
Abstract
Transgenic mice have been instrumental in dissecting the role of various neuronal proteins under both physiological and pathological conditions. Pronuclear injection is the most widely used protocol for the generation of transgenic mice. Here, we describe all steps involved from DNA purification to the set up of a mouse colony including vasectomy, injection of the DNA into a donor zygote, transfer of injected zygotes into recipient foster mice, screening of offspring and establishment of transgenic mouse lines. We discuss the use of neuron-specific promoters to express proteins with a role in Alzheimer disease. Transgenic expression of a truncated form of the microtubule-associated protein tau (Δtau) is used as an example for the anticipated results.
Similar content being viewed by others



Introduction
The generation of transgenic mice is a relatively cost-intensive and time-consuming process. As a conservative estimate, it takes approximately a year from making the DNA construct for pronuclear injection to establishing a new transgenic mouse strain with some phenotypic characterization. Transgenic mouse production requires thorough planning to ensure sufficient space in an existing facility or to set up a new facility, to obtain the four groups of mice needed for transgenic mouse production and to purchase and install the equipment needed for pronuclear injection and other experimental animal manipulations and making the different types of capillaries.
In recent years, alternative methods have been developed to make transgenic mice including intracytoplasmic sperm injection, transfection of spermatozoa with exogenous DNA/DMSO complexes or retroviral-mediated approaches. These are intended to make transgenesis easier in terms of time, expertise and equipment needed; however, retroviral-mediated approaches, for example, tend to cause multiple chromosomal integration of the transgene1. Intracytoplasmic sperm injection may be chosen to introduce large genomic transgenes such as yeast artificial chromosomes where the efficiency of transgenesis by pronuclear approach is low2. For standard expression constructs, however, the classical pronuclear injection approach described here continues to be the major technique used since it was established in the early 1980s (see refs. 3, 4, 5, 6, 7, 8) as it is a straightforward method to consistently obtain mice with a single integration site. Targeted transgenesis either knocks out or modifies/replaces genes in situ. This approach involves a preselection using embryonic stem cells, as shown for example, for the catalytic subunit of protein phosphatase 2A, where a complete knockout caused an embryonically lethal phenotype9. The insertion of the green fluorescent protein into the endogenous mouse tau locus (a so-called knock-in approach) abolished tau expression, but at the same time, the neurons were fluorescently labeled with green fluorescent protein10. For very detailed information about all aspects of mouse manipulation including the use of embryonic stem cells to produce knockout mice, the reader is referred to the standard manual initially published by Nagy et al.11 Compared with the embryonic stem cell-mediated approach of gene targeting, by the approach described here, the transgene inserts randomly into the genome and expression is therefore subject to position effects. Furthermore, the injected DNA usually integrates as concatemers with a non-predictable copy number. All this explains the usually highly variable expression patterns and levels in different transgenic founders.
By appropriate choice of the promoter for transgenic expression, literally every cell type or organ can be targeted. In the past decade, we have concentrated on the generation of transgenic mice with expression in the nervous system12,13,14,15; however, we have also targeted other organs16,17,18. For transgenic expression in neurons, a host of expression vectors is available, but in practical terms only a limited number are used. We have been mainly using the mouse Thy1.2 (mThy1.2) expression vector for expression in neurons of brain and spinal cord14,15,19,20,21,22. The mThy1.2 promoter has also been successfully used to fluorescently label neurons and provide a Golgi-like vital stain23. In the field of Alzheimer disease research, the two major histopathological lesions, the Aβ plaques and the neurofibrillary tangles, have been reproduced by transgenic neuronal expression of the Aβ precursor protein APP and the microtubule-associated protein tau, respectively21,22. The following promoters have been used by us and others to express APP: platelet-derived growth factor (PDGF)24,25, hamster prion protein (PrP) cosmid26, mPrP27 and mThy1.2 (see refs. 28, 29, 30); and tau: human Thy1 (hThy1)/β-globin pA/splice cassette12, 3-hydroxy-3-methylglutaryl-coenzyme-A (mHMG CoA) reductase31, mPrP32,33, mPrP with autoregulatory transactivator loop34, mThy1.2 (see refs. 19,29,35,36,37), platelet-derived growth factor38, Ca2+/calmodulin-dependent kinase II (CaMKII) (see ref. 39), endogenous tau locus40 and eventually a bigenic, regulatable approach, CaMKII-transactivator (TA)/tet-off41.
A direct comparative analysis of the neuron-specific enolase, hThy1, rhombotin and neurofilament L promoter has been performed by one research group to express APP. It was found that the hThy1 promoter gave the highest APP mRNA levels throughout the brain, most notably in hippocampus and cerebral cortex42. We made the same observations using all four promoters for tau expression36, and found subsequently that the mThy1.2 expression vector is superior to the hThy1 vector12,19.
Although the production of transgenic mice is a technically demanding and time-consuming process, most strains are well worth the effort as exemplified by the widespread use of the hamster PrP cosmid-driven APP transgenic strain Tg2576 (ref. 26), with 250 PubMed entries at the end of 2006.
Stepwise training program
Many research groups are reluctant to generate transgenic mice by themselves. To get started, we recommend a stepwise training program.
-
The vasectomy of mice is a simple, isolated procedure. It can be performed sequentially, by operating on males on consecutive days. Mating with NMRI foster mice reveals a successful operation (a) by the presence of a copulation plug and (b) the failure of generating offspring.
-
Isolation and purification of zygotes. A few matings are sufficient to just obtain enough zygotes to go through all the steps of taking out the oviduct, flushing of the zygotes, collecting them and purifying them from follicular cells, with the help of hyaluronidase. This test run also helps to optimize the timing of the hormone treatment and to get familiar with the optics of the stereoscopic and microinjection microscope. Culture zygotes overnight. Most zygotes should reach the two-cell stage, if not, the culture conditions (such as the medium) need to be adjusted.
-
Training of some steps of the reimplantation procedure, such as finding the infundibulum using euthanized mice.
-
Reimplantation of uninjected zygotes. It needs some training to perform the reimplantation, to find the infundibulum and position it such that the zygotes can be easily reimplanted. We suggest a few test runs with reimplanting euthanized mice with medium. The reimplantation should be performed with a reimplantation capillary that has been previously tested with zygotes for an optimal diameter and capillary opening. We suggest producing several reimplantation capillaries for later use. After the test runs with sacrificed mice, zygotes should be reimplanted into narcotized mice, as it is important to master the reimplantation procedure in the living mouse, without hurting blood vessels, and to provide a proof of a successful reimplantation by generating offspring.
-
Finally, the researcher should try to isolate zygotes, inject them and reimplant them all on the same day. It takes some time to do the injections without lysing the zygotes. If too many of them lyse, we recommend that an attempt is made to obtain around 20 injected zygotes. These should be reimplanted and if there are enough plugged fosters available, it is a good idea to reimplant the leftovers of uninjected zygotes, to perfect the procedure.
Materials
Reagents
-
Mice. The absolute minimal numbers of mice that are needed to generate transgenic mice are listed below. For routine generation of transgenic animals, the numbers need to be doubled (see REAGENT SETUP)
Caution
Experiments involving mice must conform to National and Institutional regulations.
-
Microinjection buffer (MI buffer): 8 mM Tris-HCl (T5941; Sigma), 0.15 mM EDTA (E5134; Sigma)
-
DNA grade agarose (200.0011; Probiogen Biochemicals)
-
Tris-borate-EDTA: 89 mM Tris-borate and 2 mM EDTA, pH 8.3. (T4415; Sigma)
-
2% (w/v) crystal violet solution (C3886; Sigma)
-
Jetsorb Gel Extraction Kit (110150; Genomed)
-
M2 medium (M7167; Sigma)
-
M16 medium (M7292; Sigma)
-
Mineral oil (M8410; Sigma)
-
Penicillin/streptomycin (100×) (15140-122; GIBCO)
-
Gentamycin (1,000×) (15750-060; GIBCO)
-
Iodine-based skin disinfectant (e.g. Braunol; Braun/R)
-
70% (v/v) ethanol (E7023; Sigma)
-
Phosphate-buffered saline (PBS) pH 7.4: 1.06 mM potassium phosphate monobasic, 2.97 mM sodium phosphate dibasic, 155.17 mM sodium chloride (10010-023; GIBCO)
-
100 mg ml−1 ketamine (Ketamil injection; Ilium)
-
20 mg ml−1 xylazine (Ilium Xylazil-20; Ilium)
-
50 U ml−1 pregnant mare's serum gonadotropin
-
50 U ml−1 hCG (human chorionic gonadotropin)
-
RedTaq with buffer (D5684; Sigma)
-
10 mM dNTP mix (954143031; Eppendorf)
-
Gene-specific forward and reverse primer
Critical
To eliminate amplification of the endogenous gene locus, the forward primer should be positioned in the promoter sequence, whereas the reverse primer should be gene-specific. Establish PCR protocol in advance by mixing the transgenic vector in genomic DNA extracted by the method intended to be used later for genotyping of the mice.
-
10 mg ml−1 hyaluronidase (H3884; Sigma)
Equipment
-
DNA electrophoresis setup
Critical
A small unit only used for preparation of DNA for pronuclear injection is recommended to avoid contamination.
-
Bright-field transillumination table
-
0.2 or 0.45 μm Ultrafree-MC spin column (UFC30HV00; Millipore)
-
1.5/2 ml tube centrifuge (5417R; Eppendorf)
-
Photometer to measure DNA concentration at 260/280 nm
-
Sterile filter Millex GP (SLGP033RB; Millipore)
-
Microcapillaries intraMARK green 50 μl (708733; Brand)
-
Diamond cutter
-
Bunsen burner
-
Mouthpiece, tubing and microcapillary holder
-
35, 60 and 100 mm cell culture dishes (93040, 93060, 93100; TPP)
-
Dissection tools: 2× Dumont #5 forceps (91150-20), surgical forceps (91106-12), iris forceps (11064-07), iris scissors (91460-11), vessel clamp (18374-43), wound clips (12040-01) and applier (12018-12; Fine Scientific Tools)
-
Warming pad or, alternatively, cool packs warmed in a 37 °C water bath
-
Electrical hair cutter
-
Red lamp for warming narcotized animals (alternatively, warming pad)
-
Fusel-free tissue paper (Kimwipe)
-
Superfrost microscope slide (Menzel)
Critical
Clean slide before use with acetone/ethanol to remove remaining oil from previous injections.
-
Stereoscopic microscope with top and bottom illumination (SZX7; Olympus)
-
Inverted microinjection microscope with DIC optic (IX71; Olympus)
-
Manual microinjector for holding oocytes (Cell Tram Oil; 5176000.025; Eppendorf)
-
2× Micromanipulator (TransferMan NK2; 5188000.012; Eppendorf)
-
Microinjector (FemtoJet; 5247000.013; Eppendorf)
-
Microcapillary with 15 μm inner diameter, 100 μm outer diameter and 35° tip angle for holding oocytes (VacuTip; 5175108.000; Eppendorf)
-
Microinjection capillaries (Femtotip; 5242952.008; Eppendorf)
-
Microloader (5242956.003; Eppendorf)
-
37 °C incubator (Haemocult)
-
PCR machine
Reagent setup
-
Vasectomized male mice Keep 10–12 vasectomized male albino mice (e.g., NMRI strain) individually in type II or II long (II L) cages. Vasectomized mice can either be bought directly from animal suppliers or the simple procedure can be performed on 2–3-month-old males (see below). Vasectomized males can be kept for up to a year, successively replacing performers that do not plug (see below).
Critical
Other outbred albino strains such as CD1 may be considered as sterile stud males depending on the local animal supplier's strain availability. The advantage of using an albino strain is that offspring accidentally derived from an NMRI × NMRI mating, owing to an incomplete vasectomy, can easily be discriminated from offspring derived from reimplanted and injected zygotes, owing to the white color.
-
Foster mother mice Forty female albino mice (e.g., NMRI) are needed to serve as pseudopregnant foster mothers. Buy the females at 6–8 weeks of age and keep in type III cages in groups of 6–8 mice. They need to be replaced regularly as they are used up, but also when they become too fat or too old.
Critical
Other outbred albino strains such as CD1 may be considered as foster mothers depending on the local animal supplier's strain availability. In our hands, the NMRI strain has shown to be very robust during surgical procedures, the infundibulum is easily accessible for zygote transfer and the foster mothers can feed huge litters. The advantage of using an albino strain is that offspring accidentally derived from an NMRI × NMRI mating, owing to an incomplete vasectomy, can easily be discriminated from offspring derived from reimplanted and injected zygotes, owing to the white color.
-
Male parental mice A total of 10–12 male B6D2F1 (F1 of strains C57Bl/6 × DBA2) mice are needed as mating partners for the production of zygotes. They should be kept individually in type II cages. Buy at 6–8 weeks of age and use for up to a year.
-
Zygote donor mice A total of 40–50 female B6D2F1 mice are needed to produce zygotes for DNA injection. These mice should be bought in batches suitable for 4–5 injection dates, that is, a total of 40–50 mice, which are kept in 5–7 type III cages. They can be used at 6–10 weeks of age and should be constantly replenished.
Critical
The optimal age of the zygote donors needs to be empirically determined by the experimenter as it depends on the strain used and the time scheme for superovulation, zygote collection and pronuclear injection (see below). Inbred stains such as C57Bl/6 may be used to generate transgenic mice on a pure background; however, it needs more injections to obtain a comparable number of transgenic lines, owing to a lower yield and a higher fragility of C57Bl/6 compared to B6D2F1 zygotes and a lower frequency of transgene integration. On the other hand, if a transgene is expressed on a mixed B6D2F1 background, ten rounds of backcrossing onto C57Bl/6 are needed to obtain a pure strain. In practical terms, if the experimenter is less experienced or the transgenic strain has to be produced very rapidly, an F1 strain such as B6D2F1 should be the strain of choice.
-
Mouse housing Depending on the local facilities, the mice may be either kept in an environment free of pathogens, a so-called SPF (specified pathogen-free) facility, in individually ventilated cage systems or in a conventional facility, possibly under optimized holding conditions. The latter is the cheapest option (with respect to equipment and labor) and also the least time consuming, but when transgenic strains are to be exchanged with other laboratories, in most cases SPF mice are requested. One option is to establish the transgenic strain first in a conventional facility followed by an embryo transfer into an SPF foster (see below).
Critical
For transgenic mouse production, sufficient space is needed in the animal facility. In addition to fosters, vasectomized males, oocyte donors and their mating partners, space is needed for subsequent strain selection. Once transgenic founders are established among the offspring of reimplanted founders, these are used to establish transgenic lines. We routinely generate and breed ten or more founder animals (F0 generation) per transgenic construct. Sufficient numbers need to be bred for screening of expression level, expression pattern and pathology, before all but two or three lines are terminated. This screening needs space that is comparable to what is eventually needed for subsequent experiments with the selected strains. For pronuclear injection experiments, we recommend a lighting cycle of 6 pm to 6 am dark.
-
M2+ medium Supplement M2 medium with penicillin/streptomycin or gentamycin and sterile filter.
Critical
Gentamycin is not stable at final concentration. Supplement the M2 medium with 20 mM HEPES pH 7.4, to buffer pH changes outside the CO2 incubator and thereby prevent shrinkage and a reduced viability of the zygotes.
-
Anesthesia Dilute ketamine and xylazine in phosphate-buffered saline to a final concentration of 12.5 and 2.5 mg ml−1, respectively, sterile filter and freeze in 1 ml aliquots. Anesthetize mice at 0.006 ml g−1 body weight. Other anesthesia protocols may be used instead, depending on the local animal ethics regulations.
-
Transfer and reimplantation capillaries Hand pull intraMARK green 50 μl microcapillaries with a Bunsen burner. Then break with a diamond cutter at a suitable length to obtain an internal diameter of 200–300 μm. Polish the capillary by rapidly passing through flame.
Procedure
Vasectomy
Timing 2–3 h
-
1
Anesthetize 10–12 males by intraperitoneal (i.p.) injection with ketamine/xylazine and place on their backs to expose the abdomen when deeply narcotized.
Critical Step
Vasectomies have to be performed at least 10–14 days before the first mating, to allow the recovery of males from the surgical procedure. It is advisable to determine sterility of the stud mice by breeding them with females. Males can be reused for subsequent injections.
-
2
Apply moderate pressure to the abdomen to push the testes into the scrotal sac (Fig. 1a). Make a 10 mm midline skin incision. The transparent scrotal sac should appear on the left and right of the midline.
Figure 1: Vasectomy. 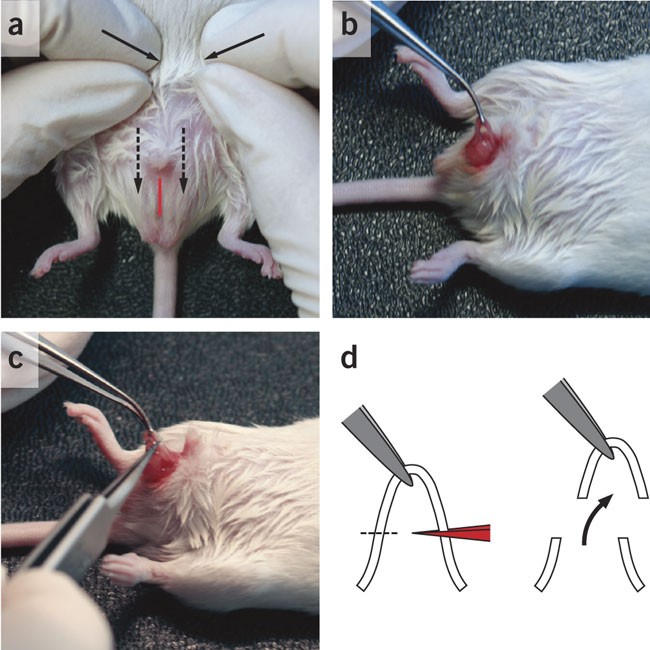
(a) An anesthetized male NMRI mouse placed on its back to expose the abdomen. Moderate pressure is applied to the abdomen (arrows) to push the testes into the scrotal sac (broken arrows). A 10 mm midline incision is made in the scrotal sac (red line). (b) The vas deferens is pulled out with a forceps and held with a forceps such that a loop is formed. (c) A fine forceps is heated until the tip is glowing red, and the loop of the vas deferens (∼ 0.5 cm) removed by cauterizing the ends. (d) Scheme illustrates positioning of the forceps, formation of loop and removal of vas deferens.
-
3
Make a 5 mm incision on the left (right) of the midline and push the testis gently to the left (right) until the vas deferens becomes visible as a white tubule together with a single vessel.
-
4
Pull the vas deferens out with a forceps (Fig. 1b). Hold the vas deferens with the forceps such that a loop is formed. Heat a fine forceps until the tip is glowing red, and remove the loop of the vas deferens (∼0.5 cm) by cauterizing the ends (Fig. 1c, d).
-
5
Perform the same procedure for the right vas deferens and sew or clip the skin.

Preparation of DNA
Timing 6–8 h
-
6
For zygote injection, the DNA construct needs to be linearized and the remaining vector sequences removed. Digest 10 μg of purified vector containing the gene of interest in a volume of 50 μl with 50 U restriction enzyme(s) to complete digestion within less than 2 h. Sequential digestion may be needed when the cutting temperatures of enzymes used differ.
Critical Step
Run 2 μl of the digest on an agarose gel to test complete digestion. Increase digestion time if needed.
-
7
Separate the whole digest on a preparative agarose gel. Instead of ethidium bromide, which is toxic even at low concentrations, add crystal violet to the gel buffer (1:1,000 dilution of 2% stock). Supplement the loading buffer and electrophoresis buffer with crystal violet, if desired, to assist in visualization of the DNA band on a bright-field transillumination table. Cut the band containing the linearized gene construct from the gel and purify the DNA using a silica bead-based kit such as the Jetsorb Gel Extraction Kit (Genomed) according to the manufacturer's protocol. Gel absorption kits are not recommended.
Critical Step
Elute the DNA with MI buffer, especially when the DNA concentration is expected to be low and the DNA cannot be much further diluted for the injection.
-
8
Ultrafiltrate the eluted DNA through a 0.45 μm Ultrafree-MC spin column. For DNA constructs below 10 kb, dilute the DNA to 3 ng μl−1 in MI buffer and pass through a 0.2 μm Ultrafree-MC spin column. For larger DNA constructs, dilute the DNA to 3 ng μl−1 in MI buffer that has been filtered through a 0.2 μm Ultrafree-MC spin column.
Pause point
The purified digest can be stored at −20 °C until use.
-
9
Thaw purified DNA immediately before injection (Step 27), centrifuge the diluted DNA at 17,000g for 5 min at 4 °C and keep on ice until use. When loading the injection capillary, take the DNA from the upper phase.
Mouse schedule
Timing 4 days
-
10
To produce zygotes for pronuclear injection, superovulate 8–10 female B6D2F1 mice and mate with B6D2F1 males. For this, inject females i.p. with 5 IU pregnant mare's serum gonadotropin (PMSG; in 100 μl) in the afternoon (∼4 pm) of day 1 (Fig. 2a). After 46–48 h, that is, in the afternoon of day 3 (∼2 pm), inject the females i.p. with 5 IU hCG to induce ovulation 10–12 h later. Following the hCG injection, put the females together with B6D2F1 males in a 1:1 ratio in single cages overnight.
Figure 2: Mating scheme and copulation plug. 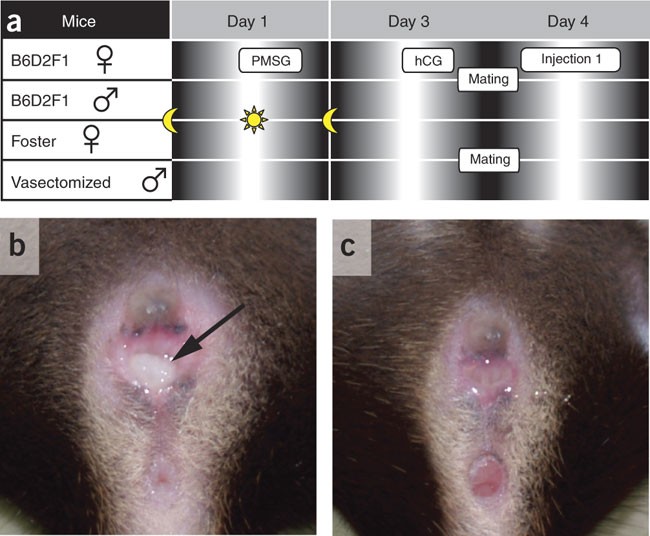
(a) Timeline of hormone treatments and matings. (b,c) Plugged (arrow) compared to unplugged female B6D2F1 mouse.
Critical Step
This timing is critical and needs to be empirically optimized, to obtain sufficient numbers of oocytes with clearly visible pronuclei on the next day.
-
11
To obtain pseudopregnant females, select NMRI females in estrus (as evidenced by a swollen, moist and pink vagina) and put two females together with one vasectomized NMRI male each overnight. We recommend a minimum of ten breedings (Fig. 2a).
Critical Step
Transferring some mulch from the female into the male home cage an hour before mating can increase the performance of the males.
-
12
On the morning of day 4, collect B6D2F1 females with vaginal plugs (Fig. 2b) for zygote preparation and eliminate the females without plugs (Fig. 2c). Similarly, collect the NMRI females with plugs, but leave them in the animal facility until needed later in the afternoon for reimplantation.

Zygote preparation
Timing 1–2 h
-
13
Prepare the transfer setup as shown in Figure 3a. Place six drops of 10 μl M2+ medium in three 35 mm dishes (Fig. 3b). Then overlay the whole dish with mineral oil by carefully flooding the dish from the center. For safety handling, place the 35 mm dishes on the lids of 100 mm dishes.
Figure 3: Transfer capillary setup and preparation of zygotes in M2+ medium. 
(a) Scheme illustrating pipetting device for handling and reimplanting zygotes. (b) Six drops of 10 μl M2+ medium placed in 35 mm dish, indicating drop 1. Drops are overlaid with mineral oil by carefully flooding the dish from the center.
Critical Step
Keeping a bigger gap between the first and last drop makes future orientation easier.
-
14
Fill two additional 60 mm dishes with medium and place all dishes into a 37 °C incubator.
-
15
Kill plugged B6D2F1 females by cervical dislocation.
-
16
Place mice in groups of three to four on their back on absorbent paper and rinse thoroughly with 70% ethanol. Pinch skin at the midline and make a small incision. Hold the skin firmly above and below the incision and pull toward the head and tail to expose the abdomen (Fig. 4a). Incise the peritoneum and expose the inner organs.
Figure 4: Harvesting of zygotes. 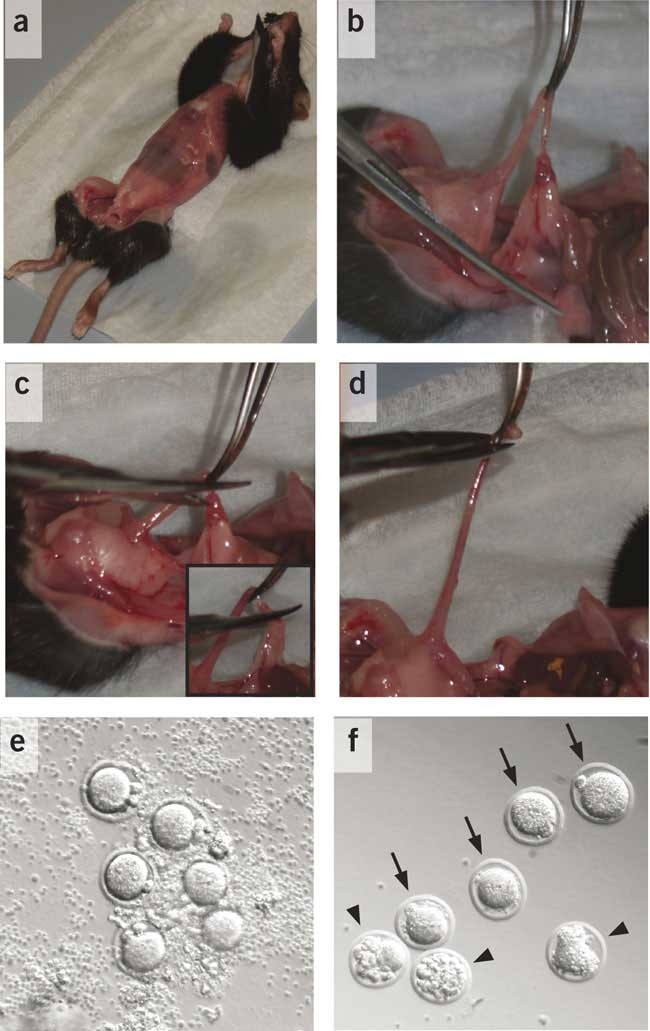
(a) Cervically dislocated mouse is skinned after pulling incised skin toward head and tail to expose the abdomen. (b) Uterus separated from mesometrium and fat tissue using closed scissors. (c) Cut made first between oviduct and ovary (see inset) and then (d) through the uterus near the oviduct. (e) Zygotes harvested with follicular cumulus cells, and (f) after successive treatments with hyaluronidase (normal zygotes, arrows; degenerated zygotes, arrowheads).
-
17
Push up the gut and grab one of the uterine horns with an iris forceps. With the second pair of forceps, separate the uterus from the mesometrium and fat tissue (Fig. 4b). Make a cut first between the oviduct and ovary (Fig. 4c) and then through the uterus near the oviduct (Fig. 4d). Transfer the oviduct (with some adjacent uterine and ovarian tissue) onto a 60 mm dish with medium.
-
18
Under the stereoscopic microscope, transfer one oviduct at a time into the second 60 mm dish with medium, tearing the oviduct where it is most swollen (site where zygotes mainly are) using a pair of Dumont #5 forceps. Squeeze zygotes gently out of the oviduct if they do not flow out by themselves.
-
19
The zygotes are in a cluster of follicular cumulus cells (Fig. 4e), which need to be removed as they are sticky and would clog the injection capillary. Clean zygotes from follicular cells by transferring all zygotes along with the adhering cells into the first of the six drops of 10 μl M2+ medium in the first 35 mm dish (Fig. 3b).
-
20
Add 1–2 μl of the hyaluronidase stock solution to the cells in the droplet. In a slow circular motion, stir the zygotes in the drop and successively transfer them clockwise through the drops, until in the final drop, the zygotes are collected and all cumulus cells are left behind (Fig. 4f).
-
21
Finally, transfer all purified zygotes to the first drop of medium in the second 35 mm dish which is placed on a lid of a 100 mm dish for easy handling, and place the dish in the 37 °C incubator.


Injecting zygotes
Timing 1–3 h
-
22
Place an 18 μl drop of M2+ medium in the center of a clean microscope slide. Using an insulin syringe, place two small drops of mineral oil on the left and right of the medium drop until they float together to form a thin film of oil covering the medium drop, thereby preventing evaporation and thus shrinkage of the oocytes and shedding of the zona pellucida (Fig. 5).
Figure 5 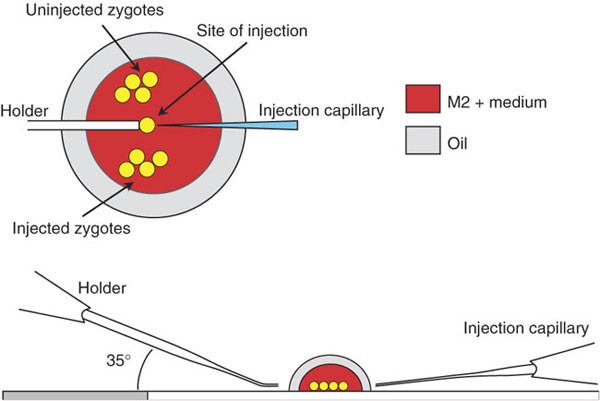
Arrangements of zygotes, holder and injection capillary on injection stage.
-
23
Place the slide under a stereoscopic microscope and transfer 20–50 zygotes, depending on experience, into the upper part of the drop (Fig. 5).
Critical Step
A careful separation of the uninjected zygotes in the upper part of the drop and subsequent placing of injected zygotes prevents a mix-up of injected and uninjected zygotes.
-
24
Place the slide onto the stage of the microinjection microscope.
-
25
Place the microcapillary for holding oocytes into the connector piece of the oil-filled manual microinjector. Fill the capillary to remove all air bubbles until a drop of oil appears at the tip. Remove the drop of oil carefully with tissue paper.
-
26
Fill the capillary up to two-thirds with M2+ medium by suction. Remove excess medium with tissue paper. Adjust the mounting angle of the holder to 35°, such that the tip runs parallel to the slide (Fig. 5). Shift the motor inwards and position the tip beside the medium drop. Set a low point to prevent breakage of the holding capillary.
-
27
Fill the injection capillary using a microloader with 0.5–1 μl of plasmid DNA. Insert the injection capillary into the connector piece and lower the capillary into the medium drop on the stage such that it is positioned in the center just below the zygotes. Again set a low point to prevent breakage of the capillary.
-
28
Clean the injection capillary following the recommendations of the manufacturer of the microinjection device. To inject a zygote, move the holder to the collected zygotes, apply a fine suction, move one zygote to the center and position the zygote in the same plane of focus as the opening of the holder, ideally, such that the polar body and the female and male pronuclei are aligned as shown in Figure 6a. Then move the injection capillary below the zygote (6 o'clock position) and bring the tip of the capillary in focus with the male pronucleus. To inject, bring the capillary in a parallel position, along the horizontal line of the aligned pronuclei (Fig. 5). Once the male pronucleus is penetrated (which may be accompanied by some indentation of the zona pellucida), inject DNA and then withdraw the capillary. A successful injection is evident by a swelling of the male pronucleus (Fig. 6b).
Figure 6: Pronuclear injection. 
(a) The holding capillary stabilizes one zygote in the center. The position of the zygote is in the same plane of focus as the opening of the holder, ideally facing it with the polar body and the female and male pronuclei aligned horizontally. (b) When the male pronucleus is penetrated with the injection capillary and the DNA is injected, the male pronucleus swells (inset). hc, holding capillary; pb, polar body; mp, male pronucleus; fp, female pronucleus; zp, zona pellucida.
Critical Step
The female pronucleus can also be injected, but it is more difficult to penetrate as it is smaller than the male pronucleus. The experimenter needs to empirically determine the best time of the day to do the microinjections as the visibility of the pronuclei changes with advanced maturation of the zygotes. When the zygote is damaged because nuclear DNA is sticking to the injection capillary, the zygote can slowly lyse and this may not be immediately evident. The injection capillaries can be cleaned by moving them along the shaft of the holding capillary. If zygotes stick to the injection capillary, they can be removed with the help of the holding capillary or by moving the injection capillary out of the medium drop through the oil and back again. When the injection capillary cannot be properly cleaned, it should be replaced by a freshly loaded capillary.


Reimplantation of injected zygotes
Timing 15–30 min/foster
-
29
Implant about 15–30 injected zygotes into the oviduct of one pseudopregnant NMRI foster mother. A bilateral reimplantation is optional. First load the injected zygotes into the transfer capillary as shown in Figure 7a, arranged like a chain of pearls, including air bubbles to visualize a successful reimplantation.
Figure 7: Reimplantation of injected zygotes. 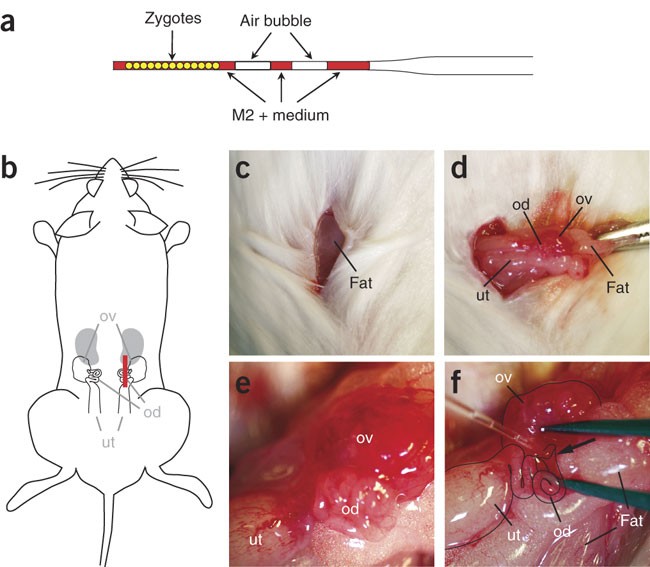
(a) The transfer capillary is loaded with injected zygotes that are arranged like a chain of pearls. The two air bubbles assist in subsequently visualizing a successful reimplantation. (b) The anesthetized plugged NMRI female is placed on a dry tissue and cleaned with 70% ethanol. (b,c) A 1-cm-long skin incision (red line) is made parallel to the dorsal midline. The fat pad attached to the ovary is visible through the transparent musculature. (d) The fat pad is grabbed with a forceps and the ovary gently pulled out together with the attached oviduct and uterus. The fat pad is fixed with a vessel clamp. (e) Coiled oviduct within the transparent bursa. (f) Transfer capillary positioned in the infundibulum (arrow). ut, uterus; od, oviduct; ov, ovary.
Critical Step
The reimplantation capillary should be thick enough to allow loading of the zygotes without damage and thin enough to be able to enter the infundibulum.
Critical Step
As an alternative to reimplanting on the injection day, the injected zygotes may be cultured overnight in M16 medium (Sigma) and reimplanted at the two-cell stage. This is the case when no pseudopregnant females are available on the day of injection.
-
30
Anesthetize a plugged NMRI female and place the narcotized mouse on a dry tissue as shown in Figure 7b. Shave a broad area of the side of the mouse, clean the area properly with 70% ethanol and transfer the mouse onto a new dry tissue. Following additional cleaning with Braunol (optional), make a 1-cm-long skin incision parallel to the dorsal midline (Fig. 7b, c). The fat pad attached to the ovary should become visible through the transparent musculature (Fig. 7c). Cut the musculature covering the ovary with a scissor and stretch the incision with a forceps to stop possible bleeding.
Critical Step
Avoid cutting blood vessels as bleeding may eventually hinder the visibility of the infundibulum and may clog the injection capillary such that the zygotes cannot be deposited in the infundibulum.
-
31
Grab the fat pad with a forceps and gently pull the ovary out until its attached oviduct and uterus are clearly visible (Fig. 7d). Fix the fat pad with a vessel clamp and position the ovary tract over the spine, and correct the positioning of this under a stereoscopic microscope if needed. The coiled oviduct within the transparent bursa should be clearly visible by now (Fig. 7e). Use two Dumont #5 forceps to tear the bursa covering the gap between ovary and the oviduct, carefully separating them to expose the infundibulum.
Critical Step
The infundibulum may be hidden in the cleft between the coiled oviduct and the ovary. When the clamp is used to position uterus, ovary and oviduct, make sure that the oviduct does not stick out in a vertical orientation, which makes reimplantation difficult but rather that it is oriented horizontally such that the reimplantation capillary can be easily inserted into the infundibulum (without a major change of plane of focus). Remove any liquid or traces of blood with tissue paper.
-
32
Pick up the reimplantation capillary with the loaded zygotes and carefully widen the cleft that buries the infundibulum with a fine forceps without causing any bleeding and insert the transfer capillary into the infundibulum (Fig. 7f). Slowly blow the zygotes into the infundibulum together until the air bubbles become visible in the ampulla, indicating a successful transfer.
Critical Step
It is important not to blow too hard as this might result in the zygotes spilling into the abdominal cavity.
-
33
Release the clamp fixing the fat pad and use blunt tools to push the ovary back into the abdomen. Sew up first the musculature and then the skin, or alternatively use wound clips for the skin.
Critical Step
Avoid coiling of the uterus when pushing it back into the body.
-
34
Keep the mice warm using a red lamp until narcosis ends and use moistening cream to prevent the eyes from drying out. Place the reimplanted mice into the home cage and transfer them back into the mouse facility.
Critical Step
It is helpful to keep up to three reimplanted fosters in one cage in case one female only delivers a few babies and/or has not enough milk.

Genotyping
-
35
Extract genomic DNA from tail biopsies of the 3–week–old mice by hot digestion as described43. Briefly, incubate short tail biopsies (∼2 mm) in 100 μl alkaline lysis buffer (25 mM NaOH, 0.2 mM disodium EDTA, pH 12.0) for 1 h at 95 °C in a thermal block (Eppendorf) with constant shaking at 1,000 r.p.m.
-
36
Subsequently, cool sample on ice and add 100 μl of neutralizing buffer (40 mM Tris-HCl, pH 5.0). Spin the samples in a bench-top centrifuge to recover evaporated liquid. The supernatant contains the DNA, and 1 μl should be used for genotyping.
Critical Step
Although the hot-shot method works for most PCR genotypings, further purification by ethanol precipitation or with columns may be needed.
Troubleshooting
Even when all the steps have been perfected, several problems can be encountered when examining the reimplanted fosters at the time of delivery or when screening offspring for transgene integration and expression:
-
No pups are born: this could be an indication that either the reimplantation procedure still needs to be perfected (in which case we recommend a few practice runs with uninjected zygotes) or that the zygotes did not survive the injection procedure. This can be easily monitored by cultivating injected zygotes overnight and determining how many are in the two-cell stage and how many are lysed. In this case, the injection procedure needs to be optimized. This is a matter of practice but one option is also to use a horizontal puller to make custom-made injection capillaries with optimized size, diameter and shape.
-
Only a few pups are born: possible reasons are that either not all zygotes have been transferred (i.e., some have been spilled into the peritoneum), that the injection procedure is not optimal or that the DNA is toxic (owing to imperfect purification or a concentration that is too high).
-
Many pups are born but only a few or even none are transgenic: This indicates that the reimplantation was successful but not the injection. Either the DNA concentration has been too low (in which case we would suggest using a twofold higher concentration) or, during the actual injection, the DNA has been injected into the cytoplasm and not the pronucleus or not enough DNA has been injected. A swelling of the pronucleus needs to be evident during the injection process, if not, the injection time and/or the injection pressure need to be increased.
-
Many transgenic pups are born but they do not express the transgene: generally, expression is monitored at the protein level. In our experience with the mThy1.2 expression vector, one in every three to four lines expresses the transgene-encoded protein at moderate to high levels, irrespective of the gene expressed. However, in general, it is not unusual that only 10% or less of the founders generate offspring that expresses the transgene. Therefore, we recommend obtaining at least ten founders for each expression construct. Some combinations of cDNA and expression vector, in our experience, lead to low-expression frequencies. In this case, we suggest to increase the numbers of injections or to use a different expression vector. We also found that some lines are difficult to reproduce even when the same plasmid preparation is reinjected. Lack of protein expression in the transgenic offspring could be due to a cloning error. An easy way to rule out this is to run a northern blot. If mRNA, but no protein, is detectable, this could be an indication of an error such as a frameshift mutation. If no mRNA is detectable, the DNA concentration used for microinjection might have been too low, leading to the integration of only a few copies, and subsequently, either no or a very low transgene expression.
-
Variability of transgene expression in transgenic strain: if transgene-positive offspring shows a high variability in expression, this can be due to genetic background (if the transgenic strain has been established on a mixed background), or multiple integration of the transgene and improper segregation. This can be easily determined by Southern blot analysis. We recommend to establish transgenic lines always starting with one F1 animal and not the F0 founder, thus resisting the urge to establish a line slightly faster by interbreeding several F1 animals.
Timing
Cloning: 14 days to >2 months (depending on the complexity of construct)
Vasectomy: 2–3 h for 10–20 males (plus 10–14 days recovery)
Preparation of DNA: 6–8 h
Mouse schedule: 4 days
Zygote preparation: 1–2 h
Injecting zygotes: 1–3 h
Reimplantation: 15–30 min per foster
F0 generation: 2 months (20 days gestation, PCR analysis at 3 weeks of age)
F1 and further generations: 6–12 months
Anticipated results
Tau is a microtubule-associated protein and it contains two major domains, a projection and a microtubule-binding domain44. To dissect the role of these domains, we expressed the projection domain of tau under the control of the mThy1.2 promoter (Δtau) (Fig. 8a). For this, the sequence encoding amino acids 256–441 of the longest human tau isoform, htau40, was removed from the full-length cDNA. After linearization with NotI and PvuI and removal of the vector sequences, the DNA was injected into fertilized B6D2F1 oocytes. Thirty-nine pups (20 female, 19 male) were born 20 days after the reimplantation and genotyped upon weaning at postnatal day 21 using the primers 5′-aagtcacccagcagggaggtgctcag-3′ and 5′-gggtgtctccaatgcctgcttcttcag-3′ (Fig. 8b). Of these, six females and four males (F0 generation) were typed transgene-positive and mated individually with C57Bl/6 mice at 6 weeks of age. Seven of the ten breeding pairs gave offspring (F1 generation). Transgenic animals from the F1 generation were again mated with C57Bl/6 mice to establish seven individual Δtau transgenic strains. Breeding of two strains was terminated after >20 offspring were all genotyped as wild type. One strain transmitted the transgene to offspring, but the litter sizes were small (<4 per litter) and the frequency of birth was low (once in 3 months). Of the remaining strains, expression of transgenic tau protein was analyzed by western blotting with a human tau-specific antibody (HT7) of brain extracts, revealing different expression levels (Fig. 8c). In the hippocampus, expression of human tau was limited to cells of the neuronal layers CA1 and CA3 (Fig. 8d). Thus, we established two low- and two high-expressing Δtau strains.

(a) DNA construct, (b) PCR genotyping of F0 generation (+, transgene-positive; −, transgene-negative; co, plasmid control), (c) Western blotting for tau of brain extracts, (d) immunohistochemical staining of tau (red) revealing expression in hippocampal CA1 and CA3 region (inset: CA1 at higher magnification). Nuclear staining with DAPI (blue).
References
Bryda, E.C., Pearson, M., Agca, Y. & Bauer, B.A. Method for detection and identification of multiple chromosomal integration sites in transgenic animals created with lentivirus. Biotechniques 41, 715–719 (2006).
Moreira, P.N., Pozueta, J., Giraldo, P., Gutierrez-Adan, A. & Montoliu, L. Generation of yeast artificial chromosome transgenic mice by intracytoplasmic sperm injection. Methods Mol. Biol. 349, 151–161 (2006).
Brinster, R.L., Chen, H.Y., Trumbauer, M.E. & Avarbock, M.R. Translation of globin messenger RNA by the mouse ovum. Nature 283, 499–501 (1980).
Gordon, J.W. & Ruddle, F.H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science 214, 1244–1246 (1981).
Brinster, R.L. et al. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell 27, 223–231 (1981).
Costantini, F. & Lacy, E. Introduction of a rabbit beta-globin gene into the mouse germ line. Nature 294, 92–94 (1981).
Wagner, E.F., Stewart, T.A. & Mintz, B. The human beta-globin gene and a functional viral thymidine kinase gene in developing mice. Proc. Natl. Acad. Sci. USA 78, 5016–5020 (1981).
Wagner, T.E. et al. Microinjection of a rabbit beta-globin gene into zygotes and its subsequent expression in adult mice and their offspring. Proc. Natl. Acad. Sci. USA 78, 6376–6380 (1981).
Gotz, J., Probst, A., Ehler, E., Hemmings, B. & Kues, W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Calpha. Proc. Natl. Acad. Sci. USA 95, 12370–12375 (1998).
Tucker, K.L., Meyer, M. & Barde, Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 4, 29–37 (2001).
Nagy, A., Gertsenstein, M., Vinterstein, K. & Behringer, R. Manipulating the Mouse Embryo—A Laboratory Manual. 3rd edn. (Cold Spring Harbor Press, Cold Spring Harbor, New York, 2003).
Gotz, J. et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 14, 1304–1313 (1995).
Rauer, M., Gotz, J., Schuppli, D., Staeheli, P. & Hausmann, J. Transgenic mice expressing the nucleoprotein of Borna disease virus in either neurons or astrocytes: decreased susceptibility to homotypic infection and disease. J. Virol. 78, 3621–3632 (2004).
Kins, S. et al. Reduced PP2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J. Biol. Chem. 276, 38193–38200 (2001).
Schild, A., Ittner, L.M. & Gotz, J. Altered phosphorylation of cytoskeletal proteins in mutant protein phosphatase 2A transgenic mice. Biochem. Biophys. Res. Commun. 343, 1171–1178 (2006).
Tam, C.W. et al. Enhanced vascular responses to adrenomedullin in mice overexpressing receptor-activity-modifying protein 2. Circ. Res. 98, 262–270 (2006).
Gotz, J., Eibel, H. & Kohler, G. Non-tolerance and differential susceptibility to diabetes in transgenic mice expressing major histocompatibility class II genes on pancreatic beta cells. Eur. J. Immunol. 20, 1677–1683 (1990).
Schild, A. et al. Impaired development of the Harderian gland in mutant protein phosphatase 2A transgenic mice. Mech. Dev. 123, 362–371 (2006).
Probst, A. et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. (Berl.) 99, 469–481 (2000).
Gotz, J. & Nitsch, R.M. Compartmentalized tau hyperphosphorylation and increased levels of kinases in transgenic mice. Neuroreport 12, 2007–2016 (2001).
Gotz, J. Tau and transgenic animal models. Brain Res. Brain Res. Rev. 35, 266–286 (2001).
Gotz, J. et al. Transgenic animal models of Alzheimer's disease and related disorders: histopathology, behavior and therapy. Mol. Psychiatry 9, 664–683 (2004).
Feng, G. et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51 (2000).
Games, D. et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373, 523–527 (1995).
Mucke, L. et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058 (2000).
Hsiao, K. et al. Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 (1996).
Borchelt, D.R. et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19, 939–945 (1997).
Sturchler-Pierrat, C. et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 94, 13287–13292 (1997).
Oddo, S. et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles. Intracellular abeta and synaptic dysfunction. Neuron 39, 409–421 (2003).
Richards, J.G. et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 23, 8989–9003 (2003).
Brion, J.P., Tremp, G. & Octave, J.N. Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer's disease. Am. J. Pathol. 154, 255–270 (1999).
Ishihara, T. et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24, 751–762 (1999).
Lewis, J. et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405 (2000).
Gotz, J. et al. Oligodendroglial tau filament formation in transgenic mice expressing G272V tau. Eur. J. Neurosci. 13, 2131–2140 (2001).
Spittaels, K. et al. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am. J. Pathol. 155, 2153–2165 (1999).
Gotz, J., Chen, F., Barmettler, R. & Nitsch, R.M. Tau filament formation in transgenic mice expressing P301L tau. J. Biol. Chem. 276, 529–534 (2001).
Allen, B. et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 22, 9340–9351 (2002).
Tanemura, K. et al. Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol. Dis. 8, 1036–1045 (2001).
Tatebayashi, Y. et al. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. USA 99, 13896–13901 (2002).
Andorfer, C. et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590 (2003).
Santacruz, K. et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 (2005).
Andra, K. et al. Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol. Aging 17, 183–190 (1996).
Truett, G.E. et al. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 29, 52–54 (2000).
Chen, F., David, D., Ferrari, A. & Gotz, J. Posttranslational modifications of tau—role in human tauopathies and modeling in transgenic animals. Curr. Drug Targets 5, 503–515 (2004).
Acknowledgements
We thank Daniel Schuppli (University of Zurich) for critically reading this manuscript, and Mian Bi, Mathias Minderer, Heidrun Woelfing, Yazi Ke and Denise Nergenau for technical assistance. L.M.I. has been supported by the DFG. J.G. is a Medical Foundation Fellow and has been supported by the University of Sydney, the National Health & Medical Research Council (NHMRC), the Australian Research Council (ARC), the New South Wales Government through the Ministry for Science and Medical Research (BioFirst Program), the Nerve Research Foundation, the Medical Foundation (University of Sydney) and the Mason Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Ittner, L., Götz, J. Pronuclear injection for the production of transgenic mice. Nat Protoc 2, 1206–1215 (2007). https://doi.org/10.1038/nprot.2007.145
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.145
This article is cited by
-
Targeting vulnerable microcircuits in the ventral hippocampus of male transgenic mice to rescue Alzheimer-like social memory loss
Military Medical Research (2024)
-
Upregulation of estrogen receptor alpha (ERα) expression in transgenic mice expressing human CYP4Z1
Breast Cancer Research and Treatment (2022)
-
Genetic quality: a complex issue for experimental study reproducibility
Transgenic Research (2022)
-
In vivo enrichment of busulfan-resistant germ cells for efficient production of transgenic avian models
Scientific Reports (2021)
-
Increased hippocampal excitability in miR-324-null mice
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
