
Abstract
Riluzole is a glutamate-modulating agent with neuroprotective properties approved for use in amyotrophic lateral sclerosis. The efficacy and safety of riluzole vs placebo as an adjunct to antidepressant medication in outpatients with major depressive disorder (MDD) was examined in a 3-site, 8-week, randomized, double-blind, placebo-controlled, fixed-dose trial using a sequential parallel comparison design comprised of two phases of 4 weeks. Patients with MDD in a current major depressive episode (N=104) with an inadequate response to either a prospective or a historical trial of an antidepressant medication were randomized in a 2 : 3 : 3 ratio to the treatment sequences of riluzole/riluzole, placebo/placebo, and placebo/riluzole, respectively. The primary outcome was change in depression severity, as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS). Secondary efficacy outcomes included the response rate, defined as at least a 50% improvement in MADRS, Clinical Global Impressions severity and improvement subscales, and patient-reported measures of depression and cognitive function. Eighty-five patients completed the randomized treatment phases. Treatment groups did not differ in mean change in MADRS scores, response rate, or in any secondary efficacy outcomes. Riluzole was generally well tolerated, with a side effect profile consistent with its clinical use. In conclusion, a fixed dose of riluzole (100 mg/day) did not show adjunctive antidepressant efficacy compared to placebo. The trial was adequately powered to detect a moderate riluzole effect, and the risk for exaggerated placebo responses was mitigated. The lack of efficacy suggests that mechanisms underlying riluzole’s neuroprotective effects are insufficient for clinical response in treatment-resistant depression.
Similar content being viewed by others

Introduction
The quest for new antidepressant brain targets beyond the monoamines is an area of intense investigation (Papakostas and Ionescu, 2015; Newport et al, 2015), owing to limitations in efficacy of existing therapies. Drugs which impact amino acid neurotransmitter systems such as glutamate have received particular scrutiny, in recognition that these systems subserve fundamental roles in regulation of synaptic plasticity and impact essential human processes of mood, cognition, and reward (Abdallah et al, 2014; Duman et al, 2016). Recent CNS drug discovery programs involving these systems include agents impacting ionotropic ((N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)) receptors, metabotropic receptors, glial cell transporters, and glutamate release inhibitors (Jaso et al, 2017).
Riluzole is an orally administered medication approved by the US Food and Drug Administration since 1994 for the treatment of amyotrophic lateral sclerosis. Its pharmacological mechanism of action and its effects on the glutamatergic system are complex. In contrast to ketamine and memantine, riluzole is not an open-channel antagonist of the NMDA receptor, though similar to lamotrigine, it was found to significantly enhance surface expression of AMPA receptor subunits in cultured hippocampal neurons (Du et al, 2007). Early reports related riluzole’s inhibition of presynaptic release of glutamate, partly due to inactivation of voltage-dependent sodium channels on glutamatergic nerve terminals, to the drug’s anti-excitoxicity actions (Doble, 1996). Subsequent studies elucidated riluzole’s neuroprotective mechanisms involving glutamate clearance from the synaptic cleft through facilitation of astrocytic glutamate reuptake (Frizzo et al, 2004), and stimulation of neurotrophic factor expression (Türck and Frizzo, 2015). Preclinical rodent models found riluzole’s effects on brain derived neurotrophic factor (BDNF) and glutamate transporter expression to be associated with antidepressant-like action (Banasr et al, 2010; Gourley et al, 2012).
Riluzole has been tested in several small open-label trials in patients with mood and anxiety disorders (Pittenger et al, 2008; Mathew et al, 2005), including as monotherapy in patients with treatment-resistant major depression (TRD) (Zarate et al, 2004) and bipolar depression (Brennan et al, 2010), and as an adjunct to a SSRI (Sanacora et al, 2007) or mood stabilizer (Zarate et al, 2005). While moderate-to-large within-subject effect sizes were observed, these studies were limited by small sample sizes (less than 20 subjects) and absence of placebo control. Subsequent small placebo-controlled studies in TRD patients reported that riluzole failed to prevent relapse following an early response to intravenous ketamine (Mathew et al, 2010; Ibrahim et al 2012), and did not show benefit for ketamine non-responders (Niciu et al, 2014). However, these studies were not designed to directly examine the antidepressant effects of riluzole and were not powered to detect moderate sized effects. In contrast, a recent placebo-controlled trial of riluzole conducted in 60 non-resistant MDD Iranian patients showed efficacy; in this 6-week inpatient study, the combination of riluzole and citalopram was found to be superior to the combination of citalopram and placebo with large effect sizes (Cohen’s d>0.8 at weeks 2, 4, and 6 of treatment, which was every time point examined) (Salardini et al, 2016).
The present study was designed to examine the adjunctive efficacy of a fixed dose of riluzole in outpatients with MDD who had failed to adequately respond to at least one adequate trial of an approved antidepressant medication in the current episode. We used a sequential parallel comparison design (SPCD) (Fava et al, 2003) to optimize efficiency and potentially diminish placebo response (Fava et al, 2016). We hypothesized that riluzole added to an antidepressant medication would be superior to add-on placebo in improving depressive symptoms in this difficult-to-treat patient population.
Materials and methods
Participants
The study enrolled patients at three academic medical centers between June 2011 and December 2014, with the final study visit in February 2015. The Institutional Review Boards at the participating sites approved the study. After complete description of the study, written informed consent was obtained from all participating subjects.
Patients were eligible to participate if they were between 18 and 65 years of age, had a primary diagnosis of MDD, assessed by the Structured Clinical Interview for DSM-IV-TR—Axis I disorders (First et al, 2007), and had an inadequate response to at least one but no greater than four adequate trials of an antidepressant using the MGH-Antidepressant Treatment History Questionnaire (ATRQ) (Chandler et al, 2010). Patients were required to have at least moderate depressive symptom severity, indexed by a Montgomery-Asberg Depression Rating Scale (MADRS) (Montgomery and Asberg, 1979) score⩾18 and an Inventory of Depressive Symptomatology-Self Rated (IDS-SR) (Rush et al, 1996) score⩾20. Patients were excluded if they were at serious suicide risk, had substance use disorders within the last 6 months, lifetime histories of bipolar disorder or psychotic disorders, or had severe and unstable medical illness. Structured psychotherapy for the treatment of depression was exclusionary if initiated within 8 weeks of randomization. All patients had a physical examination, routine hematologic and biochemical tests, urine toxicology, and electrocardiogram (ECG) to detect unstable medical illnesses or substance use.
Study Procedures
Initial group assignment
Patients meeting initial eligibility criteria were assigned to one of two groups (A or B), depending on whether or not they were receiving a FDA-approved antidepressant medication at Screening. Patients not taking an antidepressant (Group A), and who were experiencing a major depressive episode for at least 10 weeks, were given a prospective 8-week open-label trial of sertraline (2 patients received citalopram as the lead-in antidepressant prior to a protocol amendment). For participants taking concurrent disallowed psychotropic medications (such as atypical antipsychotics or mood stabilizers), a taper was conducted such that these medications were discontinued while open-label treatment with sertraline was initiated at 50 mg/day for two weeks, after which the dose was increased in weekly increments of 50 mg for the next two weeks to a dose of 150 mg per day. If tolerated, the patient was maintained at 150 mg/day for at least four weeks. In case of poor tolerability, the sertraline dose could be lowered to a minimum of 50 mg/day. Group A patients were eligible for the double-blind phase following the 8-week lead-in period if they met depressive severity thresholds (MADRS⩾18 and IDS-SR⩾20) and had<50% decrease in the IDS-SR score from Screening.
Group B participants were individuals not responding, based on the ATRQ, to an ongoing treatment with an SSRI, serotonin-norepinephrine reuptake inhibitor (SNRI), or bupropion for a minimum of 8 weeks, with a stable dose for at least 4 weeks prior to randomization. All patients were required to be free of other psychotropic medications for the duration of the study with the exception of a stable dose of a non-benzodiazepine hypnotic (eg, zolpidem 10 mg nightly) or a benzodiazepine, if on a stable dose for at least two weeks prior to baseline at doses no greater than lorazepam 2 mg/day or equivalent.
Sequential parallel comparison design (SPCD)
SPCD was used for the 8-week randomized, double-blind phase, which comprised two blocks of approximately 28 days each. Patients were randomly assigned to adjunctive treatment with riluzole (50 mg twice per day) or placebo, in a 2 : 3 : 3 ratio to the treatment sequences of riluzole/riluzole, placebo/placebo, and placebo/riluzole, respectively. Patients in the placebo/riluzole group were randomized to receive adjunctive placebo for the first 4-week block and adjunctive riluzole for the second 4-week block. To minimize patient expectancies regarding changes in study drug allocation during the double-blind period, the consent form did not mention the two blocks, but rather, described the odds of receiving placebo or riluzole at any point during the 8-week randomized phase of the study.
The pharmacist at each site assigned randomization numbers in consecutive order. Subjects were randomized to the three treatment groups using computer-generated codes in a randomly permuted block procedure consisting of blocks of 8 patients, stratified by pre-randomization group (A or B) and site. Only the site pharmacists and the study statistician had access to the randomization code. All other study personnel were masked to treatment assignment. Pharmacists at each study site prepared riluzole tablets (50 mg) and matching placebo in capsules that were identical in size, appearance, and taste. Patients were instructed to take one capsule of study medication in the morning and evening, while remaining on the same dosage of concurrent antidepressant medication. In case of intolerable side effects, study clinicians could lower the dosage of study medication to one capsule daily. Patients unable to tolerate the minimum allowed daily dosage were withdrawn from the study.
Patients were evaluated every 7 days by the study psychiatrist and by a clinical rater. A comprehensive hepatic panel and CBC were obtained after 4 weeks and at endpoint, and assessed by clinicians not involved in the patient’s treatment or ratings. Adherence was monitored by pill count and inspection of medication diary; patients who did not take between 80 and 120% of study medication for two consecutive visits were considered non-adherent and withdrawn from study medication.
Outcome Measures
The primary outcome was the change in the MADRS score during each 4-week phase of the double-blind treatment period. Trained raters masked to treatment-group assignment performed clinical assessments. Raters were experienced research staff extensively trained in the use of the instruments and who passed certification criteria by an external ratings vendor. MADRS raters were trained to reliability across the three sites prior to the study.
Secondary outcomes included the response rate, defined as a reduction in the MADRS score by 50% or more, and remission rate, defined as a MADRS score⩽9. Additional measures were the Clinical Global Impressions severity (CGI-S) and improvement (CGI-I) subscales, IDS-SR, and the MGH Cognitive and Physical Functioning Questionnaire (CPFQ) (Fava et al, 2009). Safety and tolerability was assessed with the Systematic Assessment for Treatment Emergent Events (SAFTEE-SI) (Levine and Schooler, 1986), and suicidal ideation and behavior was tracked with the Columbia-Suicide Severity Rating Scale (Posner et al, 2011).
Statistical Analysis
Data were examined prior to analysis using descriptive statistics. Randomized groups were compared on continuous variables using ANOVA and on categorical predictors using χ2-tests or Fisher’s exact tests. Continuous measures were assessed for normality. Our primary analysis was intent-to-treat using all available data on all subjects. For subjects who dropped out in Block 1 (the first 4 weeks of double-blind treatment), the last observation was carried forward to the last visit in Block 1 (visit 4), while these subjects’ data were not used in calculating the part of the test statistic corresponding to Block 2. For subjects who dropped out in Block 2 (the second 4 weeks of double-blind treatment), last observation was carried forward in Block 2 and all data from this subject were used in the analyses. Sensitivity analyses for MADRS included analyses using missing values on the response variable replaced by predicted values from linear or generalized linear models of the longitudinal data over time. All tests are two-sided at alpha=0.05.
The primary hypothesis regarding improvement in MADRS was tested using the approach described in Tamura and Huang (2007) for continuous data where effect estimates from the two phases were weighted to assess overall riluzole-placebo differences. While the data from all the randomized subjects were used in the first phase, only the data from placebo non-responders were used in the second phase. The two seemingly unrelated regression models for the two phases had change in MADRS for the corresponding block as the dependent variable, the treatment indicator for riluzole as the main predictor of interest, and controlled for site and baseline MADRS at the beginning of each block. The test statistic was a z-score based on weighted average of the treatment effect estimates in the two phases with the weight chosen to maximize statistical power relative to the sample sizes for each arm and to detect clinically meaningful drug effects in each phase of the study (w=0.75). The analysis was performed in SAS using PROC MODEL and the SUR option as specified in the Appendix of Tamura and Huang (2007).
For binary outcomes (response and remission rates) we used a weighted average of the effects from the two phases of double-blind treatment. We used as test-statistic z=h/SE of h, where h=w(p1−q1)+(1−w)(p2−q2), where p1, p2 were the response rates to riluzole in the first and second phases, and q1, q2 were the rates for placebo. We used w=0.75. Secondary hypotheses were tested using the same approach as in the primary analysis. To examine safety and tolerability we compared adverse events in both the riluzole and placebo treated groups.
The a priori power calculations indicated that 150 subjects needed to be randomized (56 on placebo/placebo, 56 on placebo/riluzole and 38 on riluzole/riluzole) to have at least 80% power to detect a weighted mean difference of ~3.0 points (2.5 points in phase 1 and 4 points in phase 2; standard deviation (SD) assumed to be 7.8) between riluzole and placebo at two-sided alpha level of 0.05. We assumed a drop-out rate of 10% and placebo response rate of 20% in phase 1, while a 15% drop-out rate was anticipated in phase 2.
Results
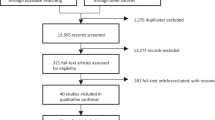
Two hundred forty-three individuals provided informed consent, of which 129 were assigned to Group A and 114 were assigned to Group B, based on the absence or presence of a concurrent antidepressant medication, respectively. Seventy-two patients in Group A began the 8-week prospective SSRI trial, of which 39 completed and continued to fulfill eligibility criteria for randomization. Sixty-five patients from Group B met eligibility criteria and underwent randomization (Figure 1). Of the 104 patients randomized and allocated to one of the three treatment groups, 85 patients completed the 8-week double-blind placebo phase.

CONSORT flow diagram of participants in a randomized placebo-controlled trial of adjunctive riluzole in patients with treatment-resistant major depressive disorder.
Baseline clinical and demographic characteristics of the three randomized groups are presented in Table 1. There were no statistically significant differences between groups at baseline. Dropout rates were not significantly different between groups (χ2=2.76, df=2, p=0.30), with a 84% completion rate in the riluzole/riluzole group, 74.4% in the placebo/riluzole group, and 87.5% in the placebo/placebo group.
Primary Outcome
For the change in MADRS score from baseline, the overall test of treatment differences between riluzole and placebo was not statistically significant (χ2=2.76, df=1, p=0.10). Neither of the estimates from the two treatment blocks was statistically significant (Table 2). There were no main effects of site or baseline MADRS score. These results were confirmed with a mixed effect model imputation analysis (χ2=2.50, df=1, p=0.11).
Secondary Outcomes
Consistent with the primary outcome, there were no significant differences in MADRS response rates between riluzole and placebo (z=−0.21, p=0.83), nor significant differences in MADRS remission rates (z=−0.03, p=0.98) (Table 3). None of the secondary efficacy outcomes were statistically significant for the overall test of treatment differences, when correcting for multiple testing, and neither of the estimates from the two blocks were statistically significant. There were no effects of site or baseline scores for any of these measures.
Adverse Events
Headache (28%), fatigue (28%), and body pain (12%) were the most commonly reported AEs in the riluzole/riluzole group, and marked the only AEs reported by >10% of riluzole-treated participants. The placebo/placebo group reported rates of 27.5, 15, and 2.5%, respectively, for these same AEs. There were no serious adverse events during the 8-week double-blind phase. One patient in the prospective sertraline trial was admitted to a psychiatric facility for relapse of alcohol use disorder and was discontinued from the study. There were no cases of treatment-emergent suicidal preparatory acts or suicide attempts, per the C-SSRS. No clinically relevant effects on weight, vital signs, or EKG were observed. No patients exposed to riluzole had abnormal ALT or AST values, defined as greater than five times the upper limit of normal. One patient in the placebo/riluzole group was discontinued from the study at Week 5 due to neutropenia after having received 4 weeks of placebo.
Discussion
In this three-site trial in patients with persistent moderate-to-severe depressive symptoms despite either a prospective or a historical trial of an antidepressant medication, we found that adjunctive riluzole (100 mg/day) did not show antidepressant effects compared to adjunctive placebo. The negative results for the primary outcome measure (MADRS) were affirmed with multiple secondary outcomes, including response and remission rates, clinician global ratings, and patient-reported measures. Patients in the riluzole group experienced transient side effects consistent with previous reports and clinical experience.
Our results contrast with a recent 6-week placebo-controlled trial of inpatients with non-resistant MDD, in which riluzole (50 mg BID) in combination with citalopram exerted superior antidepressant effects compared with the combination of citalopram and placebo (Salardini et al, 2016), with significant drug-placebo differences observed by week 2. While the trials are not directly comparable because of design differences, these results support the duration of treatment blocks (4 weeks) in our study. Our study contradicts positive open-label reports of riluzole monotherapy (Zarate et al, 2004) and adjunctive therapy (Sanacora et al, 2007) in MDD. In the study most similar in design to the current trial, we previously reported a 40 and 30% response and remission rate, respectively, with a fixed dose of riluzole (50 mg BID) as adjunctive therapy to SSRIs (Sanacora et al, 2007).
The two-stage SPCD design provided sufficient power to detect a moderate effect of riluzole with a substantially lower number of patients than a conventional parallel-group add-on study and may mitigate the placebo response rate. The SPCD is designed such that signal detection is enhanced in the second block of the randomized phase, where only non-responders to placebo in the first block are included in the analyses of the second block. However, riluzole effects among non-responders to placebo in phase 2 were opposite to the expected direction (a MADRS mean±SD change=0.84±5.79 in the placebo/riluzole group vs 3.87±6.49 in the placebo/placebo group). Similarly in phase 1, improvement in MADRS scores on riluzole was numerically less than on placebo in the riluzole/riluzole group (3.20±3.86) compared to 4.83±7.85 in the placebo/placebo and 5.77±7.83 in the placebo/riluzole group. Thus, although we fell short of our recruitment target (104 subjects were randomized while 150 were required according to the original power calculations), the lack of significant findings is not due to insufficient power, since there was no indication that riluzole improved outcome greater than placebo in the two 4-week periods.
The neurobiological mechanisms underlying riluzole’s activity remain incompletely understood despite its use in ALS and neurodegenerative disorders for over two decades. Riluzole reversed cellular, metabolic, and behavioral alterations associated with chronic stress, in part through stimulation of EAAT2 expression, enhancement of glial cell metabolism, and brain-derived neurotrophic factor (BDNF) signaling (Banasr et al, 2010). Longer-term riluzole treatment, over a period of 17 weeks, was recently reported to improve memory performance in aged rats, potentially through an increase in EAAT2 expression in hippocampus (Pereira et al, 2017); clinically, riluzole enhanced expression of the neuronal metabolite N-acetyl-aspartate (NAA) in hippocampus in patients with GAD, most prominently in those with favorable responses (Abdallah et al, 2013). It is unknown how these purported mechanisms translate into intervention for chronic and recurrent depressive disorders. Notably among the secondary outcomes of our study, riluzole did not show benefit on the CPFQ, a patient-reported measure of cognition and sharpness/mental acuity. It has been hypothesized that the efficacy of glutamate-based pharmacotherapies may vary as a function of illness duration (early vs late in disease) or medication history, as described for a novel glutamatergic therapy in schizophrenia (Kinon et al, 2015).
Notable strengths of the study include the modest attrition rate and relatively low placebo response rate. Placebo response rates were lower than expected for phase 1 (15% for placebo/placebo group and 12.8% for placebo/riluzole group) and, of course, for phase 2, where the degree of change on the MADRS was 20% lower than the one observed in phase 1. The inclusion of patients with both prospective and historical inadequate response to antidepressant medication enhanced the study’s generalizability. The absence of an efficacy signal for the patient-reported outcomes along with the clinician-administered MADRS bolsters the confidence of the negative results.
There were several limitations of our trial. We used a fixed dose of riluzole (100 mg/day), which is the standard dosing regimen used in ALS. However, dose-dependent effects of riluzole were found for the expression of BDNF and GLT1 (Gourley et al, 2012), and the positive open-label monotherapy trial in MDD (Zarate et al, 2004) reported a considerably higher mean daily dose of 166 mg, raising the question of whether higher doses in the current trial would have been effective. Second, we did not collect riluzole blood levels. However, an analysis of riluzole blood serum levels in a clinical trial in pediatric OCD conducted at the National Institute of Mental Health did not show any relationship of levels and either beneficial or adverse effects (P. Grant, personal communication). Third, riluzole has 60% absolute bioavailability and is a substrate of P-glycoprotein, responsible for efflux of drug out of the brain (Milane et al, 2007). It is unknown whether individual differences in brain bioavailability impacted outcomes due to pharmacokinetic interactions at the level of the blood–brain barrier. Finally, the small sample size did not permit examination of moderators and trajectories of response over time.
In conclusion, treatment-resistant patients in a major depressive episode did not show an antidepressant response to a fixed dose of riluzole. Future research of glutamate modulating agents should account for individual differences in drug metabolism and consider alternative dosing and trial duration strategies. Identifying markers of target engagement are also essential for progress in this area.
Funding and disclosure
This work was supported by the National Institute of Mental Health (R01MH085055, R01MH085054, R01MH085050). This work was also supported with resources and the use of facilities at the Michael E. DeBakey VA Medical Center, Houston, TX, the Connecticut Department of Mental Health and Addiction Services, and the West Haven VA PTSD Center. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the National Institutes of Health, Department of Veterans Affairs or the United States government. Sanofi S.A. donated a supply of riluzole and matching placebo for the study.
In the past three years, Dr Mathew has received funding from the National Institute of Mental Health and Janssen Research & Development and has received consulting fees from Acadia, Alkermes, Allergan, Cerecor, Naurex, Otsuka, Teva, and Valeant. He receives support from the Johnson Family Chair of Research at Baylor College of Medicine.
Dr Sanacora holds shares in BioHaven Pharmaceuticals Holding Company and is a co-inventor on a US patent (#8 788 979) that is related to the use of glutamate modulating medications in the treatment of neuropsychiatric disorders held by Yale University. In the past 3 years he has received consulting fees from Allergan, Alkermes, AstraZeneca, BioHaven Pharmaceuticals, Hoffman La-Roche, Janssen, Merck, Naurex, Sage Pharmaceuticals, Servier Pharmaceuticals, Taisho Pharmaceuticals, Takeda, Teva, Valeant, and Vistagen Therapeutics. In addition, he has received research contracts from AstraZeneca, Bristol-Myers Squibb, Eli Lilly& Co., Johnson&Johnson, Hoffman La-Roche, Merck, Naurex, and Servier over the past 24 months.
Dr Fava’s lifetime disclosures can be viewed on line at: http://mghcme.org/faculty/faculty-detail/maurizio_fava.
Research Support: Abbott Laboratories; Acadia Pharmaceuticals; Alkermes; American Cyanamid;Aspect Medical Systems; AstraZeneca; Avanir Pharmaceuticals; AXSOME Therapeutics; BioResearch; BrainCells; Bristol-Myers Squibb; CeNeRx BioPharma; Cephalon; Cerecor; Clintara; Covance; Covidien; Eli Lilly and Company;EnVivo Pharmaceuticals; Euthymics Bioscience; Forest Pharmaceuticals; FORUM Pharmaceuticals; Ganeden Biotech; GlaxoSmithKline; Harvard Clinical Research Institute; Hoffman-LaRoche; Icon Clinical Research; i3 Innovus/Ingenix; Janssen R&D; Jed Foundation; Johnson & Johnson Pharmaceutical Research & Development; Lichtwer Pharma GmbH; Lorex Pharmaceuticals; Lundbeck; MedAvante; Methylation Sciences; National Alliance for Research on Schizophrenia & Depression (NARSAD); National Center for Complementary and Alternative Medicine (NCCAM);National Coordinating Center for Integrated Medicine (NiiCM); National Institute of Drug Abuse (NIDA); National Institute of Mental Health (NIMH); Neuralstem; NeuroRx; Novartis AG; Organon Pharmaceuticals; PamLab.; Pfizer; Pharmacia-Upjohn; Pharmaceutical Research Associates.; Pharmavite;PharmoRx Therapeutics; Photothera; Reckitt Benckiser; Roche Pharmaceuticals; RCT Logic (formerly Clinical Trials Solutions); Sanofi-Aventis US; Shire; Solvay Pharmaceuticals; Stanley Medical Research Institute (SMRI); Synthelabo; Takeda Pharmaceuticals;Tal Medical; VistaGen); Wyeth-Ayerst Laboratories.
Advisory Board/Consultant: Abbott Laboratories; Acadia; Affectis Pharmaceuticals AG; Alkermes; Amarin Pharma; Aspect Medical Systems; AstraZeneca; Auspex Pharmaceuticals; Avanir Pharmaceuticals; AXSOME Therapeutics; Bayer AG; Best Practice Project Management; Biogen; BioMarin Pharmaceuticals; Biovail Corporation; BrainCells; Bristol-Myers Squibb; CeNeRx BioPharma; Cephalon; Cerecor; CNS Response; Compellis Pharmaceuticals; Cypress Pharmaceutical; DiagnoSearch Life Sciences (P); Dinippon Sumitomo Pharma; Dov Pharmaceuticals; Edgemont Pharmaceuticals; Eisai; Eli Lilly and Company; EnVivo Pharmaceuticals; ePharmaSolutions; EPIX Pharmaceuticals; Euthymics Bioscience; Fabre-Kramer Pharmaceuticals; Forest Pharmaceuticals; Forum Pharmaceuticals; GenOmind; GlaxoSmithKline; Grunenthal GmbH; Indivior; i3 Innovus/Ingenis; Intracellular; Janssen Pharmaceutica; Jazz Pharmaceuticals; Johnson & Johnson Pharmaceutical Research & Development; Knoll Pharmaceuticals Corp.; Labopharm; Lorex Pharmaceuticals; Lundbeck; MedAvante; Merck & Co.; MSI Methylation Sciences; Naurex; Nestle Health Sciences; Neuralstem; Neuronetics; NextWave Pharmaceuticals; Novartis AG;Nutrition 21; Orexigen Therapeutics; Organon Pharmaceuticals; Osmotica; Otsuka Pharmaceuticals; Pamlab; Pfizer; PharmaStar; Pharmavite.; PharmoRx Therapeutics; Precision Human Biolaboratory; Prexa Pharmaceuticals; PPD; Puretech Ventures; PsychoGenics; Psylin Neurosciences; RCT Logic (formerly Clinical Trials Solutions); Rexahn Pharmaceuticals; Ridge Diagnostics; Roche; Sanofi-Aventis US; Sepracor; Servier Laboratories; Schering-Plough Corporation; Shenox Pharmaceuticals; Solvay Pharmaceuticals; Somaxon Pharmaceuticals; Somerset Pharmaceuticals; Sunovion Pharmaceuticals; Supernus Pharmaceuticals; Synthelabo; Taisho Pharmaceutical; Takeda Pharmaceutical Company Limited; Tal Medical; Tetragenex Pharmaceuticals; TransForm Pharmaceuticals; Transcept Pharmaceuticals; Vanda Pharmaceuticals; VistaGen.
Speaking/Publishing: Adamed, Co; Advanced Meeting Partners; American Psychiatric Association; American Society of Clinical Psychopharmacology; AstraZeneca; Belvoir Media Group; Boehringer Ingelheim GmbH; Bristol-Myers Squibb; Cephalon; CME Institute/Physicians Postgraduate Press; Eli Lilly and Company; Forest Pharmaceuticals; GlaxoSmithKline; Imedex; MGH Psychiatry Academy/Primedia; MGH Psychiatry Academy/Reed Elsevier; Novartis AG; Organon Pharmaceuticals; Pfizer; PharmaStar; United BioSource,Corp.; Wyeth-Ayerst Laboratories.
Equity Holdings: Compellis; PsyBrain.
Royalty/patent, other income: Patents for Sequential Parallel Comparison Design (SPCD), licensed by MGH to Pharmaceutical Product Development (PPD); and patent application for a combination of Ketamine plus Scopolamine in Major Depressive Disorder (MDD), licensed by MGH to Biohaven.
Dr Gueorguieva discloses consulting fees for Palo Alto Health Sciences and Mathematica Policy Research and a provisional patent submission by Yale University: Chekroud, AM., Gueorguieva, R., & Krystal, JH. ‘Treatment Selection for Major Depressive Disorder’ [filing date 3rd June 2016, USPTO docket number Y0087.70116US00]. The remaining authors declare no potential conflict of interest.
References
Abdallah CG, Coplan JD, Jackowski A, Sato JR, Mao X, Shungu DC et al (2013). A pilot study of hippocampal volume and N-acetylaspartate (NAA) as response biomarkers in riluzole-treated patients with GAD. Eur Neuropsychopharmacol 23: 276–284.
Abdallah CG, Jiang L, De Feyter HM, Fasula M, Krystal JH, Rothman DL et al (2014). Glutamate metabolism in major depressive disorder. Am J Psychiatry 171: 1320–1327.
Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, Behar KL et al (2010). Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry 15: 501–511.
Brennan BP, Hudson JI, Jensen JE, McCarthy J, Roberts JL, Prescot AP et al (2010). Rapid enhancement of glutamatergic neurotransmission in bipolar depression following treatment with riluzole. Neuropsychopharmacology 35: 834–846.
Caddy C, Amit BH, McCloud TL, Rendell JM, Furukawa TA, McShane R et al (2015). Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev 9: CD011612.
Chandler GM, Iosifescu DV, Pollack MH, Targum SD, Fava M (2010). Validation of the Massachusetts General Hospital Antidepressant Treatment History Questionnaire (ATRQ). CNS Neurosci Ther 16: 322–325.
Doble A (1996). The pharmacology and mechanism of action of riluzole. Neurology 47: S233–S241.
Du J, Suzuki K, Wei Y, Wang Y, Blumenthal R, Chen Z et al (2007). The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology 32: 793–802.
Duman RS, Aghajanian GK, Sanacora G, Krystal JH (2016). Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med 22: 238–249.
Fava M, Evins AE, Dorer DJ, Schoenfeld DA (2003). The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychother Psychosom 72: 115–127.
Fava M, Iosifescu DV, Pedrelli P, Baer L (2009). Reliability and validity of the Massachusetts General Hospital Cognitive and Physical Functioning Questionnaire. Psychother Psychosom 78: 91–97.
Fava M, Memisoglu A, Thase ME, Bodkin JA, Trivedi MH, de Somer M et al (2016). Opioid modulation with buprenorphine/samidorphan as adjunctive treatment for inadequate response to antidepressants: a randomized double-blind placebo-controlledtrial. Am J Psychiatry 173: 499–508.
First M, Williams J, Spitzer R, Gibbon M (2007)Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Clinical Trials Version (SCID-CT). Biometrics Research, New York State Psychiatric Institute: New York, USA.
Frizzo ME, Dall’Onder LP, Dalcin KB, Souza DO (2004). Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol 24: 123–128.
Gourley SL, Espitia JW, Sanacora G, Taylor JR (2012). Antidepressant-like properties of oral riluzole and utility of incentive disengagement models of depression in mice. Psychopharmacology 219: 805–814.
Ibrahim L, Diazgranados N, Franco-Chaves J, Brutsche N, Henter ID, Kronstein P et al (2012). Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: Results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology 37: 1526–1533.
Jaso BA, Niciu MJ, Iadarola ND, Lally N, Richards EM, Park M et al (2017). Therapeutic modulation of glutamate receptors in major depressive disorder. Curr Neuropharmacol 15: 57–70.
Kinon BJ, Millen BA, Zhang L, McKinzie DL (2015). Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry 78: 754–762.
Levine J, Schooler NR (1986). SAFTEE: a technique for the systematic assessment of side effects in clinicial trials. Psychopharmacol Bull 22: 343–381.
Mathew SJ, Amiel JM, Coplan JD, Fitterling HA, Sackeim HA, Gorman JM (2005). Open-label trial of riluzole in generalized anxiety disorder. Am J Psychiatry 162: 2379–2381.
Mathew SJ, Murrough JW, aan het Rot M, Collins KA, Reich DL, Charney DS (2010). Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: A pilot randomized, placebo-controlled continuation trial. Int J Neuropsychopharmacol 13: 71–82.
Montgomery SA, Asberg M (1979). A new depression scale designed to be sensitive to change. Br J Psychiatry 134: 382–389.
Milane A, Fernandez C, Vautier S, Bensimon G, Meininger V, Farinotti R (2007). Minocycline and riluzole brain disposition: interactions with p-glycoprotein at the blood-brain barrier. J Neurochem 103 (1): 164–173.
Newport J, Carpenter LL, McDonald WM, Potash JB, Tohen M, Nemeroff CB (2015). Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry 172: 950–966.
Niciu MJ, Luckenbaugh DA, Ionescu DF, Richards EM, Vande Voort JL, Ballard ED et al (2014). Riluzole likely lacks antidepressant efficacy in ketamine non-responders. J Psychiatr Res 58: 197–199.
Papakostas GI, Ionescu DF (2015). Towards new mechanisms: an update on therapeutics for treatment-resistant major depressive disorder. Mol Psychiatry 20: 1142–2250.
Pereira AC, Gray JD, Kogan JF, Davidson RL, Rubin TG, Okamoto M et al (2017). Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol Psychiatry 22: 296–305.
Pittenger C, Coric V, Banasr M, Bloch M, Krystal JH, Sanacora G (2008). Riluzole in the treatment of mood and anxiety disorders. CNS Drugs 22: 761–786.
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA et al (2011). The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168: 1266–1277.
Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH (1996). The Inventory of Depressive Symptomatology (IDS): Psychometric properties. Psychol Med 26: 477–486.
Salardini E, Zeinoddini A, Mohammadinejad P, Khodaie-Ardakani MR, Zahraei N, Zeinoddini A et al (2016). Riluzole combination therapy for moderate-to-severe major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Psychiatr Res 75: 24–30.
Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V et al (2007). Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry 61: 822–825.
Tamura RN, Huang X (2007). An examination of the efficiency of the sequential parallel design in psychiatric clinical trials. Clin Trials 4: 309–317.
Türck P, Frizzo ME (2015). Riluzole stimulates BDNF release from human platelets. Biomed Res Int 2015: 189307.
Zarate CA Jr, Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D et al (2004). An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry 161: 171–174.
Zarate CA Jr, Quiroz JA, Singh JB, Denicoff KD, De Jesus G, Luckenbaugh DA et al (2005). An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry 57: 430–432.
Acknowledgements
We thank the members of the study Steering Committee: Thomas Kosten, John H. Krystal, Andrew Nierenberg, Carlos A. Zarate, Jr., and the Data Safety and Monitoring Board of the NIMH for oversight of the study. We also acknowledge the following individuals for their contributions: BCM—Syed Iqbal, Jeremy Joseph, Kemp Knott, Shirali Patel, Alex Proctor; Yale—Elizabeth Cooper, Lisa Roach, Madonna Fasula, Wendol Williams, Samuel Wilkinson, Jane Wyniri.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Mathew, S., Gueorguieva, R., Brandt, C. et al. A Randomized, Double-Blind, Placebo-Controlled, Sequential Parallel Comparison Design Trial of Adjunctive Riluzole for Treatment-Resistant Major Depressive Disorder. Neuropsychopharmacol. 42, 2567–2574 (2017). https://doi.org/10.1038/npp.2017.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2017.106
This article is cited by
-
Randomized trial of ketamine masked by surgical anesthesia in patients with depression
Nature Mental Health (2023)
-
Pharmacological targeting of cognitive impairment in depression: recent developments and challenges in human clinical research
Translational Psychiatry (2022)
-
High-fat diet induces depression-like phenotype via astrocyte-mediated hyperactivation of ventral hippocampal glutamatergic afferents to the nucleus accumbens
Molecular Psychiatry (2022)
-
Update on GPCR-based targets for the development of novel antidepressants
Molecular Psychiatry (2022)
-
The stressed synapse 2.0: pathophysiological mechanisms in stress-related neuropsychiatric disorders
Nature Reviews Neuroscience (2022)
