than HAQ nomenclature. The major P. aeruginosa AHQ signal molecules are 2-heptyl-3-hydroxy-4(1H)-quinolone (the Pseudomonas quinolone signal (PQS)) and 2-heptyl-4-quinolone (HHQ, Fig. 1)16,21,22,23. While HHQ is the immediate precursor of PQS, it can also function as a QS signal molecule not only in P. aeruginosa but also in Burkholderia pseudomallei19 and probably in other AHQ-producing bacteria, although this remains to be confirmed.

Structures of the AHQs, PQS and HHQ.
AHQ biosensor assay
Here we present simple biosensor-based protocols that we previously published that facilitate the rapid, cost-effective detection and tentative identification of AHQs, in particular PQS and HHQ, in bacterial cells and culture supernatants16,17,19 (M.P.F., S.P.D., S. Crusz, S.R. Chhabra, M.C. and P.W., unpublished manuscript). This method utilizes a P. aeruginosa biosensor designed to respond to AHQs by emitting bioluminescence and producing the green pigment pyocyanin. The assay can be configured for use as a detection system following TLC of cell or supernatant extracts or as a 96-well microtiter plate assay. Although the combined use of the TLC and AHQ biosensor overlay, and synthetic standards for PQS and HHQ are more than sufficient to establish the identity of these two AHQs in P. aeruginosa cultures, for other bacteria and indeed other AHQs that activate the biosensor, additional MS and NMR analysis will be required to unequivocally identify the AHQ compound.
In P. aeruginosa, AHQ biosynthesis depends on the pqsABCDE operon and mutation of pqsA renders the organism AHQ-negative19,21,24. Both PQS and HHQ act as autoinducers as they are required to drive their own synthesis by binding to the LysR-type regulator, PqsR(MvfR), such that PqsR(MvfR) binding to the pqsA promoter is enhanced17,23,25. To construct an AHQ biosensor, we first deleted pqsA in strain PAO1 before introducing a luxCDABE-based pqsA promoter fusion onto the chromosome of the pqsA mutant17 (M.P.F., S.P.D., S. Crusz, S.R. Chhabra, M.C. and P.W., unpublished manuscript) using the mini-CTX luxCDABE plasmid system described by Becher and Schweizer26. The surrogate pqsA promoter was inserted into a non-coding site in the chromosome where it can be activated independently of the native gene promoter. The use of the entire luxCDABE operon dispenses with the need to add exogenously a long-chain fatty aldehyde substrate for the luciferase7. Figure 2 shows the mechanism by which an AHQ such as PQS activates the biosensor and induces bioluminescence. As the pqsA mutant does not produce any AHQs, it is dark and light is emitted only upon exposure to an exogenous AHQ source. Bioluminescence can be detected on TLC plate overlays using an X-ray film or a photon video camera or quantified by using a luminometer for liquid-based assays. As P. aeruginosa is not naturally bioluminescent, the biosensor background light level is very low and it responds to a wide dynamic range of both PQS and HHQ, from nanomolar to micromolar concentrations (M.P.F., S.P.D., S. Crusz, S.R. Chhabra, M.C. and P.W., unpublished manuscript). In addition, although less amenable to rapid quantification, the bioreporter produces the blue–green pigment pyocyanin in response to both PQS and HHQ, which is readily apparent on the TLC overlays. Furthermore, the AHQ biosensor is most sensitively activated by PQS and HHQ, both of which have C7 alkyl chains. Analogs of both compounds with alkyl side chains from C1 to C11 are capable of activating the reporter albeit at higher concentrations (M.P.F., S.P.D., S. Crusz, S.R. Chhabra, M.C. and P.W., unpublished manuscript).

(a) PQS binds to PqsR(MvfR) and enhances PqsR binding to the pqsA promoter, resulting in expression of the luxCDABE operon and production of the proteins required for light generation. (b) LuxAB form the α- and β-subunits of the luciferase enzyme that catalyzes the oxidation of reduced flavin mononucleotide (FMNH2) and a long-chain fatty aldehyde (R-CHO), generated by the LuxCDE proteins. This results in the emission of a blue-green light at 490 nm. As the biosensor bacteria are naturally dark and cannot produce AHQs, light is emitted only upon exposure to an exogenous AHQ source.
AHQs such as PQS and HHQ can be detected in crude cell-free culture supernatants using the lux-based AHQ biosensor in a liquid microtiter plate assay. If AHQ levels are low or where information on cell or vesicle-associated AHQs is required, they can simply be concentrated by extraction with solvents such as dichloromethane or ethyl acetate, as AHQs readily partition into the organic phase. After removal of the solvent by rotary evaporation, the residue can be rehydrated in an aqueous buffer before AHQ biosensor analysis. Although the P. aeruginosa AHQ bioreporter can detect HHQ and PQS at nanomolar concentrations (detection limit 12 nM for both AHQs), it responds in a dose-dependent manner to both compounds (M.P.F., S.P.D., S. Crusz, S.R. Chhabra, M.C. and P.W., unpublished manuscript). The AHQ biosensor is most sensitively activated by PQS and HHQ both of which have C7 alkyl chains. Analogs with C5, C9 and C11 are also capable of activating the biosensor albeit less sensitively (M.P.F., S.P.D., S. Crusz, S.R. Chhabra, M.C. and P.W., unpublished manuscript). Consequently, the light output from the bioreporter in response to a P. aeruginosa culture supernatant reflects the combined concentrations of HHQ and PQS as well the other AHQs listed above. However, the latter, at least in P. aeruginosa, are mostly present at substantially lower concentrations except for the corresponding C9 analogs, 2-nonyl-4-quinolone (HNQ) and 2-nonyl-3-hydroxy-4-quinolone (C9-PQS) (see ref. 27). The AHQ liquid bioassay therefore provides a positive indication for the presence of PQS, HHQ and closely related AHQs. However, it is only a semiquantitative indication of the total AHQs present in a given spent bacterial culture supernatant. In P. aeruginosa, for example, the data obtained will reflect predominantly the PQS and HHQ concentrations. The assay is however fully quantitative if used for bacteria, which make only a single AHQ and for synthetic standards. A typical dose–response curve of maximal light output from the AHQ biosensor in response to a range of PQS and HHQ concentrations is shown in Figure 3. The liquid assay is therefore highly appropriate where comparative overall, rather than absolute, AHQ concentrations for individual AHQs are required. The liquid assay cannot provide information on the concentrations of the individual AHQs present in a mixture as this would require the introduction of additional separation and purification stages by, for example, HPLC. An additional limitation of the biosensor is that it will not respond to all AHQ analogs although this could be considered as a major advantage with respect to HHQ and PQS analysis.

Dose–response curves showing the light output from the AHQ biosensor (PAO1 pqsA CTX-lux::pqsA) in response to increasing concentrations of (a) PQS and (b) HHQ. The AHQ biosensor produces light on exposure to AHQs in a dose-dependent manner. The detection limit for PQS and HHQ is 12 nM for both AHQs.
To determine whether both PQS and HHQ are present together with other AHQs, cell-free supernatant, cell or vesicle extracts can be extracted with ethyl acetate and subjected to TLC. After chromatography, the TLC plate is overlaid with a thin nutrient agar containing the bioreporter strain, which is subsequently monitored for bioluminescence and pyocyanin production (Fig. 4). Tentative identification of positive spots can be made by comparison of their relative migration (Rf) values with synthetic standards. Unequivocal chemical identification of any positive compounds can be made by MS and/or NMR analysis. The method can also be made more quantitative by spotting a range of concentrations of a synthetic AHQ(s) on the TLC plate.

(a) TLC plate run with standards of PQS and HHQ and supernatant extracts of PAO1, PAO1 pqsA and PAO1 pqsH and visualized under UV at 312 nm. (b) Overlay of TLC plate with soft top agar containing the biosensor bacteria showing production of the green pigment, pyocyanin, in response to AHQs, (c) Overlay of TLC plate with soft top agar containing biosensor bacteria showing production of light in response to AHQs visualized using a luminograph photon video camera. (d) Overlay of TLC plate with soft top agar containing biosensor bacteria showing production of light in response to AHQs visualized using X-ray film exposed to the overlaid TLC plate for 5 min. TLC lanes: (1): PQS 10 mM, 2 μl, (2): HHQ 10 mM, 2 μl, (3): PAO1 supernatant extract, 5 μl, (4): PAO1 pqsA supernatant extract, 5 μl, (5): PAO1 pqsH supernatant extract, 5 μl. The AHQ biosensor emits light over spots identified as PQS and HHQ in PAO1 and HHQ only in PAO1 pqsH. No light spots are observed for the AHQ-negative pqsA mutant. The spot migrating below HHQ in lanes 3 and 5 is primarily the C9 analog of HHQ, that is, HNQ.
Alternative AHQ detection and quantification assays
The AHQ biosensor provides a simple, rapid and sensitive means to detect AHQs and in particular PQS and HHQ without requiring any sophisticated instrumentation provided they are biologically active. Definitive chemical analysis can be achieved using reverse phase high-performance liquid chromatography (RP-HPLC) and liquid chromatography (LC)-tandem mass spectrometry19,20. For RP-HPLC analysis of AHQs, acidified ethyl acetate extracts of cell-free culture supernatants can be eluted isocratically using 80% (vol/vol) acetonitrile in water as the mobile phase and a reverse phase C18 column19 or using a linear 30–100% acetonitrile/water gradient plus 1% acetic acid on a reverse phase C8 column20 monitoring at 240–250 nm. The same conditions can be used for LC-MS/MS, and an extensive mass spectrometric analysis of the P. aeruginosa AHQs using positive electrospray ionization is presented by Lépine et al20. For quantitative PQS analysis, a stable isotope dilution assay has been described using single ion monitoring versus MS/MS in which deuterated PQS (5,6,7,8-tetradeutero-2-heptyl-3-hydroxy-4-quinolone) is employed as an internal standard27. The methods described by Lépine et al.20 are sensitive, can provide definitive chemical identification and quantification of individual AHQs irrespective of their biological activity. However, they require access to sophisticated expensive instrumentation, which requires specialist expertise to operate and to interpret the spectra, depend on the availability of the synthetic deuterated PQS standard and can be very time consuming for large numbers of samples.
Applications
The AHQ biosensor assays described here can be used to detect, tentatively identify and quantify AHQs in bacterial culture supernatants and in fluids or on surfaces contaminated with AHQ producers. To date, AHQ producers have been found in the genera Pseudomonas, Burkholderia and Alteromonas but are likely to be much more widespread given the presence of homologs of AHQ biosynthetic genes in other Gram-negative bacteria19. These assays are highly amenable to screening large numbers of environmental and clinical bacterial isolates for AHQ production and could be configured for use as clinical or industrial diagnostic tools. In the TLC configuration, the biosensor assay will provide information on the AHQ spectrum, and both liquid and TLC assays can be used to evaluate the impact of growth conditions, for example, the biofilm mode of growth or specific mutations on AHQ production. The biosensor assay can also be used to confirm the biological activity of synthetic PQS, HHQ and other closely related compounds.
Materials
Reagents
-
Stocks of the bacterial strains listed in the table below (available from PW): all bacterial stocks are maintained at −80 °C in 25% (vol/vol) glycerol
Table 1 Table 2 -
PQS synthetic standard 10 mM in methanol (PQS MW 259)
-
HHQ synthetic standard 10 mM in methanol (HHQ MW 243)
-
PQS and HHQ can be synthesized as described by Pesci et al.22 and by Diggle et al.17. Alternatively, small amounts can be purchased from this laboratory
-
Bacto-tryptone (Becton, Dickinson and Co.)
-
Tryptone (Oxoid)
-
Yeast extract (Oxoid)
-
Agar technical no. 3 (Oxoid)
-
Sodium chloride (Sigma)
-
Potassium dihydrogen phosphate monobasic (KH2PO4) (Sigma)
-
Tetracycline, 50 mg ml−1 in methanol
-
Methanol, HPLC grade (Fisher Scientific)
-
Ethyl acetate, HPLC grade (Fisher Scientific)
-
Acetone, HPLC grade (Fisher Scientific)
-
Dichloromethane, HPLC grade (Fisher Scientific)
-
Glacial acetic acid, analytical grade (Fisher Scientific)
-
Luria-Bertani (LB) medium: 1% (wt/vol) bacto-tryptone, 0.5% (wt/vol) yeast extract, 1% (wt/vol) sodium chloride in distilled water
-
LB agar: LB media with addition of 2% (wt/vol) agar technical no. 3
-
Soft top agar: 0.65% (wt/vol) agar technical no. 3, 1% (wt/vol) tryptone, 0.5% (wt/vol) sodium chloride
-
Potassium dihydrogen phosphate solution: 5% (wt/vol) KH2PO4 in distilled water
-
Acidified ethyl acetate: 0.01% (vol/vol) glacial acetic acid in ethyl acetate
Equipment
-
−80 °C freezer
-
Shaking incubator (e.g., Innova 4000; New Brunswick Scientific)
-
Centrifuge (e.g., Avanti 30; Beckman)
-
Spectrophotometer (e.g., Novaspec II; Pharmacia)
-
Rotary evaporator (e.g., R-114; Buchi)
-
−20 °C freezer
-
Hybridization oven (Stuart Scientific)
-
UV transilluminator (e.g., Vilber Lourmat TFX-20.M, 312 nm)
-
Luminograph photon video camera (e.g., LB 980; EG & G Berthold)
-
X-ray film (e.g., Pierce CL-Xposure 18 × 24 cm film)
-
96-well plate luminometer (Anthos Lucy1 or Tecan GENios Pro)
-
96-well black, clear bottom microtiter plates (Costar-Corning Inc.)
-
Tube luminometer (e.g., EG&G Berthold Junior LB9509)
-
Luminometer tubes (e.g., EG&G Berthold Junior Tubes 12 × 75 mm)
-
Microwave
-
Bunsen burner
-
Bench top vortexer
-
250 ml Erlenmeyer flasks (sterile)
-
Glass separating funnels
-
50 ml glass round-bottom flasks
-
Container to hold TLC plate for overlay
-
Normal phase 20 × 20 cm silica 60F254 TLC plates (Merck)
-
TLC developing tank (Sigma-Aldrich, 27.5 × 27.5 × 7.5 cm)
-
Universal containers (sterile) (Bibby-Sterilin)
-
Centrifuge tubes (sterile) (conical polypropylene; BD)
-
Petri dishes (sterile) (Bibby-Sterilin)
-
10 μl inoculation loops (sterile) (SLS)
-
Autoclave tape (e.g., Comply; 3M)
-
Sample vials 2 ml (SLS)
-
Disposable syringe (sterile) (Terumo)
-
0.20 μm disposable sterile filters (e.g., Minisart High Flow; Sartorius)
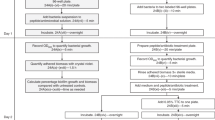
Procedure
Preparing bacterial cultures for AHQ extraction
Timing 2 days
-
1
Under sterile conditions streak out a 10 μl loop of the test bacterium (e.g., P. aeruginosa PAO1) and the control strains (PAO1 pqsA (AHQ negative) and PAO1 pqsH (HHQ positive PQS negative)) onto fresh LB agar plates. Grow overnight at 37 °C.
-
2
Next day, inoculate a single colony of each strain into separate universal containers, each universal containing 5 ml LB medium. Culture overnight at 37 °C with shaking at 200 r.p.m. While LB medium is used here, other nutrient media can be substituted if desired. Appropriate antibiotics or other selection reagents can be added to the growth medium as required for individual strains.
-
3
Determine the optical density at 600 nm (OD600) of 1 ml of each overnight culture and standardize the cultures to approximately OD 1.0 by diluting with fresh LB. Standardizing the cultures to OD600 1.0 under specific growth conditions allows cultures to be directly compared with one another for AHQ production. If comparability between strains is not important, standardization of cultures can be omitted to save time.
-
4
Aseptically transfer 0.25 ml of the standardized cultures to 25 ml of LB medium in a 250 ml Erlenmeyer flask. A small volume of culture in a large flask allows good aeration of the medium. Incubate at 37 °C, with shaking at 200 r.p.m. for 8 h. After growth, check the OD600 again to ensure the cultures have grown to an equivalent optical density if comparison between strains is required. To perform the microtiter plate assay, proceed as described in Box 1. To proceed with the TLC assay, continue with Step 5.
AHQ extraction of bacterial cultures
Timing 5 h
-
5
Transfer a defined volume of each culture (in this example 10 ml) to a 50 ml centrifuge tube and centrifuge at 10,000g, 4 °C for 10 min.
-
6
Extract the cells (option A) or the supernatants (option B) depending on whether cell-associated or extracellular AHQs are being assayed. It is possible to carry out both options concurrently.
-
A
Cell extraction
-
i
Decant the supernatants (save for supernatant extract (option B)) and gently resuspend the cells in 10 ml of fresh LB medium.
-
ii
Centrifuge at 10,000g, 4 °C for 10 min and discard the supernatant. Repeat the wash steps (6A(i) and (ii)) to remove all traces of the supernatant from the cells.
-
iii
Add 10 ml of methanol to the resulting cell pellets and vortex vigorously until resuspended.
-
iv
Allow to stand for 10 min to allow the cells to lyse before centrifuging again at 10,000g, 4 °C for 10 min.
-
v
Filter the extract through sterile Minisart (Sartorius) 0.2 μM filters into new centrifuge tubes to remove all cell debris from the extraction mixtures.
Pause point
Cell extraction mixtures can be stored in the freezer at −20 °C for several days if required.
-
i
-
B
Supernatant extraction
-
i
Filter the supernatants through sterile Minisart (Sartorius) 0.2 μM filters into clean centrifuge tubes to remove any unpelleted cells from the extraction mixtures.
-
ii
Add 10 ml of acidified ethyl acetate to the supernatants and vortex vigorously for 30 s so that the two phases are well mixed. As PQS is a ferric iron chelator17, the ethyl acetate is acidified to release the metal ion to facilitate partitioning of PQS into the organic solvent.
-
iii
Transfer the extraction mixtures into two separating funnels that have been previously washed with acetone and allow the extraction mixtures to settle and the two phases to separate.
-
iv
Transfer the top organic layers to clean centrifuge tubes. Repeat Steps 6B(ii) and (iii) on the bottom layers twice before discarding. Pool the collected organic layers.
Pause point
Supernatant extraction mixtures can be stored in the freezer at −20 °C for several days if required.
-
i
-
A
-
7
Transfer the extraction mixtures to 50 ml round-bottom flasks that have been previously washed with acetone and rotary evaporate the mixtures to dryness.
-
8
Add 0.5 ml of methanol to the round-bottom flasks and agitate for 30 s before transferring the liquid to 2 ml glass sample vials. Repeat this step with two further additions of 0.5 ml methanol and pool each sample in each vial.
Pause point
Cell and supernatant extraction mixtures can be stored in the freezer at −20 °C for several days if required.
-
9
Dry down the extraction mixtures in the sample vials under a stream of nitrogen gas. Store the samples in the freezer at −20 °C until required.
Pause point
Dry cell and supernatant extraction residues can be stored in the freezer at −20 °C for several months if required.
Preparation of TLC plates and running of samples
Timing 3 h
-
10
Prepare normal phase silica 20 × 20 cm 60F254 TLC plates by soaking in a 5% (wt/vol) solution of KH2PO4 for 30 min before activating at 100 °C for 1 h. A hybridization oven can be used to do this.
Pause point
The TLC plates are now ready for use and can be stored for several weeks if kept clean, dry and at room temperature (20–22°C).
Critical Step
Preparation of the biosensor culture (Steps 15 and 16) can be started at the same time as the test bacterium i.e. 2 days before running the TLC plate.
-
11
Draw a faint line in pencil approximately 4 cm from the bottom of the activated silica TLC plate; this will be used as a guide for spotting sample extracts.
-
12
Reconstitute sample extracts (from Step 9) in 100 μl of methanol and spot 5 μl of each onto the TLC plate. The quantity of extract to be spotted can be altered as desired. As positive controls, 2 μl of 10 mM stock solutions of synthetic PQS and HHQ (or other AHQs) in methanol can be spotted onto the TLC plate. Space each spot at 2 cm intervals along the line and optionally a hairdryer can be used to dry samples during spotting to give a tighter spot.
-
13
When dry, place the TLC plate into a developing tank and run the TLC using a mixture of dichloromethane:methanol (95:5) as the mobile phase until the solvent front is 1–2 cm from the top of the plate. The TLC plate can be visualized using a UV transilluminator at 312 nm and photographed at this point.
-
14
Allow the TLC plate to dry and apply autoclave tape to both the underside of the TLC plate and around the edges so that the tape creates a well at least 0.5 cm deep around the TLC plate into which the nutrient agar containing the biosensor will be poured. Make sure the autoclave tape is firmly pressed down and forms a tight seal. This procedure is described step by step in Figure 5.
Figure 5: Preparation of the biosensor bacteria agar TLC plate overlay. 
(a) Attach a strip of autoclave tape to the aluminum backing along each edge of the TLC plate, pressing down firmly to ensure a good seal. (b) Neatly trim the excess autoclave tape using scissors. (c) Pinch the autoclave tape at each corner of the TLC plate, creating a barrier of autoclave tape along each side of the TLC plate, as shown for corners (1) and (2) to create a well. (d) Place the TLC plate prepared as shown in c into a suitable dish and pour into the well the molten soft top agar containing the biosensor bacteria.
Pause point
The TLC plate can be stored at room temperature (20–22 °C) in a dry place for up to 1 week

Overlay of TLC plates with the AHQ reporter
Timing 2 days
-
15
Using aseptic technique, streak out a 10 μl loop of the AHQ biosensor (PAO1 pqsA CTX-lux∷pqsA) onto fresh LB agar plates containing 125 μg ml−1 tetracycline and grow overnight at 37 °C.
-
16
The next day, inoculate a single colony into a universal container containing 5 ml LB medium and grow overnight at 37 °C with shaking at 200 r.p.m.
Critical Step
Steps 15 and 16 can be performed before running the TLC plate if required to save time.
-
17
The next day, melt 100 ml of soft top agar in a microwave and allow to cool to ∼50 °C. Add 1 ml of the overnight culture to the soft top agar and mix gently.
Critical Step
Make sure that the agar has cooled sufficiently before adding the reporter bacteria. Too high a temperature will harm or kill the bacteria and attenuate growth. A temperature of 50 °C allows easy pouring of agar and gives sufficient time to pour the plate before the agar sets.
-
18
Pour the mixture slowly into the well made around the TLC plate, being careful to minimize bubble formation in the agar and on the TLC plate.
Critical Step
Do not delay too long or the agar will begin to solidify before pouring. If bubbles form, these can either be agitated to the edge of the plate before the agar sets or a Bunsen burner can be used to (carefully!) flame the surface of the agar to remove them.
-
19
Allow the agar to cool and set aseptically around a Bunsen flame before static incubation at 37 °C for 6 h to view light production, or overnight to view pyocyanin production.
-
20
Visualize the plates for light production using a luminograph photon video camera or develop using X-ray film (e.g., Pierce CL-XPosure film). Alternatively, simply view production of the blue/green phenazine pigment pyocyanin by eye. The results of this procedure are shown in Figure 4.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Preparing bacterial cultures for AHQ extraction, Steps 1–4: 2 days
AHQ extraction of bacterial cultures, Steps 5–9: 5 h
Preparation of TLC plates and running of samples, Steps 10–14: 3 h. Steps 15 and 16 can be carried out in advance of Steps 10–14.
Overlay of TLC plates with the AHQ reporter, Steps 15–20: 2 days (to view light output) or 3 days (to view pyocyanin production)
Remember that a substantial proportion of the quoted times for both assays involve 'dormant' periods, for example, for growth of cultures, running of TLCs, etc. The actual workload involved is much less than that implied by the total time frame.
Anticipated results
TLC assay
The TLC biosensor assay when coupled with synthetic standards will indicate the presence or absence of the AHQ molecules such as PQS and HHQ in a sample prepared from P. aeruginosa. For AHQs produced by other bacteria, a positive response from the biosensor for a spot with a given Rf value is a tentative indication of the presence of an AHQ, and further MS or NMR analysis will be required to unequivocally identify the AHQ. In the example shown in Figure 4, cultures of PAO1, the AHQ-negative pqsA mutant of PAO1 and the PQS-negative, HHQ-positive pqsH mutant were extracted for AHQs, the extracts run on TLC and then overlaid with agar containing the AHQ biosensor bacteria. PAO1 is a P. aeruginosa wild-type strain and produces an extensive range of AHQ molecules including both PQS and HHQ whereas the pqsA mutant is deficient for AHQ biosynthesis. The AHQ culture supernatant extracts, run on TLC and compared with PQS and HHQ standards and visualized under UV light, are shown in Figure 4a. Using this method, under UV light (312 nm) the PQS synthetic standard fluoresces pink and has a greater Rf value than the purple HHQ standard as it is more polar and so runs further up the plate. These spots are present in the PAO1 extract but not the PAO1 pqsA extract. Confirmation that these spots are PQS and HHQ is shown when the plates are overlaid with the bioreporter together with PQS and HHQ synthetic standards. The reporter bacteria produce light in response to PQS and HHQ on the plate over the respective standards, and also over the corresponding spots from the PAO1 supernatant sample extracts. The reporter bacteria only respond to AHQs; therefore, there are no light spots over the sample extracts from the PAO1 pqsA mutant. The pqsH mutant shows primarily a spot corresponding to HHQ. The spot running beneath HHQ in both cell and supernatant extracts is a mixture of HHQ-related molecules including HNQ and the N-oxide derivative of HHQ (2-heptyl-4-quinolone-N-oxide), which do not completely resolve on this TLC and are also found in P. aeruginosa supernatants21. Note that there is a greater response to HHQ than PQS, which may be a consequence of stronger interactions between PQS and the silica stationary phase, as the latter is a strong metal ion chelator17. Pyocyanin production by the biosensor follows a similar pattern to light production. If desired, definitive identification of the AHQs detected via this TLC assay can subsequently be achieved using MS or NMR techniques.
Microplate assay
The liquid AHQ biosensor assay will provide a cumulative response to all of the biologically active AHQS in a culture sample. Figure 6 shows the mean maximal light output from cell-free culture supernatants prepared from PAO1 and the PAO1 pqsA mutant before and after extraction with ethyl acetate, respectively. The reporter bacteria only respond to AHQs; therefore, there is only baseline light output from the concentrated supernatant extracts of the PAO1 pqsA mutant and negative control (bioreporter alone). The PAO1 samples both elicit a large increase in bioluminescence by the bioreporter, confirming the presence of AHQ molecules in the sample and giving an indication of their comparative cumulative levels.

Mean maximal light output from the AHQ biosensor in the absence or presence of cell-free culture supernatants (crude or concentrated ethyl acetate extracts) prepared from PAO1 or the pqsA mutant (negative control), respectively. The biosensor emits light in response to the PAO1 but not the pqsA culture supernatant.
References
Withers, H., Swift, S. & Williams, P. Quorum sensing as an integral component of gene regulatory networks in Gram-negative bacteria. Curr. Opin. Microbiol. 4, 186–193 (2001).
Winzer, K., Hardie, K.R. & Williams, P. LuxS and autoinducer-2: their contribution to quorum sensing and metabolism in bacteria. Adv. Appl. Microbiol. 53, 291–296 (2003).
Lazdunski, A.M., Ventre, I. & Sturgis, J.N. Regulatory circuits and communication in Gram-negative bacteria. Nat. Rev. Microbiol. 2, 581–592 (2004).
Diggle, S.P., Cornelis, P., Williams, P. & Cámara, M. 4-Quinolone signalling in Pseudomonas aeruginosa: old molecules, new perspectives. Int. J. Med. Microbiol. 296, 83–91 (2006).
Yim, G., Wang, H. & Davies, J. The truth about antibiotics. Int. J. Med. Microbiol. 296, 163–170 (2006).
Williams, P. et al. Small molecule-mediated density-dependent control of gene expression in prokaryotes: bioluminescence and the biosynthesis of carbapenem antibiotics. FEMS Microbiol. Lett. 100, 161–168 (1992).
Winson, M. et al. Engineering the luxCDABE genes from Photorhabdus luminescens to provide a bioluminescent reporter for constitutive and promoter probe plasmids and mini-Tn5 constructs. FEMS Microbiol. Lett. 163, 193–202 (1998).
Andersen, J. et al. gfp-based N-acyl homeserine-lactone sensor systems for detection of bacterial communication. Appl. Environ. Microb. 67, 575–585 (2001).
Shaw, P. et al. Detecting and characterizing N-acyl-homeserine lactone signal molecules by thin-layer chromatography. Proc. Natl. Acad. Sci. USA 94, 6036–6041 (1997).
McClean, K. et al. Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomeserine lactones. Microbiology 143, 3703–3711 (1997).
Bassler, B.L., Greenberg, E.P. & Stevens, A.M. Cross-species induction of luminescence in the quorum sensing bacterium Vibrio harveyi . J. Bacteriol. 179, 4043–4045 (1997).
Wratten, S.J., Wolfe, M.S., Andersen, R.J. & Faulkner, D.J. Antibiotic metabolites from a marine Pseudomonad. Antimicrob. Agents Chemother. 11, 411–414 (1977).
Machan, Z.A., Taylor, G.W., Pitt, T.L., Cole, P.J. & Wilson, R. 2-Heptyl-4-hydroxyquinoline N-oxide, an antistaphylococcal agent produced by Pseudomonas aeruginosa . J. Antimicrob. Chemother. 30, 615–623 (1992).
Hooi, D.S.W., Bycroft, B.W., Chhabra, S.R., Williams, P. & Pritchard, D.I. Differential immune modulatory activity of Pseudomonas aeruginosa quorum-sensing signal molecules. Infect. Immun. 72, 6463–6470 (2004).
Brendenbruch, F., Geffers, R., Nimtz, M., Buer, J. & Häussler, S. The Pseudomonas aeruginosa quinolone signal (PQS) has an iron chelating activity. Environ. Microbiol. 8, 1318–1329 (2006).
Diggle, S.P. et al. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol. Microbiol. 50, 29–43 (2003).
Diggle, S.P. et al. The Pseudomonas aeruginosa 4-quinolone signal molecules HHQ and PQS play multifunctional roles in quorum sensing and iron entrapment. Chem. Biol. 14, 87–96 (2007).
Long, R.A., Qureshi, A., Faulkner, D.J. & Azam, F. 2-n-Pentyl-4-quinolinol produced by a marine Alteromonas sp. and its potential ecological and biogeochemical roles. Appl. Environ. Microb. 69, 568–576 (2003).
Diggle, S.P. et al. Functional genetic analysis reveals a 2-alkyl-4-quinolone signalling system in the human pathogen Burkholderia pseudomallei and related bacteria. Chem. Biol. 13, 701–710 (2006).
Lépine, F., Milot, S., Déziel, E., He, J. & Rahme, L.G. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa . J. Am. Soc. Mass Spectrom. 15, 862–869 (2004).
Déziel, E. et al. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc. Natl. Acad. Sci. USA 101, 1339–1344 (2004).
Pesci, E.C. et al. Quinolone signalling in the cell-to-cell communication system of Pseudomonas aeruginosa . Proc. Natl. Acad. Sci. USA 96, 11229–11234 (1999).
Xiao, G. et al. MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol. Microbiol. 62, 1689–1699 (2006).
Gallagher, L.A., McKnight, S.L., Kuznetsova, M.S., Pesci, E.C. & Manoil, C. Functions required for extracellular quinolone signalling by Pseudomonas aeruginosa . J. Bacteriol. 184, 6472–6480 (2002).
Wade, D.S. et al. Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa . J. Bacteriol. 187, 4372–4380 (2005).
Becher, A. & Schweizer, P. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 29, 948–952 (2000).
Lépine, F., Déziel, E., Milot, S. & Rahme, L.G. A stable isotope dilution assay for the quantification of the Pseudomonas qunolone signal in Pseudomonas aeruginosa cultures. Biochim. Biophys. Acta 1622, 36–41 (2003).
Acknowledgements
We thank Ram Chhabra for his helpful comments on the manuscript. Funding for this work in the authors laboratory was provided by a grant from the Biotechnology and Biological Sciences Research Council, UK, and we gratefully acknowledge the support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Fletcher, M., Diggle, S., Cámara, M. et al. Biosensor-based assays for PQS, HHQ and related 2-alkyl-4-quinolone quorum sensing signal molecules. Nat Protoc 2, 1254–1262 (2007). https://doi.org/10.1038/nprot.2007.158
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.158
This article is cited by
-
Miniaturized dispersive liquid-liquid microextraction and MALDI MS using ionic liquid matrices for the detection of bacterial communication molecules and virulence factors
Analytical and Bioanalytical Chemistry (2018)
-
Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis
Nature Communications (2017)
-
Molecular Signature of Pseudomonas aeruginosa with Simultaneous Nanomolar Detection of Quorum Sensing Signaling Molecules at a Boron-Doped Diamond Electrode
Scientific Reports (2016)
-
Synthesis and electrochemical detection of a thiazolyl-indole natural product isolated from the nosocomial pathogen Pseudomonas aeruginosa
Analytical and Bioanalytical Chemistry (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
