
Abstract
Phosphodiesterase type 4 (PDE4) is a family of enzymes that selectively degrade intracellular cAMP. PDE4 inhibitors have been shown to regulate the rewarding and reinforcing effects of cocaine, but the underlying mechanisms remain poorly understood. Here we show that pretreatments with the PDE4 inhibitor rolipram attenuated cocaine-induced locomotor sensitization in mice. Repeated cocaine exposure in vivo caused a decrease in inhibitory postsynaptic currents (IPSCs) and an increase in the AMPAR/NMDAR ratio in ventral tegmental area (VTA) dopamine neurons in midbrain slices ex vivo. Cocaine exposure disrupted the balance between excitation and inhibition as shown by an increase in the excitation to inhibition (E/I) ratio. Rolipram pretreatments in vivo prevented cocaine-induced reductions in GABAergic inhibition but did not further increase cocaine-induced potentiation of excitation, leading to the restoration of a balance between excitation and inhibition and normalization of the E/I ratio. In support of this idea, we found that repeated cocaine exposure led to an increase in the single-unit action potential firing rate in vivo in VTA dopamine neurons, which was blocked by rolipram pretreatments. These results suggest that repeated cocaine exposure in vivo disrupts the balance between excitation and inhibition in VTA dopamine neurons, while PDE4 inhibition reestablishes the balance between excitation and inhibition through distinct mechanisms.
Similar content being viewed by others

Introduction
Phosphodiesterases (PDEs) are a family of enzymes that hydrolyze intracellular cAMP and cGMP (Conti et al, 2003). There are 11 subtypes of PDEs (PDE1-11); amongst them, PDE4 selectively hydrolyzes cAMP (Lugnier, 2006; Zhang, 2009). Non-selective PDE and PDE4-specific inhibitors reduce drug intake and/or drug seeking for psychostimulants, alcohol, and opioids (Olsen and Liu, 2016; Wen et al, 2015), suggesting that inhibitors of PDE4 or other PDE isoforms alter common drug-induced neuroadaptations in the reward circuitry of the brain. Selective PDE4 inhibitors such as rolipram significantly reduce cocaine-induced increases in locomotor activity, behavioral sensitization, conditioned place preference (CPP) and self-administration (Janes et al, 2009; Knapp et al, 1999; Thompson et al, 2004). PDE4 has thus emerged as a potential therapeutic target for the treatment of substance abuse and substance use disorders (Olsen and Liu, 2016; Wen et al, 2015).
The mechanisms by which PDE4 inhibitors reduce behavioral effects of drugs of abuse remain poorly understood. Presynaptic Gi/o-coupled receptors, such as CB1 and D2, have been implicated in the actions of drugs of abuse (Johnson and Lovinger, 2016; Wang and Lupica, 2014; Wang et al, 2015; Xi et al, 2006). We have shown that PDE4 inhibitors blocked the acquisition of cocaine CPP and blocked endocannabinoid-mediated long-term depression of inhibitory synaptic transmission (I-LTD) in the VTA (Zhong et al, 2012). However, it remained unknown whether and how administration of the PDE4 inhibitors in vivo affected cocaine-induced long-term synaptic plasticity. Repeated cocaine exposure in vivo leads to a decrease in GABAergic inhibition of VTA dopamine neurons (Bocklisch et al, 2013; Liu et al, 2005), which primes excitatory synapses for LTP induction (Liu et al, 2005). Single or repeated cocaine exposure in vivo potentiates excitatory synaptic transmission as shown by an increase in the AMPAR/NMDAR ratio in VTA dopamine neurons ex vivo (Argilli et al, 2008; Bellone and Luscher, 2006; Borgland et al, 2004; Liu et al, 2005; Mameli et al, 2007; Ungless et al, 2001). In the present study, we examined whether the administration of the PDE4 inhibitor rolipram in vivo altered cocaine-induced effects on inhibitory and excitatory synaptic plasticity in VTA dopamine neurons.
Balance between excitation and inhibition is essential for normal function of the brain. The imbalance between excitation and inhibition has been implicated in a variety of neurological disorders including epilepsy, schizophrenia, and autism spectrum disorders (Nelson and Valakh, 2015; Tao et al, 2014). We examined whether repeated cocaine exposure in vivo altered the excitation to inhibition (E/I) ratio in VTA dopamine neurons, and if so, whether rolipram affects cocaine-induced changes in the E/I ratio. An altered balance of excitation and inhibition may directly impact action potential firing in vivo, leading to an altered output of behaviorally relevant neuronal populations. We performed in vivo single-unit recordings to determine whether cocaine exposure and rolipram treatments altered action potential firing in VTA dopamine neurons. Our results indicate that repeated cocaine exposure in vivo increases the E/I ratio in VTA dopamine neurons ex vivo and action potential firing in vivo. Rolipram pretreatments, however, prevent cocaine-induced reductions in GABAergic inhibition and do not further increase cocaine-induced potentiation of excitation, leading to a restoration of the balance between excitation and inhibition in the VTA, thereby suggesting a putative cellular mechanism for the PDE inhibition-induced attenuation of cocaine-induced behavioral effects.
Materials and methods
Methods and materials are described in detail in Supplementary Materials and Methods.
Animals
Roughly equal numbers of male and female adult C57BL/6 mice (8–10 weeks) were used. Homozygous GAD-Cre mice (Gad2-IRES-Cre) were obtained from the Jackson Laboratory and maintained on the same C57BL/6 background.
Animal Surgery and intra-VTA AAV Microinjections
Gad2-IRES-Cre mice (8–10 weeks old) of either sex were anesthetized and placed in a stereotaxic device. AAV DJ-EF1-DIO hChR2-(H134R)-eYFP (Stanford Neuroscience Gene Vector and Virus Core) was microinjected bilaterally into the VTA. Mice were allowed to recover for 1 week before starting experiments.
Slice Electrophysiology
Horizontal midbrain slices (200 μm thick) were cut using a vibrating slicer (Leica VT1200s, Nussloch, Germany), as described in our recent studies (Chen et al, 2016; Yu et al, 2013). EPSCs and IPSCs were recorded as we have described (Pan et al, 2008a). For recording of excitation-inhibition ratio (E/I ratio), VTA dopamine neurons were identified by electrophysiological properties (Chieng et al, 2011; Johnson and North, 1992; Jones and Kauer, 1999) and voltage-clamped alternatively at the reversal potentials for EPSCs (0 mV) and IPSCs (−60 mV). For optogenetic stimulation of hChR2-positive GABAergic neurons in the VTA, laser pulses (1 ms, 473 nM) were delivered every 30 s. AMPA/NMDA receptor (AMPAR/NMDAR) ratio was measured based on published studies (Liu et al, 2005; Saal et al, 2003).
In Vivo Electrophysiology
Mice were anesthetized and positioned in a stereotaxic frame, and craniotomies were performed to allow in vivo single-unit recordings of VTA dopamine neurons. Electrodes (10–15 MΩ) were filled with 2 M NaCl containing 1.5% neurobiotin. Dopamine neurons were identified by a broad triphasic extracellular action potential of a width >2 ms and a relatively slow firing rate (<10 Hz; Chaudhury et al, 2013; Ungless et al, 2004). To confirm cell type and electrode placement, neurons were juxtacellularly labeled with neurobiotin.
Immunofluorescence Staining
Mice were anesthetized and perfused transcardially with paraformaldehyde. Coronal VTA sections (30 μm) were cut and incubated with primary and secondary antibodies. Confocal imaging was performed using a Nikon TE2000-U inverted microscope equipped with the C1 Plus confocal system.
Data Analysis and Statistics
All results are expressed as mean±SEM. sIPSCs were analyzed using Mini-analysis (Synaptosft; Zhong et al, 2015). E/I ratio was calculated as the current ratio of EPSCs and IPSCs (EI=EPSCs−60 mV /IPSCs0 mV). The Shapiro–Wilk test was used to test the normality of the sample distributions, and samples were considered to be normally distributed at p>0.05. Results were analyzed with Student’s t-test or two-way ANOVA or repeated ANOVA followed by Tukey’s post hoc analysis. Results were considered to be significant at p<0.05.
Results
Rolipram Attenuated Cocaine-Induced Locomotor Sensitization
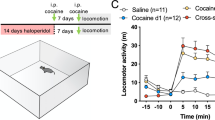
We examined the effects of the PDE4 inhibitor rolipram on cocaine-induced locomotor sensitization in male (n=20) and female (n=19) mice. Mice were first allowed to habituate to the open-field chamber for 2 days. Female mice exhibited significantly higher basal locomotor activity than male mice (t37=5.4, p<0.001; Supplementary Figure 1a). Starting from day 3, the male and female mice were randomly divided into four groups, and the mice received daily injections of saline or cocaine (15 mg/kg, i.p.) for 5 days. Rolipram (1 mg/kg, i.p.) or vehicle was injected 30 min before cocaine or saline injection. Three-way ANOVA of locomotor activity from day 3 to day 7 indicated that cocaine produced significant increases in locomotor activity in both sexes, a significantly greater increase in locomotor activity in female mice than in male mice, and that rolipram pretreatments significantly attenuated the cocaine-induced increase in locomotor activity in both male and female mice; there was no significant interaction between rolipram and sex (Supplementary Figure 1b,c,d). The detailed statistics summary is shown in Supplementary Table 1.
Rolipram Pretreatments Blocked the Reduction of GABAergic Inhibition Induced by Repeated Cocaine Exposure In Vivo
Repeated cocaine exposure in vivo leads to a decrease in IPSCs in VTA dopamine neurons, and this reduced GABAergic inhibition may contribute to the increase in the excitability of these neurons (Bocklisch et al, 2013; Liu et al, 2005). Having shown that rolipram pretreatments attenuated cocaine-induced locomotor sensitization (Supplementary Figure 1), we examined whether spontaneous IPSCs (sIPSCs) were altered in VTA dopamine neurons in slices prepared from these mice. One day after the locomotor test, mice shown in Supplementary Figure 1 were killed and midbrain slices were prepared. sIPSCs were recorded from VTA dopamine neurons in slices from these eight groups of mice. Male and female mice did not exhibit significant differences of the mean frequency and amplitude of sIPSCs among corresponding groups (Supplementary Figure 2); the data from both sexes were pooled in each group for subsequent analysis. Two-way ANOVA showed that cocaine and rolipram treatments had significant effects on the mean frequency of sIPSCs (cocaine: F(1,57)=5.7, p=0.020; rolipram: F(1,57)=20.7, p<0.001; cocaine × rolipram interaction: F(1,57)=8.6, p=0.005; Figures 1a and b), and the mean amplitude of sIPSCs (cocaine: F(1,57)=6.9, p=0.011; rolipram: F(1,57)=6.4, p=0.014; cocaine x rolipram interaction: F(1,57)=4.7, p=0.035; Figures 1a and c). Tukey’s post hoc tests indicated that cocaine injections led to significant decreases in the mean frequency (p<0.001; Figure 1b) and amplitude of sIPSCs (p<0.001; Figure 1c) in vehicle-pretreated mice. The cocaine-induced decreases in the frequency and amplitude of sIPSCs were blocked in slices from mice that received rolipram pretreatment (p<0.001; Figures 1b and c). The cumulative distribution for inter-event intervals of sIPSCs was right-shifted in the vehicle/cocaine group compared to that in the vehicle/saline group, and this shift was blocked by rolipram pretreatment (Figure 1d). The cumulative distribution for the amplitude of sIPSCs was left-shifted in the vehicle/cocaine group, and this shift was blocked by rolipram pretreatments (Figure 1e). Together, these results indicate that repeated cocaine exposure in vivo led to decreases in sIPSC frequency and amplitude, and these decreases were blocked by rolipram pretreatments.

Rolipram pretreatments blocked the reduction of GABAergic inhibition to dopamine neurons induced by repeated cocaine exposure in vivo. (a), Representative sIPSCs recorded from VTA dopamine neurons in slices prepared from saline- or cocaine-injected mice pre-treated with vehicle or rolipram. (b, c) The averaged frequency (b) and amplitude (c) of sIPSCs in VTA dopamine neurons in these four groups of mice. The mean frequency and amplitude of sIPSCs were significantly decreased in cocaine-injected vehicle-treated mice (***p<0.001, n=15–16), and this decrease was blocked by rolipram pretreatments (***p<0.001, n=14–16). (d, e), The cumulative probability plots indicated that cocaine exposure led to shifts in the distribution of the inter-event intervals (d) and amplitude (e) in vehicle-treated mice; these shifts were blocked by rolipram pretreatments (p<0.001, n=13–16).
Acute Rolipram Perfusion Increased GABAergic Inhibition of VTA Dopamine Neurons
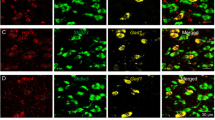
VTA dopamine neurons receive inhibitory synaptic inputs from local GABAergic neurons in the VTA as well as GABAergic axonal projections from the nucleus accumbens (NAc) (Bocklisch et al, 2013). We examined whether acute rolipram perfusion altered the strength of inhibitory inputs from VTA GABAergic neurons to VTA dopamine neurons by selectively expressing channelrhodopsin2 (hChR2) in GABAergic neurons. AAV DJ-EF1-DIO-hChR2-(H134R)-eYFP (denoted hChR2-eYFP for simplicity) was bilaterally microinjected into the VTA of Gad2-IRES-Cre mice. After recovery for 1 week, immunohistochemical staining indicated that the hChR2-eYFP was co-labeled with NeuN, a neuronal marker, but not with tyrosine hydroxylase (TH), a marker for dopamine neurons. The hChR2-eYFP was expressed in 95.1±4.6% of TH- neurons and was largely limited to the VTA (Supplementary Figure 3), indicating robust targeting of non-dopaminergic, putative GABAergic neurons in the VTA (Figures 2a, n=3 mice/group). hChR2-eYFP was expressed in axonal terminals in the NAc, which is consistent with long-range GABA projections from the VTA GABAergic neurons to NAc (Brown et al, 2012). Importantly, we did not observe any hChR2-eYFP expression in the somata of NAc neurons as shown by Nissl co-staining (Supplementary Figure 4). The lack of retrograde hChR2-eYFP expression suggests that optogenetic stimulation activates hChR2 expressed on local VTA GABAergic neurons.

Optogenetic interrogation of rolipram mechanisms on inhibitory synaptic plasticity in VTA dopamine neurons. (a), Expression of AAV DJ-EF1-DIO-hChR2(H134R)-eYFP in VTA GABAergic neurons in Gad2-IRES-Cre mice following intra-VTA viral injection. The majority of tyrosine hydroxylase negative (TH−) neurons in the VTA were infected with the AAVs. (b), Laser stimulation (473 nm, 1 ms pulse) induced an inward current in VTA dopamine neurons, which was abolished by the GABAA receptor blocker picrotoxin (p<0.001, n=3). Representative recordings of the laser-evoked currents at the indicated membrane potentials. Linear regression fit of I–V curve indicates the reversal potential of laser-evoked IPSCs is −21 mV (n = 7). (c), Bath perfusion of rolipram induced a significantly greater increase in the amplitude of laser-evoked IPSCs in slices from cocaine-injected mice than in saline-injected mice (p=0.031, n=6–7).
We first recorded IPSCs in VTA dopamine neurons evoked by optogenetic stimulation of axonal terminals from local GABAergic neurons. Dopamine neurons were identified by electrophysiological properties as well as the lack of eYFP fluorescence. Single laser pulses (473 nm, 1 ms duration) induced an inward current in VTA dopamine neurons at −60 mV, which was abolished by the GABAA receptor blocker picrotoxin (50 μM, n=3; Figure 2b). The currents were voltage-dependent and had a reversal potential of -20.5±3.6 mV (n=7; Figure 2b), consistent with the calculated ECl− (−21.0 mV) for the high-Cl− (54 mM) internal solution we used (Supplementary Materials and Methods). Thus, laser stimulation induced GABAA receptor-mediated IPSCs in VTA dopamine neurons. IPSCs evoked by electrical stimulation had a similar reversal potential (−20.9±3.8 mV; n=8), and were blocked by picrotoxin (n=2; Supplementary Figure 5a). We also made recordings from eYFP+ GABAergic neurons in the VTA. The same single laser pulses induced currents that showed strong inward rectification, reversed at ~0 mV and were not altered by picrotoxin (n=2, Supplementary Figure 5b). Thus, these currents are mediated by direct activation of hChR2 expressed on GABAergic neurons.
We examined whether acute application of rolipram altered laser-evoked IPSCs in slices prepared from mice that received five daily saline or cocaine injections in vivo. Bath perfusion of rolipram (10 μM) produced significant increases in the amplitude of laser-evoked IPSCs in slices prepared from saline-injected mice (t10=2.8, p=0.020) and cocaine-injected mice (t12=5.8, p<0.001; Figure 2c; Supplementary Figure 6). The IPSC amplitude from cocaine-exposed animals was significantly greater than that of saline-exposed animals after the application of rolipram (t11=2.5, p=0.031; Figure 2c). Thus, application of rolipram produces greater potentiation of IPSCs in slices prepared from cocaine-injected mice than that in saline-injected mice. These results suggest that in vivo cocaine exposure, which suppresses inhibitory inputs onto VTA dopamine neurons, permits greater PDE4 inhibition-induced potentiation compared to saline exposure.
Effects of Cocaine Exposure and Rolipram Pretreatments on the AMPAR/NMDAR Ratio in VTA Dopamine Neurons
We determined whether the AMPAR/NMDAR ratio was affected in mice that received saline or cocaine injections with rolipram or vehicle pretreatments. A new cohort of mice received 5 daily injections of saline or cocaine (15 mg/kg, i.p.). Rolipram (1 mg/kg, i.p.) or vehicle was injected 30 min before cocaine or saline injection. One day after locomotor activity was assessed, the mice were euthanized and midbrain slices were prepared. The AMPAR/NMDAR ratio was measured in VTA dopamine neurons prepared from these eight groups of mice. There were no significant sex differences of the AMPAR/NMDAR ratio between corresponding groups (Supplementary Figure 7), so the data from both sexes were pooled in each group. We found that cocaine (F(1,43)=21.0, p=0.001) and rolipram (F(1,43)=18.2, p<0.001) had significant effects on the AMPAR/NMDAR ratio, and there was a significant interaction between cocaine and rolipram (F(1,43)=19.4, p<0.001; Figure 3a). Tukey’s post hoc tests indicated that repeated cocaine injections led to a significant increase in the AMPAR/NMDAR ratio compared with saline injections (p<0.001; Figure 3a). Interestingly, compared with the vehicle group, rolipram pretreatments alone increased the AMPAR/NMDAR ratio in saline-injected mice (p<0.001) but did not cause any further increase in the AMPAR/NMDAR ratio in cocaine-injected mice (p=0.871; Figure 3a). Together, these results indicate that rolipram pretreatments alone increase the AMPAR/NMDAR ratio and do not further increase the cocaine-induced potentiation in the AMPAR/NMDAR ratio in VTA dopamine neurons.

Effects of cocaine exposure and rolipram pretreatments on the AMPAR/NMDAR ratio in VTA dopamine neurons. (a) Representative AMPAR- and NMDAR-mediated evoked EPSCs recorded from VTA dopamine neurons. Compared with vehicle, rolipram pretreatments increased the AMPAR/NMDAR ratio in slices from saline-injected mice (***p<0.001, n=11) and did not further increase the AMPAR/NMDAR ratio in slices from cocaine-injected mice (p=0.871, n=11-12). (b) Acute rolipram perfusion caused a significantly greater increase in the amplitude of evoked EPSCs in slices from saline-injected mice than that in cocaine-injected mice (p=0.024, n=6–7). The effect of rolipram in slices from saline- or cocaine-injected mice was blocked by the PKA inhibitor H89 (saline, p<0.001, n=7; cocaine, p=0.011, n=6).
To investigate potential mechanisms involved, we investigated the effects of acute bath application of rolipram on evoked EPSCs. Mice received five daily saline or cocaine injections without vehicle or rolipram pretreatment. One day after the last saline or cocaine injection, mice were euthanized and midbrain slices were prepared. Whole-cell recordings of dopamine neurons voltage-clamped at −60 mV were performed. In slices prepared from saline-injected mice, bath application of rolipram significantly increased the amplitude of evoked EPSCs (t12=5.5, p<0.001; Figure 3b; Supplementary Figure 8). In slices prepared from cocaine-injected mice, rolipram application also induced a significant increase in the amplitude of evoked EPSCs (t10=2.9, p=0.016; Figure 3b; Supplementary Figure 8), with smaller EPSC potentiation relative to saline-treated animals (t11=2.6, p=0.024; Figure 3b). The effects of rolipram in both groups were blocked by the PKA inhibitor H89 (saline, 89.1±2.7%, t12=6.3, p<0.001; cocaine, 95.7±6.3%, t10=3.1, p=0.011; Figure 3b), suggesting that rolipram increased EPSCs via cAMP-PKA signaling. Thus, repeated cocaine injections in vivo induces synaptic potentiation of glutamatergic inputs onto VTA dopamine neurons, and rolipram pretreatments do not produce further potentiation of excitatory synaptic inputs.
Rolipram Pretreatments Blocked Cocaine-Induced Disruption of Excitatory/Inhibitory Balance
We determined whether repeated cocaine exposure and rolipram pretreatments in vivo altered the balance of excitation and inhibition in VTA dopamine neurons. Mice received 5 daily injections of saline or cocaine (15 mg/kg, i.p.). Rolipram (1 mg/kg, i.p.) or vehicle was injected 30 min before each cocaine or saline injection. One day after the behavioral test, the mice were euthanized and midbrain slices were prepared. We recorded evoked EPSCs and IPSCs sequentially from the same VTA dopamine neurons in slices prepared from these four groups of mice. The NMDAR antagonist CPP (5 μM) was present throughout the experiment. VTA dopamine neurons were voltage-clamped alternatively at the reversal potential for IPSCs (−60 mV) and EPSCs (0 mV) to isolate EPSCs and IPSCs, respectively. Indeed, IPSCs recorded at 0 mV were abolished by the GABAA receptor blocker picrotoxin (50 μM), while EPSCs recorded at −60 mV were abolished by the AMPAR antagonist CNQX (20 μM; Figure 4a). We next determined whether cocaine and rolipram treatments altered the E/I ratio in these four groups of mice. Two-way ANOVA indicated that cocaine and rolipram pretreatments had significant effects on the E/I ratio (cocaine: F(1,37)=12.7, p=0.001; rolipram: F(1,37)=8.9, p=0.005; cocaine × rolipram interaction: F(1,37)=10.0, p=0.004; Figures 4b and c). Tukey’s post hoc tests indicated that the E/I ratio was significantly increased in the vehicle/cocaine group compared to the vehicle/saline group (p<0.001; Figure 4c). The increase in E/I ratio induced by in vivo cocaine exposure was blocked by rolipram pretreatment (p<0.001; Figure 4c). Thus, rolipram pretreatment prevents the disruption of excitatory/inhibitory balance in VTA dopamine neurons induced by repeated cocaine exposure.

Rolipram pretreatments restored the cocaine-induced imbalance of excitation and inhibition in the VTA. (a) EPSCs and IPSCs were isolated by voltage-clamping VTA dopamine neurons at the reversal potentials of IPSCs (−60 mV) and EPSCs (0 mV), respectively. EPSCs were blocked by the AMPAR antagonist CNQX (n=3), while IPSCs were blocked by the GABAA receptor blocker picrotoxin (n=3). (b) Sample EPSCs and IPSCs recorded in slices from saline- or cocaine-injected mice that received vehicle or rolipram pretreatments. (c) Repeated cocaine injections led to an increase in the E/I ratio (***p<0.001, n=10–11). This increase was blocked by rolipram pretreatments (***p<0.001, n=9-11).
Rolipram Pretreatment Blocked Cocaine-Induced Increase in In Vivo Action Potential Firing in VTA Dopamine Neurons
Previous studies have shown that repeated cocaine exposure in vivo leads to increased excitability in VTA dopamine neurons (Bocklisch et al, 2013; Liu et al, 2005). We examined whether cocaine-induced behavioral sensitization was associated with changes in action potential firing in VTA dopamine neurons in vivo. One day after the locomotor activity tests, mice were anesthetized with urethane and in vivo single-unit recordings were performed. Dopamine neurons were identified by firing characteristics (see Materials and Methods) and were validated by juxtacellular labelling with neurobiotin and post hoc TH immunostaining (Chaudhury et al, 2013; Ungless et al, 2004) (Figures 5a and b). We found that cocaine exposure and rolipram pretreatments had significant effects on the frequency of action potential firing (cocaine: F(1,66)=27.4, p<0.001; rolipram: F(1,66)=5.4, p=0.024; cocaine x rolipram interaction: F(1,66)=21.2, p<0.001; Figure 5c), and percentage of spikes in bursts in VTA dopamine neurons (cocaine: F(1,43)=12.5, p=0.001; rolipram: F(1,43)=4.5, p=0.040; cocaine x rolipram interaction: F(1,43)=18.1, p<0.001; Figure 5d). Tukey’s post hoc tests indicated that cocaine exposure significantly increased the frequency of action potential firing and the percentage of spikes in bursts in vehicle-pretreated mice (both p<0.001). These increases were blocked by rolipram pretreatments (p>0.05). There was no significant difference in the firing frequency and the percentage of spikes in bursts between vehicle- and rolipram-pretreated mice that received saline injections (p>0.05). Cocaine exposure and rolipram pretreatments had no significant effects on the proportion of dopamine neurons showing burst firing (cocaine: F(1,15)=0.2, p=0.705; rolipram: F(1,15)=0.4, p=0.525; cocaine x rolipram interaction: F(1,15)=4.0, p=0.068; Figure 5e). Thus, cocaine locomotor sensitization was accompanied by an increase in the activity of VTA dopamine neurons, and this increase was blocked by rolipram pretreatments.

Rolipram pretreatments attenuated cocaine-induced increase in single-unit action potential firing of VTA dopamine neurons in vivo. (a) Repeated cocaine exposure led to an increased rate of action potential firing by VTA dopamine neurons. Dopamine neurons were identified by a broad triphasic extracellular action potential of a width >2 ms and a relatively slow firing rate. (b) A recorded dopamine neuron was confirmed by neurobiotin (green) and TH (red) co-localization. (c) Repeated cocaine exposure in vivo led to an increase in the frequency of action potential firing by VTA dopamine neurons (***p<0.001, n=16–17). This increase was blocked by rolipram pretreatments (***p<0.001, n=15–17). (d) Repeated cocaine exposure caused an increase in the proportion of spikes that occurred in a burst (***p<0.001, n=10–11). This increase was blocked by rolipram pretreatments (***p<0.001, n=10–11). (e) Repeated cocaine exposure and rolipram pretreatment did not cause changes in the proportion of dopamine neurons showing burst firing (p>0.05, n=4). A full color version of this figure is available at the Neuropsychopharmacology journal online.
Discussion
In this study, we investigated the behavioral and electrophysiological effects of rolipram, a PDE4 inhibitor, on cocaine-induced alterations in drug-associated behavior and VTA dopamine neuron neurophysiology. Further, we show that repeated cocaine injections led to an increase in the strength of excitatory inputs but a decrease in the strength of inhibitory inputs to VTA dopamine neurons, tilting the E/I balance towards over-excitation. Rolipram pretreatments re-establish the excitatory/inhibitory balance via rescue of cocaine-induced inhibition of IPSCs.
Female mice exhibit greater basal locomotor activity and augmented cocaine locomotor responses (Balda et al, 2009; Van Swearingen et al, 2013; Walker et al, 2001). Consistent with these studies, we found that female mice exhibited greater locomotor activity than males both basally and in response to cocaine injections. The effects of rolipram were independent of sex since rolipram pretreatments decreased locomotor sensitization to a similar degree in both males and females. However, we did not find significant sex differences of sIPSCs and AMPAR/NMDAR ratio in corresponding groups. Previous studies did not detect significant sex differences of basal mEPSCs, action potentials and passive electrophysiological properties in the NAc shell of mice (Willett et al, 2016). On the other hand, mEPSC frequency in the NAc core was higher in females, and cocaine treatment induced a greater increase in mEPSC frequency in the NAc core than that in males (Wissman et al, 2011). It is thus possible that the sex differences of cocaine-induced locomotor sensitization may not be reflected by changes in excitatory and inhibitory transmission in VTA dopamine neurons, but by changes in the NAc core or other brain regions.
Repeated cocaine exposure in vivo leads to a reduction of GABAergic inhibition to VTA dopamine neurons as shown by decreases in spontaneous and miniature IPSCs (Bocklisch et al, 2013; Liu et al, 2005). We showed that rolipram pretreatments in vivo blocked cocaine-induced decreases in the frequency and amplitude of spontaneous IPSCs in VTA dopamine neurons ex vivo. How might rolipram block the cocaine-induced reduction of GABAergic inhibition? Our previous studies have implicated endocannabinoid-mediated long-term depression of inhibitory synapses (I-LTD) in cocaine-induced reduction of GABAergic inhibition in VTA dopamine neurons (Pan et al, 2008a, 2008b). Endocannabinoid-mediated I-LTD is mediated by a decrease in cAMP/PKA signaling (Chiu et al, 2010; Pan et al, 2008b) since cannabinoid receptors (CB1) are Gαi/o protein-coupled receptors whose activation leads to decreased cAMP/PKA activity (Howlett, 2005). PDE4 inhibitors increase cAMP levels in the brain (Conti et al, 2003; Lugnier, 2006; Zhang, 2009). Indeed, we found that acute bath perfusion of the PDE4 inhibitors rolipram and Ro 20-1724 block I-LTD in VTA dopamine neurons via an enhancement of cAMP/PKA signaling (Zhong et al, 2012). It is thus likely that rolipram pretreatments blocked the cocaine-induced reduction of GABAergic inhibition through blockade of I-LTD-like synaptic modifications. To investigate this further, we utilized optogenetics to interrogate the specific effects of rolipram on inhibitory transmission from GABAergic neurons to dopamine neurons in the VTA. By using Cre-inducible AAVs carrying a ‘double-floxed’ inverted open reading frame, we selectively expressed hChR2-eYFP in VTA GABAergic neurons in Gad2-IRES-Cre mice that received saline or cocaine injections for 5 days. A brief laser stimulation of hChR2-eYFP in VTA GABAergic neurons induced IPSCs in VTA dopamine neurons. Bath perfusion of rolipram caused greater enhancement of IPSCs in slices from cocaine-injected mice than in slices from saline-injected mice. These results suggest that inhibitory synapses had been depressed in vivo via I-LTD-like synaptic modifications, permitting a greater potentiation by rolipram in slices. Alternatively, repeated cocaine injections may produce an increase in PDE4 activity and/or expression in GABAergic neurons in the VTA, thus inhibition of PDE4 with rolipram would therefore cause greater potentiation of IPSCs in these slices. As CB1-mediated I-LTD is presynaptic (Heifets and Castillo, 2009), it is likely that rolipram regulates IPSCs via a presynaptic mechanism.
There is ample evidence that single or repeated cocaine injections in vivo leads to an increase in the AMPAR/NMDAR ratio in VTA dopamine neurons (Argilli et al, 2008; Bellone and Luscher, 2006; Borgland et al, 2004; Liu et al, 2005; Mameli et al, 2007; Ungless et al, 2001). We found that repeated cocaine injections for 5 days led to an increase in the AMPAR/NMDAR ratio in VTA dopamine neurons ex vivo, while rolipram pretreatments alone increased the AMPAR/NMDAR ratio in slices from saline-injected mice but did not further increase the AMPAR/NMDAR ratio in slices from cocaine-injected mice. The cocaine-induced increase in the AMPAR/NMDAR ratio requires D1 dopamine receptors (Argilli et al, 2008; Brown et al, 2010). The D1 receptor is a Gαs-coupled receptor which, when activated, leads to activation of cAMP/PKA signaling (Anderson and Pierce, 2005; Neve et al, 2004; Tritsch and Sabatini, 2012). By blocking cAMP degradation, PDE4 inhibition also enhances cAMP/PKA signaling. PKA activation is required for the cocaine-induced increase in the AMPAR/NMDAR ratio in VTA dopamine neurons (Argilli et al, 2008; Brown et al, 2010; Liu et al, 2016). Indeed, we found that the potentiation of AMPAR-EPSCs by bath perfusion of rolipram was blocked by the PKA inhibitor H89. PKA phosphorylates GluA1 at S845, increasing the conductance and open probability of the AMPAR channel (Banke et al, 2000). Additionally, PKA enhances synaptic strength via presynaptic mechanisms (Chavez-Noriega and Stevens, 1994). We therefore suspect that rolipram potentiates AMPAR-EPSCs by pre- and postsynaptic mechanisms. Thus, activation of cAMP/PKA signaling appears to be required for the increase in the AMPAR/NMDAR ratio induced by repeated cocaine injections and rolipram pretreatments.
VTA dopamine neurons are heterogeneous (Brischoux et al, 2009) and show projection-specific synaptic plasticity in response to cocaine treatment (Lammel et al, 2011). A limitation of the present study is that dopamine neurons were recorded without knowing their projection targets. The majority of recordings were done in the parabrachial pigmented nucleus (PBP), a major subdivision of the VTA. Dopamine neurons in the lateral PBP often project to the lateral shell of the NAc, while those in the medial PBP project mainly to the medial shell of the NAc (Lammel et al, 2008, 2011). It is likely that dopamine neurons recorded in this study would project mainly to the shell of the NAc. Cocaine exposure is known to modulate excitatory synapses onto NAc shell-projecting dopamine neurons in the VTA (Lammel et al, 2011).
A recent study has shown that although the strength of total synaptic inputs to visual cortical neurons varied between cells, the ratio of excitatory and inhibitory input remained constant (Xue et al, 2014). The imbalance between excitation and inhibition has been implicated in a variety of neurological disorders including epilepsy, schizophrenia, and autism spectrum disorders (Nelson and Valakh, 2015). A balanced inhibition not only prevents epileptic activity-induced excitotoxicity but is also thought to contribute to sharpening the selectivity of excitatory sensory responses (Nelson and Valakh, 2015; Tao et al, 2014; Zhou et al, 2014). We showed here that repeated cocaine exposure in vivo led to an imbalance between excitation and inhibition, manifested by an increase in the AMPAR/NMDAR ratio and a decrease in sIPSC amplitude, which may have led to the increased E/I ratio we observed. Interestingly, rolipram pretreatments restored the E/I ratio, putatively through enhancement of GABAergic inhibition. The cocaine-induced reduction of inhibition could lead to enhanced synaptic “noise” and aberrant reinforcement learning, whereas rolipram pretreatments blocked such changes.
A direct consequence of imbalance between excitation and inhibition may be an increase in excitability (Bocklisch et al, 2013; Liu et al, 2005). Midbrain dopamine neurons selectively respond to reward and reward-predicting stimuli by an increase in action potential firing (Schultz, 2001). We found that repeated cocaine exposure led to an increase in the frequency of single unit activity in VTA dopamine neurons in vivo, while rolipram pretreatments blocked the cocaine-induced increase in action potential firing. The restoration of E/I balance by rolipram pretreatments might explain how rolipram blocked the cocaine-induced increase in single-unit firing in VTA dopamine neuron in vivo. One caveat is that AP firing is not only affected by excitatory and inhibitory synaptic inputs but also by intrinsic excitability. Withdrawal from repeated cocaine exposure in vivo produces similar potentiation of glutamatergic excitation but reduction of GABAergic inhibition to medium spiny neurons in the NAc shell (Ishikawa et al, 2013a; Ishikawa et al, 2013b; Otaka et al, 2013; Wolf, 2016). However, cocaine exposure in vivo or withdrawal leads to enhanced or depressed in vivo AP firing in the NAc under different experiment conditions (Guillem et al, 2014; Peoples et al, 2007). Thus, the changes in firing rate cannot be solely attributed to the shift of the balance between excitation and inhibition. Nevertheless, we found that in the presence of glutamate and GABAA receptor antagonists, AP firing in VTA dopamine neurons was not significantly different in midbrain slices prepared from saline or cocaine-injected mice that had received vehicle or rolipram pretreatments (Supplementary Figure 9). Thus, it is likely that the shift of the balance between excitation and inhibition might at least partially explain why cocaine exposure and rolipram pretreatments alter VTA dopamine neuron AP firing in vivo.
PDE4 inhibitors have been shown to reduce drug intake and/or drug seeking for psychostimulants, alcohol, and opioids (Hu et al, 2011; Lai et al, 2014), suggesting that PDE4 could be a therapeutic target for several classes of substance use disorders (Olsen and Liu, 2016; Wen et al, 2015). Most relevant to the present study are the findings that rolipram pretreatments block cocaine-induced increases in locomotor activity, behavioral sensitization, CPP and self-administration (Janes et al, 2009; Knapp et al, 1999; Thompson et al, 2004). We have shown that intra-VTA injection of rolipram attenuated cocaine CPP, suggesting that the VTA is involved in the action of rolipram (Zhong et al, 2012). Here we extended the above studies in an important way by showing that repeated cocaine exposure disrupted the balance between excitation and inhibition, with PDE4 inhibition putatively restoring this balance by enhancing GABAergic inhibition. Our study has revealed previously unrecognized cellular mechanisms for the actions of the PDE4 inhibitors.
Funding and Disclosure
This research was funded by NIH Grants DA035217 and MH101146. C.V. is a member of the Medical Scientist Training Program at the Medical College of Wisconsin, which is partially supported by a training grant from NIGMS T32-GM080202. The authors declare no conflict of interest.
References
Anderson SM, Pierce RC (2005). Cocaine-induced alterations in dopamine receptor signaling: implications for reinforcement and reinstatement. Pharmacol Ther 106: 389–403.
Argilli E, Sibley DR, Malenka RC, England PM, Bonci A (2008). Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci 28: 9092–9100.
Balda MA, Anderson KL, Itzhak Y (2009). Development and persistence of long-lasting behavioral sensitization to cocaine in female mice: Role of the nNOS gene. Neuropharmacology 56: 709–715.
Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF (2000). Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20: 89–102.
Bellone C, Luscher C (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci 9: 636–641.
Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M et al (2013). Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 341: 1521–1525.
Borgland SL, Malenka RC, Bonci A (2004). Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci 24: 7482–7490.
Brischoux F, Chakraborty S, Brierley DI, Ungless MA (2009). Phasic excitation of dopamine neurons in ventral VTA by noxious stimuli. Proc Natl Acad Sci U S A 106: 4894–4899.
Brown MT, Bellone C, Mameli M, Labouebe G, Bocklisch C, Balland B et al (2010). Drug-driven AMPA receptor redistribution mimicked by selective dopamine neuron stimulation. PLoS ONE 5: e15870.
Brown MT, Tan KR, O'Connor EC, Nikonenko I, Muller D, Luscher C (2012). Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature 492: 452–456.
Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW et al (2013). Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493: 532–536.
Chavez-Noriega LE, Stevens CF (1994). Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci 14: 310–317.
Chen Y, Liu X, Vickstrom CR, Liu MJ, Zhao L, Viader A et al (2016). Neuronal and astrocytic monoacylglycerol lipase limit the spread of endocannabinoid signaling in the cerebellum. eNeuro 3: e0048-16.2016.
Chieng B, Azriel Y, Mohammadi S, Christie MJ (2011). Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol 589: 3775–3787.
Chiu CQ, Puente N, Grandes P, Castillo PE (2010). Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J Neurosci 30: 7236–7248.
Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C (2003). Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem 278: 5493–5496.
Guillem K, Ahmed SH, Peoples LL (2014). Escalation of cocaine intake and incubation of cocaine seeking are correlated with dissociable neuronal processes in different accumbens subregions. Biol Psychiatry 76: 31–39.
Heifets BD, Castillo PE (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol 71: 283–306.
Howlett AC (2005). Cannabinoid receptor signaling. Handb Exp Pharmacol 168: 53–79.
Hu W, Lu T, Chen A, Huang Y, Hansen R, Chandler LJ et al (2011). Inhibition of phosphodiesterase-4 decreases ethanol intake in mice. Psychopharmacology (Berl) 218: 331–339.
Ishikawa M, Otaka M, Huang YH, Neumann PA, Winters BD, Grace AA et al (2013a). Dopamine triggers heterosynaptic plasticity. J Neurosci 33: 6759–6765.
Ishikawa M, Otaka M, Neumann PA, Wang Z, Cook JM, Schluter OM et al (2013b). Exposure to cocaine regulates inhibitory synaptic transmission from the ventral tegmental area to the nucleus accumbens. J Physiol 591: 4827–4841.
Janes AC, Kantak KM, Cherry JA (2009). The involvement of type IV phosphodiesterases in cocaine-induced sensitization and subsequent pERK expression in the mouse nucleus accumbens. Psychopharmacology (Berl) 206: 177–185.
Johnson KA, Lovinger DM (2016). Presynaptic G Protein-Coupled Receptors: Gatekeepers of Addiction? Front Cell Neurosci 10: 264.
Johnson SW, North RA (1992). Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol 450: 455–468.
Jones S, Kauer JA (1999). Amphetamine depresses excitatory synaptic transmission via serotonin receptors in the ventral tegmental area. J Neurosci 19: 9780–9787.
Knapp CM, Foye MM, Ciraulo DA, Kornetsky C (1999). The type IV phosphodiesterase inhibitors, Ro 20-1724 and rolipram, block the initiation of cocaine self-administration. Pharmacol Biochem Behav 62: 151–158.
Lai M, Zhu H, Sun A, Zhuang D, Fu D, Chen W et al (2014). The phosphodiesterase-4 inhibitor rolipram attenuates heroin-seeking behavior induced by cues or heroin priming in rats. Int J Neuropsychopharmacol 17: 1397–1407.
Lammel S, Hetzel A, Hackel O, Jones I, Liss B, Roeper J (2008). Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron 57: 760–773.
Lammel S, Ion DI, Roeper J, Malenka RC (2011). Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron 70: 855–862.
Liu QS, Pu L, Poo MM (2005). Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature 437: 1027–1031.
Liu X, Chen Y, Tong J, Reynolds AM, Proudfoot SC, Qi J et al (2016). Epac Signaling Is Required for Cocaine-Induced Change in AMPA Receptor Subunit Composition in the Ventral Tegmental Area. J Neurosci 36: 4802–4815.
Lugnier C (2006). Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther 109: 366–398.
Mameli M, Balland B, Lujan R, Luscher C (2007). Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317: 530–533.
Nelson SB, Valakh V (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87: 684–698.
Neve KA, Seamans JK, Trantham-Davidson H (2004). Dopamine receptor signaling. J Recept Signal Transduct Res 24: 165–205.
Olsen CM, Liu QS (2016). Phosphodiesterase 4 inhibitors and drugs of abuse: current knowledge and therapeutic opportunities. Front Biol 11: 376.
Otaka M, Ishikawa M, Lee BR, Liu L, Neumann PA, Cui R et al (2013). Exposure to cocaine regulates inhibitory synaptic transmission in the nucleus accumbens. J Neurosci 33: 6753–6758.
Pan B, Hillard CJ, Liu QS (2008a). Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci 28: 1385–1397.
Pan B, Hillard CJ, Liu QS (2008b). D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J Neurosci 28: 14018–14030.
Peoples LL, Kravitz AV, Guillem K (2007). The role of accumbal hypoactivity in cocaine addiction. Sci World J 7: 22–45.
Saal D, Dong Y, Bonci A, Malenka RC (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582.
Schultz W (2001). Reward signaling by dopamine neurons. Neuroscientist 7: 293–302.
Tao HW, Li YT, Zhang LI (2014). Formation of excitation-inhibition balance: inhibition listens and changes its tune. Trends Neurosci 37: 528–530.
Thompson BE, Sachs BD, Kantak KM, Cherry JA (2004). The Type IV phosphodiesterase inhibitor rolipram interferes with drug-induced conditioned place preference but not immediate early gene induction in mice. Eur J Neurosci 19: 2561–2568.
Tritsch NX, Sabatini BL (2012). Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76: 33–50.
Ungless MA, Magill PJ, Bolam JP (2004). Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science 303: 2040–2042.
Ungless MA, Whistler JL, Malenka RC, Bonci A (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583–587.
Van Swearingen AE, Walker QD, Kuhn CM (2013). Sex differences in novelty- and psychostimulant-induced behaviors of C57BL/6 mice. Psychopharmacology (Berl) 225: 707–718.
Walker QD, Cabassa J, Kaplan KA, Li ST, Haroon J, Spohr HA et al (2001). Sex differences in cocaine-stimulated motor behavior: disparate effects of gonadectomy. Neuropsychopharmacology 25: 118–130.
Wang H, Lupica CR (2014). Release of endogenous cannabinoids from ventral tegmental area dopamine neurons and the modulation of synaptic processes. Prog Neuropsychopharmacol Biol Psychiatry 52: 24–27.
Wang H, Treadway T, Covey DP, Cheer JF, Lupica CR (2015). Cocaine-induced endocannabinoid mobilization in the ventral tegmental area. Cell Rep 12: 1997–2008.
Wen RT, Feng WY, Liang JH, Zhang HT (2015). Role of phosphodiesterase 4-mediated cyclic AMP signaling in pharmacotherapy for substance dependence. Curr Pharm Des 21: 355–364.
Willett JA, Will T, Hauser CA, Dorris DM, Cao J, Meitzen J (2016). No evidence for sex differences in the electrophysiological properties and excitatory synaptic input onto nucleus accumbens shell medium spiny neurons. eNeuro 3: e0147-15.2016.
Wissman AM, McCollum AF, Huang GZ, Nikrodhanond AA, Woolley CS (2011). Sex differences and effects of cocaine on excitatory synapses in the nucleus accumbens. Neuropharmacology 61: 217–227.
Wolf ME (2016). Synaptic mechanisms underlying persistent cocaine craving. Nat Rev Neurosci 17: 351–365.
Xi ZX, Gilbert JG, Peng XQ, Pak AC, Li X, Gardner EL (2006). Cannabinoid CB1 receptor antagonist AM251 inhibits cocaine-primed relapse in rats: role of glutamate in the nucleus accumbens. J Neurosci 26: 8531–8536.
Xue M, Atallah BV, Scanziani M (2014). Equalizing excitation-inhibition ratios across visual cortical neurons. Nature 511: 596–600.
Yu F, Zhong P, Liu X, Sun D, Gao HQ, Liu QS (2013). Metabotropic glutamate receptor I (mGluR1) antagonism impairs cocaine-induced conditioned place preference via inhibition of protein synthesis. Neuropsychopharmacology 38: 1308–1321.
Zhang HT (2009). Cyclic AMP-specific phosphodiesterase-4 as a target for the development of antidepressant drugs. Curr Pharm Des 15: 1688–1698.
Zhong P, Liu Y, Hu Y, Wang T, Zhao YP, Liu QS (2015). BDNF interacts with endocannabinoids to regulate cocaine-induced synaptic plasticity in mouse midbrain dopamine neurons. J Neurosci 35: 4469–4481.
Zhong P, Wang W, Yu F, Nazari M, Liu X, Liu QS (2012). Phosphodiesterase 4 inhibition impairs cocaine-induced inhibitory synaptic plasticity and conditioned place preference. Neuropsychopharmacology 37: 2377–2387.
Zhou M, Liang F, Xiong XR, Li L, Li H, Xiao Z et al (2014). Scaling down of balanced excitation and inhibition by active behavioral states in auditory cortex. Nat Neurosci 17: 841–850.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, X., Zhong, P., Vickstrom, C. et al. PDE4 Inhibition Restores the Balance Between Excitation and Inhibition in VTA Dopamine Neurons Disrupted by Repeated In Vivo Cocaine Exposure. Neuropsychopharmacol 42, 1991–1999 (2017). https://doi.org/10.1038/npp.2017.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2017.96
This article is cited by
-
The Genetically Informed Neurobiology of Addiction (GINA) model
Nature Reviews Neuroscience (2023)
-
cAMP-mediated upregulation of HCN channels in VTA dopamine neurons promotes cocaine reinforcement
Molecular Psychiatry (2023)
-
Inhibition of CSF1R, a receptor involved in microglia viability, alters behavioral and molecular changes induced by cocaine
Scientific Reports (2021)
-
Ibudilast, a neuroimmune modulator, reduces heavy drinking and alcohol cue-elicited neural activation: a randomized trial
Translational Psychiatry (2021)
-
Cyclic nucleotide phosphodiesterases: potential therapeutic targets for alcohol use disorder
Psychopharmacology (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}