
Abstract
The consumption of caffeine modulates working and reference memory through the antagonism of adenosine A2A receptors (A2ARs) controlling synaptic plasticity processes in hippocampal excitatory synapses. Fear memory essentially involves plastic changes in amygdala circuits. However, it is unknown if A2ARs in the amygdala regulate synaptic plasticity and fear memory. We report that A2ARs in the amygdala are enriched in synapses and located to glutamatergic synapses, where they selectively control synaptic plasticity rather than synaptic transmission at a major afferent pathway to the amygdala. Notably, the downregulation of A2ARs selectively in the basolateral complex of the amygdala, using a lentivirus with a silencing shRNA (small hairpin RNA targeting A2AR (shA2AR)), impaired fear acquisition as well as Pavlovian fear retrieval. This is probably associated with the upregulation and gain of function of A2ARs in the amygdala after fear acquisition. The importance of A2ARs to control fear memory was further confirmed by the ability of SCH58261 (0.1 mg/kg; A2AR antagonist), caffeine (5 mg/kg), but not DPCPX (0.5 mg/kg; A1R antagonist), treatment for 7 days before fear conditioning onwards, to attenuate the retrieval of context fear after 24–48 h and after 7–8 days. These results demonstrate that amygdala A2ARs control fear memory and the underlying process of synaptic plasticity in this brain region. This provides a neurophysiological basis for the association between A2AR polymorphisms and phobia or panic attacks in humans and prompts a therapeutic interest in A2ARs to manage fear-related pathologies.
Similar content being viewed by others
Introduction
The encoding of fear-related memory is well established to involve abnormal plastic changes of information processing in amygdala circuits (Johansen et al, 2011; Mahan and Ressler, 2012). In other brain regions, synaptic plasticity is controlled by the adenosine neuromodulation system (Fredholm et al, 2005), which involves a coordinated action of inhibitory A1 receptors (A1Rs) and facilitatory A2A receptors (A2ARs) to fine tune brain neurotransmission (Cunha, 2008). In hippocampal circuits, A2ARs are found in synapses (Rebola et al, 2005a), namely in glutamatergic synapses (Rebola et al, 2005b), and are selectively engaged to control synaptic plasticity (Rebola et al, 2008; Costenla et al, 2011). The importance of this modulation system is best heralded by the observation that the overactivation of hippocampal A2ARs is necessary and sufficient to trigger spatial memory dysfunction (Li et al, 2015a; Pagnussat et al, 2015). Furthermore, conditions associated with memory deterioration trigger an upregulation of A2ARs in the hippocampus leading to abnormal synaptic plasticity (Costenla et al, 2011; Kaster et al, 2015), and A2AR blockade prevent memory impairment in conditions such as stress, aging, or Alzheimer’s disease (eg Batalha et al, 2013; Laurent et al, 2016; Oor et al, 2015; Prediger et al, 2005), an effect mimicked by caffeine (a nonselective adenosine receptor antagonist) both in animal models and in humans (reviewed in Cunha and Agostinho, 2010; Chen, 2014).
Interestingly, the acute administration of caffeine disrupts fear memory (Corodimas et al, 2000) and A2AR polymorphisms are associated with panic disorders (Deckert et al, 1998; Hamilton et al, 2004), but it is unknown whether A2ARs control fear memory and synaptic plasticity in amygdala circuits. Thus, we now explored the involvement of A2ARs in the control of synaptic plasticity in the amygdala and their possible role in the control of fear memory.
Materials and methods
For detailed Materials and Methods, see ‘Supplementary Methods’.
Mice and Drug Treatments
All experiments were approved by the Ethical Committee of the Center for Neuroscience and Cell Biology (Orbea 78-2013). Male C57Bl/6 mice (2–3 months) were daily intraperitoneally injected either with caffeine (5 mg/kg; Sigma, Sintra, Portugal; a dose preventing memory deficits without altering locomotion; Prediger et al, 2005) or with supramaximal but selective doses of the A1R antagonist DPCPX (1,3-dipropyl-8-cyclopenthylxanthine, 0.5 mg/kg; Tocris, Bristol, UK), or the A2AR antagonist SCH58261 (5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine; 0.1 mg/kg; Tocris), which are devoid of locomotor or nociceptive effects (Bastia et al, 2002), but effectively control neuronal dysfunction (Nakamura et al, 2002; Kaster et al, 2015). Drug treatments started 10 days before behavioral testing until the mice were killed.
Density and Localization of Adenosine Receptors
Western blot analysis with goat or mouse anti-A2AR antibodies (1 : 1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA or Millipore, Madrid, Spain, respectively), which selectivity was confirmed by the lack of signal in A2AR-knockout (KO) mice (Rebola et al, 2005a), or receptor binding analysis with 3 nM of 3H-SCH58261 (specific activity of 77 Ci/mmol; prepared by GE Healthcare and offered by Dr E Ongini, Schering-Plough, Italy) or 6 nM of 3H-DPCPX (specific activity of 109.0 Ci/mmol; DuPont NEN, Boston, MA, USA) was carried out in total membranes and membranes from synaptosomes (Costenla et al, 2011; Kaster et al, 2015), whereas the immunocytochemical detection of A2ARs in glutamatergic nerve terminals was carried out as described previously (Costenla et al, 2011; Rebola et al, 2005b), using goat anti-A2AR (1 : 200; Santa Cruz Biotechnology) and guinea-pig anti-vesicular glutamate transporter type 1 (vGluT1; 1 : 1000, Chemicon, Temecula, CA, USA) antibodies.
Electrophysiological Recordings in Corticoamygdala Synapses
Electrophysiological recordings in brain slices were carried out as described previously (Costenla et al, 2011) by extracellularly recording population spikes in the lateral nuclei of the amygdala upon stimulation of the external capsule (EC). Long-term potentiation (LTP) was induced with three pulses of 100 Hz delivered with an interval of 5 s.
Generation and Administration of Lentiviral Vectors
An shA2AR (nts 419–437; see Figure 3) was inserted into a lentivector together with an enhanced green fluorescent protein (EGFP) reporter gene, as described previously (Alves et al, 2008). A hairpin designed to target the coding region of red fluorescent protein (nts 22–41) was used as an internal control (sh-control). These lentivectors (1 μl at 750 000 ng of p24 antigen per ml) were stereotaxically delivered at an infusion rate of 0.1 μl/min in the following coordinates: anteroposterior: −1.1 mm; lateral: ±2.8 mm; ventral: −4.6 mm, and the injection site was confirmed on killing of the mice. A2AR downregulation was probed after 3 weeks by qPCR.
Auditory Fear Conditioning
Fear conditioning was performed as described previously (Goosens et al, 2000) in context A with three presentations of an auditory conditioned stimulus (CS; 80 dB for 30 s at 4 kHz) paired with a footshock unconditioned stimulus (US; 0.3 mA for 2 s, delivered 28 s after the beginning of CS) with a 60 s intertrial interval. At days 2 and 8, mice were returned to context A to test their freezing behavior for 8 min. At days 3 and 9, mice were placed in a different chamber (context B), the CS was presented after 3 min, and the freezing behavior was measured for 8 min.
Other Behavioral Analyses
The spontaneous locomotion of mice was measured 1 day after the fear conditioning protocols, in an open field test as described previously (Wei et al, 2014; Kaster et al, 2015). Nociceptive responses were evaluated 1 day after the open field test by using the hot-plate test (Le Bars et al, 2001).
Statistical Analysis
Results are presented as mean±SEM. Behavioral data were analyzed with a one-way ANOVA followed by a Tukey’s multiple comparison post hoc test or with a two-way ANOVA followed by Bonferroni post hoc tests, when more than one variable and condition (eg, genotype and time) were analyzed. Binding, western blot, and electrophysiological data were analyzed with unpaired Student’s t tests. The significance level was 95%.
Results
A2ARs are Localized to Glutamatergic Terminals in the Amygdala
We first probed whether A2ARs were located in glutamatergic synapses in the amygdala as occurs in the hippocampus (Rebola et al, 2005b). As shown in Figure 1a, the binding density of 3H-SCH58261 was larger (n=6, p<0.05) in synaptosomal membranes (30.90±3.47 fmol/mg protein) than in total membranes from the amygdala (20.75±2.13 fmol/mg protein). Furthermore, the density of A2ARs as evaluated by western blot was also 18.6±5.2% larger (n=6, p<0.05) in synaptosomal compared with that in total membranes from the amygdala (Figure 1b), showing that A2ARs are indeed enriched in amygdala synapses. A double immunocytochemical labeling of A2ARs and of a glutamatergic marker (vGluT1) in amygdala nerve terminals (Figure 1c) revealed that 40.4±3.5% (n=5) of the vGluT1-positive terminals were endowed with A2ARs (arrows indicate regions of overlap). Overall, these findings show that A2ARs are present in the amygdala, and found in glutamatergic synapses.

Adenosine A2A receptors (A2ARs) are enriched in synapses and located in glutamatergic synapses in the amygdala. The comparison of the binding of a supramaximal concentration of a selective A2AR antagonist, 3H-SCH58261 (3H-5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine, 3 nM) (a) or of the immunoreactivity of A2ARs (b) was larger in membranes from synaptosomes (purified synapses) than in total membranes of the amygdala. Data are mean±SEM of six mice; *p<0.05, unpaired Student’s t-test. Representative photographs of an immunocytochemical analysis of purified nerve terminals from the amygdala (c), which revealed a colocalization (yellow) of A2AR immunoreactivity (green) in nerve terminals immunopositive for vGluT1 (vesicular glutamate transporters type 1, a marker of glutamatergic nerve terminals; red), as indicated by the arrows (scale bar: 50 μm). This experiment is representative of five experiments with similar results. The selectivity of the A2AR antibody was confirmed by the lack of western blot and immunocytochemical signal in synaptosomes from A2AR knockout mice. A full color version of this figure is available at the Neuropsychopharmacology journal online.
A2ARs Control Synaptic Plasticity in the Amygdala
Changes in synaptic transmission in the amygdala are thought to underlie the acquisition and expression of long-term fear memories (Blair et al, 2001; Goosens and Maren, 2001; Johansen et al, 2011). Thus, we next tested the ability of A2ARs to control synaptic plasticity in excitatory synapses in the lateral amygdala, one of the primary sites of CS–US (conditional-unconditional stimuli) convergence during fear conditioning, upon stimulation of afferents in the EC in slices. The bath superfusion with SCH58261 (50 nM) failed to modify both basal neurotransmission (Figure 2a) and short-term plasticity (ie, paired pulse stimulation; Figure 2b). Instead, SCH58261 (50 nM) selectively impaired synaptic plasticity, shown by the reduction of LTP amplitude (128.8±6.8% potentiation over baseline in the presence of SCH58261 compared with 171.0±13.3% in its absence, n=6, p<0.05) (Figure 2c). Another selective A2AR antagonist, ZM241385 (50 nM), also reduced LTP amplitude (119.3±12.9%) compared with control (160.2±12.1%, n=5, p<0.05) (Figure 2e).

Adenosine A2A receptors (A2ARs) selectively control long-term synaptic plasticity in the amygdala. Upon extracellular recording of population spike responses in the lateral amygdala triggered by stimulation of the external capsula, the selective A2AR antagonist SCH58261 (amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine, 50 nM, in pink) did not change either basal synaptic transmission (a) or short-term plasticity evaluated as the paired pulse ratio with different interpulse intervals from 10 to 100 ms (b); in contrast, SCH58261 decreased the long-term potentiation (LTP) of the population spike responses triggered by a high-frequency stimulation (HFS) train (three pulses of 100 Hz delivered with an interval of 5 s), as shown in the time course (c) or in the pair of superimposed population spike responses before the train (baseline) and 60 min after HFS (d). Similarly, another chemically distinct but equally selective A2AR antagonist ZM241385 (4-(2-[7-amino-2-(2-furyl{1,2,4}-triazolo{2,3-a{1,3,5}triazin-5-yl-aminoethyl}])phenol, 50 nM, in purple) also decreased LTP amplitude compared with its absence (control, black symbols) (e). Data are mean±SEM of six to nine mice per group when testing SCH58261 and n=5–6 when testing ZM241385. *p<0.05 compared with the respective control (ie, lack of drugs, open symbols), unpaired Student’s t-test. A full color version of this figure is available at the Neuropsychopharmacology journal online.
Downregulation of A2ARs in the Basolateral Amygdala Impairs Long-Term Fear Memory
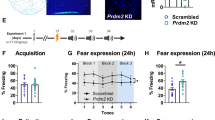
As the basolateral complex of the amygdala is a key circuit for the expression of fear memories through alterations of synaptic plasticity (Blair et al, 2001; Goosens and Maren, 2001; Johansen et al, 2011), we tested if the selective elimination of A2ARs in the amygdala was sufficient to affect conditioned fear. We developed lentivectors encoding shRNAs to selectively neutralize A2ARs (shA2AR) while simultaneously expressing EGFP (Figure 3a). These lentivectors have neuronal tropism and limited spread in the brain parenchyma (Lundberg et al, 2008). The lentivirus infected only neurons (colocalization with NeuN and no colocalization with GFAP, data not shown, see Viana da Silva et al, 2016), and covered the majority (71.0±8.8%, n=4) of the basal nucleus and spread only to the lateral nucleus of the amygdala (7.56±1.03%, n=4), as judged by the superimposable staining of EGFP (Figure 3b) and of acetylcholinesterase (Figure 3c), which is abundantly expressed in these nuclei (Berdel et al, 1996). The dissection of the amygdala 4 weeks after transfection revealed a near 70% decrease of A2AR mRNA levels compared with amygdala tissue collected from sh-control mice (Figure 3d). An immunohistochemical confirmation of A2AR protein levels could not be performed because A2AR density in the amygdala is below the threshold of detection, although the striatal injection of the lentivector downregulated A2AR protein by 55.4±4.9% upon transfection of 27.2±2.7% striatal neurons (n=4); instead, as previously shown in the hippocampus (Viana da Silva et al, 2016), we functionally confirmed the efficiency of shA2AR lentivectors to downregulate amygdala A2ARs by showing that shA2AR treatment abrogated A2AR modulation of amygdala LTP: as shown in Figure 3e, SCH58261 was devoid of effects on LTP amplitude in slices from shA2AR mice, but decreased LTP in slices from sh-control mice (Figure 3f); this enables using shA2AR lentivectors to probe the involvement of amygdala A2ARs in fear memory.

Lentivectors expressing a short hairpin RNA (shRNA) targeting adenosine A2A receptors (A2ARs) effectively downregulate A2ARs and their bilateral injection in the basolateral complex of the amygdala decreases conditioned fear acquisition and expression. Lentivector constructs containing a sequence to neutralize A2ARs (shA2AR) together with enhanced green fluorescent protein (EGFP) reporter (a) effectively transduced the basolateral complex, as gauged by the superimposition of EGFP labeling (b) with acetylcholinesterase staining (c), a marker of the basolateral complex (scale bar: 200 μm). When analyzed 4 weeks after shA2AR transduction in the amygdala (71.0±8.8% of area transfected), there was a near 70% decrease of A2AR mRNA levels in the amygdala (d). The data are mean±SEM of three of four mice; *p<0.05 with a Student’s t-test. shA2AR caused a nonsignificant reduction (p=0.09, n=3, unpaired Student’s t-test) of the number of glutamatergic nerve terminals (vGluT1 (vesicular glutamate transporter type 1)-immunopositive nerve terminals) endowed with A2ARs in the basolateral amygdala (BLA) (28.96±1.20%) compared with control mice (38.32±4.12%), but this only informs on the number of glutamatergic terminals endowed with A2ARs rather than on the amount of A2ARs in each terminal. The functional efficiency of shA2AR in the amygdala was confirmed by the elimination of the impact of SCH58261 (5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine; 50 nM) on long-term potentiation (LTP) amplitude in slices collected 3 weeks after the injection of shA2AR in the basolateral complex of the amygdala (f), whereas SCH58261 decreased LTP amplitude in slices from animals transfected with lentivectors lacking the silencing shRNA sequence to neutralize A2ARs (shCTR) (e). The data are mean±SEM of four mice per group. (g) shA2AR and control mice displayed similar spontaneous locomotion in the open field. (h) The acquisition of a freezing response (time freezing during the 8 min test) to three repeated presentations of a 30-s tone conditioned stimulus (CS) paired with an unconditioned stimulus (US, 2-s foot-shock) 4 weeks after bilateral injection of shA2AR in the basolateral complex (filled symbols) showed a lower increase of freezing with each successive CS–US paired trial compared with mice treated with a lentivector without the shRNA silencing sequences to neutralize A2ARs (control, open symbols). (i) Context testing 1 or 7 days after fear acquisition showed a lower time of freezing in shA2AR-treated mice compared with sh-control, which displayed a behavior superimposable to that of naïve mice. (j) Tone-induced freezing was also lower in shA2AR-treated mice compared with sh-control when tested 2 days, but not 8 days, after fear conditioning. Data are mean±SEM of five mice per group. *p<0.05 compared with the respective control (open symbols), two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test. A full color version of this figure is available at the Neuropsychopharmacology journal online.
Mice injected in the amygdala with shA2AR had a similar spontaneous locomotion in the open field (Figure 3g) but displayed significantly less freezing during fear conditioning compared with sh-control mice (Figure 3h). ANOVA analysis of the time of freezing confirmed an effect of trials (F2,16=21.22, p<0.0001), of the administration of shA2AR (F1,8=6.61, p<0.05), and a trial × shA2AR interaction (F2,16=7.45, p<0.05). During context re-exposure (Figure 3i), shA2AR-treated mice froze less than control mice either the next day (F1,4=19.87, p<0.05) or 7 days after fear conditioning (F1,4=5.95, p<0.05). When tested for tone-induced fear (Figure 3j), shA2AR-treated mice displayed a lower response at day 3 (F1,4=6.51, p<0.05), and similar responses at day 9 (F1,4=0.99, p=0.39). Notably, shA2AR-treated animals performed similarly to control animals in two tests probing spatial reference memory, namely the two-trial Y-maze and the object displacement test (data not shown).
Adaptive Changes of the Adenosine Neuromodulation System After Fear Conditioning
We next determined whether alterations of amygdala A2AR density and function accompany the plastic changes that occur in amygdala circuits during the implementation of fear memories. The density of A2ARs in amygdala membranes was higher (p<0.0001, n=9) in fear conditioned mice (33.0±4.9 fmol/mg protein) compared with control mice (21.5±2.4 fmol/mg/protein) 2 days after fear conditioning (Figure 4a). The increase was even more pronounced 8 days after fear conditioning (69.4±5.3 fmol/mg protein in fear conditioned mice and 23.3±3.6 fmol/mg in control mice, n=9; p<0.0001) (Figure 4b). In spite of this increased density, the immunohistochemical detection of A2ARs in amygdala sections was still near background (data not shown). A2AR density also increased in other brain regions involved in the encoding of emotional traits, such as the hippocampus (n=9) and ventral striatum (n=8), whereas there was no alteration of A2AR density in regions not directly implicated in encoding fear memories such as the dorsal striatum (n=8) (Figures 4a and b).

Conditioned fear triggers an increased density and a gain of function of adenosine A2A receptors (A2ARs) in the amygdala. The comparison of the binding density of the selective A2AR antagonist 3H-SCH58261 (3H-5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine, 3 nM) to membranes prepared from control mice (open bars) and from fear conditioned mice 2 days (a) or 8 days (b) after fear conditioning (filled bars) revealed an increased A2AR density in the amygdala (Amyg), hippocampus (HIP), and ventral striatum (vSTR), but not in the dorsal striatum (dSTR), of fear-stressed mice. This fear stress-induced upregulation seems selective for A2ARs as there was no similar modification of A1R density, as evaluated by the binding density of the selective A1R antagonist 3H-DPCPX (3H-1,3-dipropyl-8-cyclopenthylxanthine; 6 nM) in membranes from fear-stressed mice 2 days (c) or 8 days (d) after fear conditioning (filled bars) compared with control mice (open bars). Data are mean±SEM of nine mice per group for analysis of A2AR density and n=4–5 mice per group for the analysis of A1R density. *p<0.05 compared with control (open symbols), unpaired Student’s t-test. (e) The blockade of A2ARs with SCH58261 (50 nM) still effectively decreased long-term potentiation (LTP) amplitude recorded extracellularly in the lateral amygdala triggered by a high-frequency stimulation (HFS) train in external capsula of slices collected 8 days after fear conditioning; (f) such an effect that was not observed in slices collected 8 days after fear-stressed mice that were injected bilaterally in the amygdala 3 weeks before with lentivectors expressing a short hairpin RNA (shRNA) to neutralize A2ARs (shA2AR). Data are mean±SEM of four to five mice per group, unpaired Student’s t-test. (g) Comparison of LTP amplitude before and 8 days after induction of fear conditioning. (h) Comparison of the impact on LTP amplitude of the different manipulations of A2ARs before and 8 days after induction of fear conditioning.
We next tested the receptor specificity of these changes by determining whether fear conditioning altered A1R density in different brain regions. As shown in Figure 4c, 2 days after fear acquisition, there was no modification of A1R density in amygdala membranes of fear conditioned compared with control mice (n=5); in contrast, there was an increased density of A1Rs in the hippocampus (n=4, p<0.05) and in the ventral striatum (n=4, p<0.05), and no alteration in the dorsal striatum (n=4) (Figure 4c). These alterations in A1Rs were transient: 8 days after fear conditioning, there was a decreased density of A1Rs in the hippocampus (n=5, p<0.05) and a tendency for a decrease in the amygdala (n=5, p=0.082) and in the ventral striatum (n=5, p=0.061) and no alterations in the dorsal striatum (n=5) (Figure 4d).
To test if this selective upregulation of A2AR in the amygdala was associated with a modified functioning of A2ARs, we tested the ability of A2ARs to control LTP in amygdala slices collected 8 days after fear conditioning. As shown in Figure 4e, SCH58261 decreased (p<0.0001) LTP amplitude (115.3±10.5%, n=5) in slices from fear conditioned mice, whereas LTP amplitude was 187.6±8.3% (n=5) in the absence of SCH58261. A comparison across different experimental groups suggested a tendency (p=0.09; Figure 4g) for a larger LTP amplitude in slices collected 8 days after fear conditioning (187.6±8.3%, n=5) compared with slices from control mice not subject to fear conditioning (166.1±8.8%, n=11), whereas the extent of LTP inhibition upon A2AR blockade in slices from fear conditioned mice (82.5±4.9%, n=5) was larger (p<0.05) compared with that observed in slices from naïve mice (52.4±3.9%, n=6; Figure 4h). This is in agreement with the proposed involvement of synaptic plastic changes in amygdala circuits to encode fear memory (Blair et al, 2001; Goosens and Maren, 2001; Johansen et al, 2011) and further documents a gain of function of A2ARs to control LTP in the amygdala of fear conditioned mice.
A2AR, but not A1R, Blockade Impairs Long-Term Fear Memory Formation
In the last experiment, we sought to determine whether the global pharmacological blockade of A2ARs controlled the acquisition of fear memory and if this was mimicked by caffeine (a nonselective adenosine receptor antagonist that is the most widely consumed psychoactive drug), but not by A1R antagonists. Mice received daily injections either of caffeine (5 mg/kg per day), SCH58261 (0.1 mg/kg per day; A2AR antagonist), or DPCPX (0.5 mg/kg per day; A1R antagonist), prior and throughout auditory fear conditioning. During fear conditioning (Figure 5a), all groups displayed increased freezing with successive CS–US pairings (F2,30=38.9, p<0.0001). The acquisition of fear was similar across all treatments (Figure 5a). This suggests that this subchronic manipulation of A1Rs and/or A2ARs did not affect shock responsiveness, perception, or formation of the CS–US association. Additionally, spontaneous locomotion and nociceptive behavior (n=8–10 vs saline-treated mice, n=11) (Supplementary Figure 1) were not altered by our pharmacological manipulations.

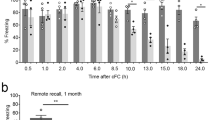
Caffeine and selective adenosine A2A receptor (A2AR), but not A1R, antagonists attenuate the expression of contextual fear. The acquisition of a freezing response (% freezing in the 8 min of test) to three repeated presentations of a 30-s tone conditioned stimulus (CS) paired with an unconditioned stimulus (US, 2-s foot-shock) are shown in (a) in control (vehicle-treated) mice (black open circles) and in mice treated intraperitoneally either with caffeine (5 mg/kg per day; squares) or the A2AR-selective antagonist SCH58261 (amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine, 0.05 mg/kg per day; triangles) or the A1R-selective antagonist DPCPX (1,3-dipropyl-8-cyclopenthylxanthine, 0.1 mg/kg per day, inverted triangles). All mice showed a comparable increase in freezing with each successive CS–US paired trial. (b) Freezing responses to the conditioning context in the absence of the CS were recorded 1 or 7 days later, showing that caffeine and SCH58261, but not DPCPX, treatments decreased context freezing. In contrast, tone CS testing, carried out in a different context, and evaluated either 2 or 8 days after fear conditioning (c) showed that none of the drug treatments significantly affected the percent time of freezing. Data are mean±SEM of 8–11 mice per group. *p<0.05 compared with control (vehicle-treated), one-way analysis of variance (ANOVA) followed by Turkey’s post hoc test. (d) Summary table of the impact of the different manipulation of A2ARs in different brain regions on the acquisition and expression of conditioned fear. (1) This study; (2) Wei et al (2014). A full color version of this figure is available at the Neuropsychopharmacology journal online.
Re-exposure of saline-treated mice to the conditioning context one day after conditioning induced freezing during 35.68±3.19% (n=11) of the 8 min exposure (Figure 5b); this was attenuated by caffeine (23.79±3.04% freezing, F1,152=28.43, p<0.0001) and SCH58261 (22.46±2.59% freezing, F1,152=39.04, p<0.0001), but not by DPCPX (34.92±3.02% freezing, F1,136=0.12, p=0.73). A similar pattern was observed when mice were re-exposed to the same context 7 days after conditioning (day 8) (Figure 5b): control mice froze during 18.43±2.09% of the 8 min exposure (n=11), which was decreased by caffeine (13.02±1.81% freezing, F1,152=11.29, p<0.001) and by SCH58261 (12.26±1.46% freezing, F1,152=16.11, p<0.0001), but not by DPCPX (19.07±1.78% freezing, F1,136=0.14, p=0.71).
As shown in Figure 5c, when mice were placed in a novel context 2 days after fear conditioning (day 3), control mice increased their freezing upon presentation of the CS (10.73±1.36% before CS to 66.65±2.66% after CS, F7,77=11.03, p<0.0001); this was not modified by caffeine (F1,152=1.77, p=0.19) or DPCPX (F1,136=0.0008, p=0.98), but was decreased by SCH58261 (F1,152=5.61, p<0.05). A similar pattern was observed when mice were exposed to the tone in a novel context 8 days after fear conditioning (day 9) (Figure 5c): control mice froze on presentation of CS (from 10.18±1.33 to 63.37±2.72%, F7,77=11.03, p<0.0001) and this was decreased by caffeine (F1,152=4.78, p<0.05) and by SCH58261 (F1,152=4.38, p<0.05), but not by DPCPX (F1,136=0.06, p=0.81).
These data indicate that caffeine and selective A2AR inhibition decreased the expression of contextual fear memory, whereas A1R blockade was devoid of effects. This ability of A2ARs to control fear memory was further tested by comparing wild type (WT) and global A2AR-KO mice. As shown in Supplementary Figure 2, A2AR-KO and WT mice displayed a similar acquisition of fear (F1,48=0.39, p=0.53 for genotype and F2,48=19.15, p<0.0001 for trials). When tested for contextual fear 1 or 7 days after conditioning, A2AR-KO mice froze less than WT mice (F1,128=13.99, p<0.0005 at day 2; F1,128=4.88, p<0.05 at day 8); in contrast, when probed in a novel context for auditory fear memory, there was no significant difference between genotypes either 2 days (F1,128=0.004, p=0.95) or 8 days (F1,128=0.11, p=0.74) after fear conditioning (Supplementary Figure 2).
Discussion
The present study identifies A2ARs as novel key regulators of synaptic plasticity in the amygdala and of fear memory. Thus, the global pharmacological inhibition of A2ARs and the downregulation of A2ARs selectively in the basolateral complex of the amygdala impaired fear memory, in accordance with the ability of A2AR blockade to selectively dampen the amplitude of long-term potentiation in this region.
The expression of contextual fear was decreased by caffeine consumption and by the genetic and pharmacological blockade of A2ARs, but was unaffected upon selective blockade of inhibitory A1Rs; given that caffeine at non-toxic doses mostly targets A1Rs and A2ARs (Fredholm et al, 2005), this indicates that the impact of caffeine consumption on contextual fear memory was likely mediated by the selective antagonism of A2ARs, as previously proposed for other behavioral responses (Cunha and Agostinho, 2010). This effect contrasts with the previously reported disruptive effects of acutely administered higher doses of caffeine (Corodimas et al, 2000), further highlighting the care to use ‘physiological’ doses of caffeine and to use schedules of administrations that mimic caffeine consumption in humans. In parallel, we also observed that A2ARs controlled synaptic plasticity processes in the lateral amygdala, the purported neurophysiological basis of conditioned fear (Johansen et al, 2011; Goosens and Maren, 2001; Blair et al, 2001). Thus, different A2AR antagonists attenuated LTP amplitude in amygdala slices, an effect inexistent upon treatment with shA2AR as well as in global A2AR-KO mice; this also testifies that, although we did not directly quantify the reduction of A2ARs in the amygdala after shA2AR treatment, the achieved downregulation of amygdala A2ARs with shA2AR was sufficient to eliminate A2AR-mediated responses. Notably, A2ARs were selectively engaged to control long-term plastic processes and were devoid of effects on the control of basal synaptic transmission or of short-term plasticity, as occurred in hippocampal (Rebola et al, 2008; Costenla et al, 2011) or striatal synapses (d’Alcantara et al, 2001; Flajolet et al, 2008). This impact on synaptic plasticity is in agreement with the enrichment of A2ARs in synapses within the amygdala and in particular with the localization of A2ARs in glutamatergic synapses, as occurs in other limbic regions such as the hippocampus (Rebola et al, 2005a, 2005b; Costenla et al, 2011). However, our data does not allow distinguishing between possible pre- and postsynaptic effects of A2ARs (Rau et al, 2015) to control LTP, which will need additional analysis using whole-cell patch-clamp recordings. Similarly, additional studies are required to test if ATP-derived adenosine is also the source of adenosine-activating A2ARs and if ATP is released during induction of synaptic plasticity in amygdala synapses, which is currently unknown. This parallel ability of A2ARs to control amygdala synaptic plasticity and fear-related responses also raises the question of disentangling if A2ARs are continuously affecting an abnormal functioning of amygdala circuits, as suggested by the sustained upregulation of A2ARs in the amygdala after fear acquisition, or if instead A2ARs are critically required for a state-dependent shift on exposure to fear, as suggested by the ability of A2ARs to control the emotional status of rodents (Wei et al, 2014; Kaster et al, 2015).
This key role of amygdala A2ARs to control fear memory was directly confirmed by our observation that the selective bilateral downregulation of A2ARs in the amygdala was sufficient to decrease fear memory. Importantly, we defined that this A2AR downregulation occurred in neurons (see Viana da Silva et al, 2016) and abolished the impact of A2ARs on amygdala LTP, but we could not disentangle the relative effect of shA2AR on pre- and postsynaptic A2ARs. However, shA2AR-treated mice displayed a decreased acquisition of conditioned fear and a decreased expression of both contextual and cued fear, whereas the global blockade of A2ARs selectively dampened contextual fear without effects on acquisition and cued fear. This was not due to an effect of caffeine or of selective A2AR antagonists on nociception, as we used a dose of SCH58261 one order of magnitude smaller than the minimum dose previously shown to have no effect on nociception (Bastia et al, 2002). Instead, the different impact of the global A2AR blockade compared with the blockade of A2ARs selectively in the amygdala indicates that the impact of A2ARs on fear memory is unlikely to be restricted to the amygdala, in accordance with the previously reported ability of hippocampal A2ARs to interfere with contextual fear and the opposite effect of amygdala and striatal A2ARs on the control of the expression of fear memory (Wei et al, 2014), as summarized in Figure 5d. In fact, the acquisition and recall of conditioned fear also involves other limbic and neocortical areas in partially redundant circuits (Orsini and Maren, 2012). Similarly, it is possible that the selective deletion of A2ARs in the amygdala might bolster the impact of otherwise less relevant A2ARs in other brain regions, as we have previously observed to occur for the recruitment of striatal DARPP-32 (Shen et al, 2013), behavioral sensitization (Shen et al, 2008), or emotional responses (Wei et al, 2014) using cell-type-selective genetic eliminations of A2ARs. In fact, several studies have dissected the involvement of different brain regions in the processing of contextual and cued fear memory (Orsini and Maren, 2012), albeit the expression of both forms of contextual fear mostly depend on amygdala circuits (Goosens and Maren, 2001). This is heralded by the differential impact on cued and contextual fear memory upon manipulation of different molecular targets (eg, Sui et al, 2006; Burghart and Bauer, 2013) or lesions/inactivation (eg, Duvarci et al, 2009; Baldi et al, 2013) in different brain regions. This apparent different involvement of A2ARs in the amygdala and in other brain regions in the acquisition and expression of contextual fear, which still remains to be detailed, does not undermine the key impact of A2AR blockade in long-term contextual fear, a conclusion of particular importance in view of the prominent role of contextualization in behavioral flexibility and psychopathology (reviewed in Maren et al, 2013).
The relevance of this novel A2AR-mediated control of conditional fear is bolstered by the reported observations that the implementation of fear memory traits is accompanied by an upregulation of A2ARs in the amygdala together with a gain of function of A2ARs controlling amygdala LTP. A2AR upregulation was also detected in other brain regions involved in the processing of emotional information, such as the hippocampus and ventral striatum, and is in agreement with the previous observation that stressful events upregulate A2ARs (Fredholm et al, 2005; Cunha and Agostinho, 2010). Furthermore, A2ARs displayed a gain of function in the control of amygdala LTP after fear stress and the blockade of A2ARs decreased excessive plasticity in the amygdala, as it was previously found to occur in the hippocampus (Costenla et al, 2011) and in the striatum (Li et al, 2015b). This makes A2ARs attractive targets to manage conditions associated with abnormal fear expression, namely upon post-traumatic stress disorders. This is supported by the association between A2AR polymorphisms with phobia and panic attacks (Deckert et al, 1998; Hamilton et al, 2004) and by the observed inverse correlation between caffeine intake and the incidence of depression (Lucas et al, 2011) and suicides (Lucas et al, 2013).
In conclusion, the present study provides combined pharmacological and genetic evidence that A2AR blockade decreases fear memory. The study also identifies the presence of A2ARs in glutamatergic terminals in the amygdala, where they selectively control synaptic plasticity processes that are considered the neurophysiological basis of conditional fear memory. Finally, the observed increased density of amygdala A2ARs and the gain of function of A2ARs to control synaptic plasticity in the lateral amygdala after fear conditioning prompt the provocative novel hypothesis that A2ARs may have a key role in the acquisition and preservation of contextual fear memories. This paves the way to consider A2AR antagonists as novel candidate drugs to manage psychiatric conditions associated with excessive expression of aversive memories such as post-traumatic stress disorders.
Funding and disclosure
RAC is a scientific consultant for the Institute for Scientific Information on Coffee. The other authors declare no conflict of interest.
References
Alves S, Régulier E, Nascimento-Ferreira I, Hassig R, Dufour N, Koeppen A et al (2008). Striatal and nigral pathology in a lentiviral rat model of Machado–Joseph disease. Hum Mol Genet 17: 2071–2083.
Baldi E, Liuzzo A, Bucherelli C (2013). Fimbria–fornix and entorhinal cortex differential contribution to contextual and cued fear conditioning consolidation in rats. Physiol Behav 114–115: 42–48.
Bastia E, Varani K, Monopoli A, Bertorelli R (2002). Effects of A1 and A2A adenosine receptor ligands in mouse acute models of pain. Neurosci Lett 328: 241–244.
Batalha VL, Pego JM, Fontinha BM, Costenla AR, Valadas JS, Baqi Y et al (2013). Adenosine A2A receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol Psychiatry 18: 320–331.
Berdel B, Moryś J, Maciejewska B, Narkiewicz O (1996). Acetylcholinesterase activity as a marker of maturation of the basolateral complex of the amygdaloid body in the rat. Int J Dev Neurosci 14: 543–549.
Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE (2001). Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 8: 229–242.
Burghardt NS, Bauer EP (2013). Acute and chronic effects of selective serotonin reuptake inhibitor treatment on fear conditioning: implications for underlying fear circuits. Neuroscience 247: 253–272.
Chen JF (2014). Adenosine receptor control of cognition in normal and disease. Int Rev Neurobiol 119: 257–307.
Corodimas KP, Pruitt JC, Stieg JM (2000). Acute exposure to caffeine selectively disrupts context conditioning in rats. Psychopharmacology 152: 376–382.
Costenla AR, Diógenes MJ, Canas PM, Rodrigues RJ, Nogueira C, Maroco J et al (2011). Enhanced role of adenosine A2A receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur J Neurosci 34: 12–21.
Cunha RA (2008). Different cellular sources and different roles of adenosine: A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity. Neurochem Int 52: 65–72.
Cunha RA, Agostinho PM (2010). Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. J Alzheimer Dis 20 (Suppl 1): 95–116.
D’Alcantara P, Ledent C, Swillens S, Schiffmann SN (2001). Inactivation of adenosine A2A receptor impairs long term potentiation in the accumbens nucleus without altering basal synaptic transmission. Neuroscience 107: 455–464.
Deckert J, Nöthen MM, Franke P, Delmo C, Fritze J, Knapp M et al (1998). Systematic mutation screening and association study of the A1 and A2A adenosine receptor genes in panic disorder suggest a contribution of the A2a gene to the development of disease. Mol Psychiatry 3: 81–85.
Duvarci S, Bauer EP, Paré D (2009). The bed nucleus of the stria terminalis mediates inter-individual variations in anxiety and fear. J Neurosci 29: 10357–10361.
Flajolet M, Wang Z, Futter M, Shen W, Nuangchamnong N, Bendor J et al (2008). FGF acts as a co-transmitter through adenosine A2A receptor to regulate synaptic plasticity. Nat Neurosci 11: 1402–1409.
Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM (2005). Adenosine and brain function. Int Rev Neurobiol 63: 191–270.
Goosens KA, Holt W, Maren S (2000). A role for amygdaloid PKA and PKC in the acquisition of long-term conditional fear memories in rats. Behav Brain Res 114: 145–152.
Goosens KA, Maren S (2001). Contextual and auditory fear conditioning are mediated by the lateral, basal and central amygdaloid nuclei in rats. Learn Mem 8: 148–155.
Hamilton SP, Slager SL, De Leon AB, Heiman GA, Klein DF, Hodge SE et al (2004). Evidence for genetic linkage between a polymorphism in the adenosine 2A receptor and panic disorder. Neuropsychopharmacology 29: 558–565.
Johansen JP, Cain CK, Ostroff LE, LeDoux JE (2011). Molecular mechanisms of fear learning and memory. Cell 147: 509–524.
Kaster MP, Machado NJ, Silva HB, Nunes A, Ardais AP, Santana M et al (2015). Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc Natl Acad Sci USA 112: 7833–7838.
Laurent C, Burnouf S, Ferry B, Batalha VL, Coelho JE, Baqi Y et al (2016). A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol Psychiatry 21: 97–107.
Le Bars D, Gozariu M, Cadden SW (2001). Animal models of nociception. Pharmacol Rev 53: 597–652.
Li P, Rial D, Canas PM, Yoo JH, Li W, Zhou X et al (2015a). Optogenetic activation of intracellular adenosine A2A receptor signaling in the hippocampus is sufficient to trigger CREB phosphorylation and impair memory. Mol Psychiatry 20: 1339–1349.
Li W, Silva HB, Real J, Wang YM, Rial D, Li P et al (2015b). Inactivation of adenosine A2A receptors reverses working memory deficits at early stages of Huntington’s disease models. Neurobiol Dis 79: 70–80.
Lucas M, Mirzaei F, Pan A, Okereke OI, Willett WC, O’Reilly ÉJ et al (2011). Coffee, caffeine, and risk of depression among women. Arch Intern Med 171: 1571–1578.
Lucas M, O'Reilly EJ, Pan A, Mirzaei F, Willett WC, Okereke OI et al (2013). Coffee, caffeine, and risk of completed suicide: results from three prospective cohorts of American adults. World J Biol Psychiatry 15: 377–386.
Lundberg C, Björklund T, Carlsson T, Jakobsson J, Hantraye P, Déglon N et al (2008). Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr Gene Ther 8: 461–473.
Mahan AL, Ressler KJ (2012). Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci 35: 24–35.
Maren S, Phan KL, Liberzon I (2013). The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci 14: 417–428.
Nakamura M, Nakakimura K, Matsumoto M, Sakabe T (2002). Rapid tolerance to focal cerebral ischemia in rats is attenuated by adenosine A1 receptor antagonist. J Cereb Blood Flow Metab 22: 161–170.
Orr AG, Hsiao EC, Wang MM, Ho K, Kim DH, Wang X et al (2015). Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat Neurosci 18: 423–434.
Orsini CA, Maren S (2012). Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev 36: 1773–1802.
Pagnussat N, Almeida AS, Marques DM, Nunes F, Chenet GC, Botton PH et al (2015). Adenosine A2A receptors are necessary and sufficient to trigger memory impairment in adult mice. Br J Pharmacol 172: 3831–3845.
Prediger RD, Batista LC, Takahashi RN (2005). Caffeine reverses age-related deficits in olfactory discrimination and social recognition memory in rats. Involvement of adenosine A1 and A2A receptors. Neurobiol Aging 26: 957–964.
Rau AR, Ariwodola OJ, Weiner JL (2015). Postsynaptic adenosine A2A receptors modulate intrinsic excitability of pyramidal cells in the rat basolateral amygdala. Int J Neuropsychopharmacol 18: pyv017.
Rebola N, Canas PM, Oliveira CR, Cunha RA (2005a). Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 132: 893–903.
Rebola N, Lujan R, Cunha RA, Mulle C (2008). Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 57: 121–134.
Rebola N, Rodrigues RJ, Lopes LV, Richardson PJ, Oliveira CR, Cunha RA (2005b). Adenosine A1 and A2A receptors are co-expressed in pyramidal neurons and co-localized in glutamatergic nerve terminals of the rat hippocampus. Neuroscience 133: 79–83.
Shen HY, Canas PM, Garcia-Sanz P, Lan JQ, Boison D, Moratalla R et al (2013). Adenosine A2A receptors in striatal glutamatergic terminals and GABAergic neurons oppositely modulate psychostimulant action and DARPP-32 phosphorylation. PLoS One 8: e80902.
Shen HY, Coelho JE, Ohtsuka N, Canas PM, Day YJ, Huang QY et al (2008). A critical role of the adenosine A2A receptor in extrastriatal neurons in modulating psychomotor activity as revealed by opposite phenotypes of striatum and forebrain A2A receptor knock-outs. J Neurosci 28: 2970–2975.
Sui L, Wang F, Liu F, Wang J, Li BM (2006). Dorsal hippocampal administration of triiodothyronine enhances long-term memory for trace cued and delay contextual fear conditioning in rats. J Neuroendocrinol 18: 811–819.
Viana da Silva S, Haberl MG, Zhang P, Bethge P, Lemos C, Gonçalves N et al (2016). Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nature Comm 7: 11915.
Wei CJ, Augusto E, Gomes CA, Singer P, Wang Y, Boison D et al (2014). Regulation of fear responses by striatal and extrastriatal adenosine A2A receptors in forebrain. Biol Psychiatry 75: 855–863.
Acknowledgements
This study was supported by DARPA (09-68-ESR-FP-010), NARSAD, FCT (PTDC/SAUNSC/122254/2010 and UID/NEU/04539/2013), QREN (CENTRO-07-ST24-FEDER-002006), CAPES, and CNPq (Ciência sem Fronteiras).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article

Cite this article
Simões, A., Machado, N., Gonçalves, N. et al. Adenosine A2A Receptors in the Amygdala Control Synaptic Plasticity and Contextual Fear Memory. Neuropsychopharmacol 41, 2862–2871 (2016). https://doi.org/10.1038/npp.2016.98
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.98
This article is cited by
-
Adenosine A2A receptors control generalization of contextual fear in rats
Translational Psychiatry (2023)
-
Adenosine A2A receptors control synaptic remodeling in the adult brain
Scientific Reports (2022)
-
Guanosine Attenuates Behavioral Deficits After Traumatic Brain Injury by Modulation of Adenosinergic Receptors
Molecular Neurobiology (2019)
-
Impact of Coffee and Cacao Purine Metabolites on Neuroplasticity and Neurodegenerative Disease
Neurochemical Research (2019)
-
Periodical reactivation under the effect of caffeine attenuates fear memory expression in rats
Scientific Reports (2018)
