
Abstract
Several lines of emerging data point to an imbalance between neuronal excitation and inhibition in at least a subgroup of individuals with autism spectrum disorder (ASD), including in those with fragile X syndrome (FXS), one of the most common genetic syndromes within ASD. In animal models of FXS and of ASD, GABA-B agonists have improved both brain and behavioral phenotypes, including social behavior. A phase 2 randomized, placebo-controlled, crossover trial found that the GABA-B agonist arbaclofen improved social avoidance symptoms in FXS. A pilot open-label trial of arbaclofen suggested similar benefits in ASD. We therefore evaluated arbaclofen in a randomized, placebo-controlled, phase 2 study of 150 participants, aged 5–21 years, with ASD. No difference from placebo was detected on the primary outcome measure, the parent-rated Aberrant Behavior Checklist Social Withdrawal/Lethargy subscale. However, a specified secondary analysis found improvement on the clinician-rated Clinical Global Impression of Severity. An exploratory post hoc analysis of participants with a consistent rater across the trial revealed greater improvement in the Vineland Adaptive Behavior Scales II socialization domain in participants receiving arbaclofen. Affect lability (11%) and sedation (9%) were the most common adverse events. In this exploratory study, secondary analyses suggest that arbaclofen may have the potential to improve symptoms in some children with ASD, but further study will be needed to replicate and extend these initial findings.
Similar content being viewed by others

Introduction
Autism spectrum disorder (ASD) is a common condition defined by difficulties in social communication and repetitive behavior (American Psychiatric Association, 2013), with the majority of those affected experiencing life-long impairment (Shattuck et al, 2012). Available treatments are not based upon an understanding of autism-specific neurobiological mechanisms. Early, intensive interventions use basic behavioral principles to improve cognitive and adaptive behavior outcomes but do not commonly lead to a change in diagnosis (Weitlauf et al, 2014). Many youth with ASD are also treated with medications to treat common comorbid symptoms, including irritability/agitation or hyperactivity (Fung et al, 2016; Reichow et al, 2013), but no medication has solid evidence for treating core ASD symptoms (McPheeters et al, 2011).
As suggested by its definition as a spectrum disorder, substantial heterogeneity is observed in core ASD symptoms. Co-occurring conditions also vary, including cognitive and language impairment, as well as neurological and psychiatric diagnoses such as epilepsy and attention deficit hyperactivity disorder (Jeste and Geschwind, 2014; Sahin and Sur, 2015). Recent genetic studies have implicated many single-nucleotide variants, as well as copy number variants, that contribute to ASD risk, but no single variant is found in more than 1–2% of cases (De Rubeis and Buxbaum, 2015; Krumm et al, 2015; Sanders et al, 2015). Numerous biochemical, physiological, and neuroimaging studies have identified group differences between ASD and control populations (Anderson, 2015; McPartland, 2016; Ruggeri et al, 2014). These biomarkers, however, also show considerable variability within people with ASD, suggesting that a variety of underlying mechanisms or pathways can lead to ASD.
Several lines of emerging data point to an imbalance between neuronal excitation and inhibition (E/I) in at least a subgroup of individuals with ASD. This hypothesis first emerged from the increased incidence of seizures in ASD (Jeste and Tuchman, 2015; Rubenstein and Merzenich, 2003). Subsequently, some but not all magnetic resonance spectroscopy, encephalography, and post-mortem studies have suggested a deficit in inhibitory signaling in ASD (Gaetz et al, 2014; Levin and Nelson, 2015; Robertson et al, 2016). Genetic findings have also implicated gamma-aminobutyric acid (GABA)- and glutamate-related genes in some patients (Kenny et al, 2014; O'Roak et al, 2012; Sanders et al, 2015). Some genetic mouse models of ASD and related syndromes also show a disruption of E/I balance (Braat and Kooy, 2015; Tabuchi et al, 2007; Won et al, 2012), although not always in the direction of excitation. As a more concrete test of the hypothesis, optogenetic enhancement of excitation versus inhibition in the medial prefrontal cortex disrupts social behavior in mice (Yizhar et al, 2011).
The E/I imbalance hypothesis could offer a potential pathway to treatment. The idea of using a GABA-B receptor agonist in ASD first emerged from observations in the drosophila model of fragile X syndrome (FXS), the most common genetic syndrome associated with ASD (Chang et al, 2008). Two studies in the genetic mouse model of FXS have subsequently found improvements in both brain and behavioral phenotypes, including social and repetitive behavior, with arbaclofen (R-baclofen, STX209), a GABA-B agonist (Henderson et al, 2012; Qin et al, 2015). Racemic baclofen, containing both R- and S-baclofen, similarly rescued brain and behavioral phenotypes, including social deficits, in a mouse model with E/I imbalance caused by NMDA receptor subunit deletion (Gandal et al, 2012). A recent study of arbaclofen in two inbred mouse strains with autism-like behavioral traits also found improvements in social behavior in one strain and decreased repetitive behaviors in both, suggesting that benefit could extend to nonsyndromal ASD (Silverman et al, 2015).
Building upon the drosophila and mouse data, we evaluated arbaclofen in a pilot randomized, placebo-controlled crossover study in children, adolescents, and adults with FXS (Berry-Kravis et al, 2012). The planned primary analysis did not show a significant difference between arbaclofen and placebo, but a post hoc analysis found differences in an FXS-specific subscale of Social Avoidance derived from the Aberrant Behavior Checklist (ABC) (Sansone et al, 2012). Based on both the general E/I imbalance hypothesis and the idea that a common pathophysiology may underlie FXS and a subgroup of idiopathic ASD, we examined response to arbaclofen in an open-label pilot study of children and adolescents with nonsyndromal ASD (Erickson et al, 2014). In this group, we observed broad improvements across multiple symptom domains, including the Social Withdrawal/Lethargy subscale of the ABC (ABC-SW/L) (Aman, 1994), as well as a favorable safety and tolerability profile.
Multiple lines of evidence therefore suggest that arbaclofen is a reasonable candidate drug for core symptoms in ASD, but two substantial challenges remain to studying such a potential medication. First, without a way to narrow the target population, it may be difficult to detect improvements that should only be expected in a portion of the heterogeneous autism spectrum. Second, as benefit in core symptoms has never been established for a medication in ASD, it is unclear what outcome measures are sensitive and specific to change in social impairment. We sought to tackle these challenges in a well-powered, randomized, placebo-controlled study of arbaclofen in children and adolescents spanning the full autism spectrum. Based upon available evidence and Food and Drug Administration (FDA) approval of other medications based upon ABC subscale scores, we chose the ABC Social Withdrawal/Lethargy subscale as our primary outcome measure (Anagnostou et al, 2014), but we also applied an array of secondary and tertiary outcome measures. Our goals were both to assess the effects of arbaclofen specifically and to provide guidance for the design of future studies targeting core symptoms in ASD.
Materials and methods
Participants
Eligible participants were 5–21 years old who met Diagnostic and Statistical Manual of Mental Disorders–4th edition–Text Revision (DSM-IV-TR) criteria for Autistic Disorder, Asperger’s Disorder, or PDD-NOS, based upon clinician interview with caregiver(s) and direct assessment of ASD symptoms on the Autism Diagnostic Observation Schedule. Based upon the previous phase 2 results in FXS (Berry-Kravis et al, 2012), study inclusion required a minimum score of 8 on the Social Withdrawal/Lethargy subscale of the Aberrant Behavior Checklist–Community edition (ABC-C), as well as a Clinical Global Impression of Severity (CGI-S) score of moderate or higher, at the screening visit and the baseline visit before randomization. Participants with a history of seizure disorder were required to be receiving treatment with antiepileptic drugs and be seizure free for at least 6 months before randomization. Participants were excluded if they were currently receiving treatment with GABA agonists (with the exception of as needed benzodiazepines for procedures such as dental visits), vigabatrin, tiagabine, riluzole, propranolol, anxiolytics/antidepressants, antipsychotics, or more than two psychoactive medications; had previously participated in a trial with arbaclofen; had a history of hypersensitivity to racemic baclofen; or had a known genetic disorder associated with ASD such as FXS, Rett syndrome, or tuberous sclerosis. Changes in medication or behavioral therapies were not allowed in the 2 months before randomization, nor were planned changes allowed for the duration of the trial. Female participants of child-bearing potential were tested and excluded if they were pregnant.
All participants or guardians provided voluntary informed consent and assent as appropriate. This study (clinicaltrials.gov identifier NCT01288716) was approved by the institutional review boards governing each site.
Design
This was an exploratory, phase II trial in individuals with ASD, using a randomized, double-blind, placebo-controlled, multisite design, at 25 sites in the United States between June 2011 and September 2012. The study drug was flexibly titrated every week during the first 4 weeks of the treatment period. The starting dose was 5 mg b.i.d. in all participants and could be increased every 7 days to 10 mg b.i.d., 10 mg t.i.d., and then to 15 mg t.i.d. The maximum dose for participants <12 years of age was limited to 10 mg t.i.d. Blinding was maintained by utilizing identical tablets containing either STX209 or placebo.
Continued up titration occurred at the Investigator’s discretion to achieve the optimal titrated dose (OTD) based on the participant’s average response compared with the baseline behavior. The OTD for each participant was the dose associated with a score of 1 on the Clinician’s Global Impression–Improvement (CGI-I) or the dose associated with the best clinical response. If a participant developed unacceptable side effects during titration, the dose could be reduced to the previously tolerated dose, and additional dose adjustment could occur throughout the 28-day up-titration and dose adjustment period. The OTD was then maintained without change from day 29 to the end of the 12-week treatment period. Participants who were randomized to receive placebo followed the same flexible-dose titration and maintenance schedule.
Participants returned for evaluations 2, 4, 8, and 12 weeks after starting treatment. Following the 12-week visit, study drug was then tapered down per protocol over a period of up to 28 days. Study drug and matching placebo were provided as 5 and 10 mg oral disintegrating tablets. Participants were randomized 1 : 1 to either arbaclofen or placebo according to a centrally generated randomization list, with stratification by age (5–11 or 12–21 years) and concomitant use of psychoactive medication. Treatment compliance was monitored with a dosing form that guardians completed on a daily basis.
Assessments
Baseline assessments included the Stanford–Binet Intelligence Scales–Fifth Edition (SB-5), the Autism Diagnostic Observation Scale (ADOS), and a review of autism spectrum disorder criteria from the DSM-IV-TR.
Global outcome measures included the CGI-I and CGI-S assessments, both rated on a 7-point Likert scale. Focused assessments included the ABC-C (Aman, 1994), a 58-item, parent-rated questionnaire yielding 5 factor scores, including Irritability, Social Withdrawal/Lethargy, Hyperactivity, Inappropriate Speech, and Stereotypy. Other measures included visual analog scales (VAS) for Disruptive and Anxiety-Driven Problem Behavior, the Vineland Adaptive Behavior Scales–Second Edition (VABS-II) (Sparrow et al, 2005b), the ADHD Rating Scale–IV (DuPaul et al, 1998), Parenting Stress Index (PSI) –Short Form, Child’s Sleep Habits Questionnaire (CSHQ), and the Sensory Profile–Short Form (SSP).
Safety assessments included physical examination, standard hematology and clinical chemistry assessments, concomitant medication usage, directed suicidality assessments, urinalysis, EKGs, and spontaneously reported adverse events.
Statistical Analysis
The primary efficacy variable was prospectively defined as change from baseline to visit 5 for the Social Withdrawal/Lethargy subscale of the ABC-C (ABC-SW/L). Analysis of covariance (ANCOVA) analysis of differences between arbaclofen and placebo groups was analyzed using a restricted maximum likelihood-based repeated measures approach in the ITT population. Unstructured within-participant covariance was used. All models included main effect terms of treatment and week as explanatory variables. Baseline score and age group were also included in the model. Other appropriate variables were analyzed with a similar approach in the ITT population. Categorical outcomes were analyzed by sign test. Secondary efficacy variables included the CGI-I score at end of treatment, change from baseline to end of treatment in VABS-II Socialization Domain, CGI-S, VAS, and ADHD-IV Total Raw Score. Exploratory efficacy assessments included changes from baseline to end of treatment for the PSI-Short Form, the raw and standardized scores for the VABS-II Maladaptive Behavior Index Communication domain, the remaining subscales of the ABC-C, the CSHQ, and the Sensory Profile–Short Form. For all secondary and exploratory comparisons, an uncorrected p-value of ⩽0.05 was required to declare nominal significance, and no adjustments for multiplicity were made for this exploratory, phase II study.
The study was powered to have an 80% likelihood of detecting an effect size of ∼0.5 on the primary end point. The planned sample size was 75 participants per treatment arm.
Post Hoc Analyses
It became apparent during data cleaning that many participants did not have the same examiner on the VABS-II at the week 12 end point as they had at baseline, as described in the protocol for this secondary measure. A post hoc analysis was conducted to examine change in VABS-II socialization in the per-protocol population that had the same examiner and rater at both time points. Follow-up post hoc analyses were performed in an attempt to identify factors that related to improvement in VABS-II socialization domain scores.
Results
Participants, Disposition, and Dosing
Baseline characteristics of the 150 participants (124 males) enrolled in the study across 24 centers in the United States are summarized in Table 1. All participants met criteria for Autistic Disorder, Asperger’s Disorder, or Pervasive Developmental Disorder–Not Otherwise Specified, according to the DSM-IV-TR (American Psychiatric Association, 2000). Based upon DSM-5 guidance regarding carrying DSM-IV diagnoses forward (American Psychiatric Association, 2013), we refer to these three diagnoses as Autism Spectrum Disorder in the main text. On the ADOS (Lord et al, 2000), 120 participants met criteria for autism, 28 for autism spectrum, and 2 were missing data because of behavioral difficulties during the assessment. On the SB-5 (Roid, 2003), 70 participants had an abbreviated IQ score of <70, and 76 participants had a score of ⩾70, with missing data on 4 participants because of behavioral difficulties. Enrollment and randomization (see Supplementary Figure S1) were stratified by age group (76 participants between the ages of 5 and 11 years and 74 between the ages of 12 and 21 years) and by the use of psychoactive medication, of which the most commonly used were psychostimulants and alpha-adrenergic agents for the treatment of inattention and hyperactivity.
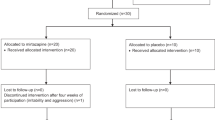
Participant disposition is illustrated in Figure 1. The overall completion rate was 93% on placebo and 80% on arbaclofen, with the difference largely attributable to the larger number of discontinuations because of adverse events in the arbaclofen treatment arm. For child participants who completed the trial, the optimal titrated dose of arbaclofen was 26.8±1.1 mg/day, and this was not significantly different from the placebo group (28.7±0.7 mg/day). For adult participants who completed the trial, the optimal titrated dose of arbaclofen was 41.7±1.3 mg/day, and this was not significantly different than the placebo group (43.7±0.7 mg/day).

CONSORT flow diagram.
Safety
Arbaclofen was generally well tolerated. The most frequent treatment-emergent adverse events (AEs) are listed in Table 2. Most AEs were mild in intensity and resolved spontaneously without dose changes. There were two events of seizure, both occurring in participants receiving placebo. Adverse events with >5% incidence and greater than twice the incidence in placebo included somnolence (9 vs 1%) and affect lability (11% vs 1%). Rhinorrhea was more commonly reported in the placebo group than in the arbaclofen group (8% vs 3%).
In the arbaclofen treatment arm, the AEs leading to discontinuation were suicidal ideation, aggression, emotional distress, sleep disorder, oculogyric crisis, dyskinesia, sensation of heaviness, and rash. In the placebo treatment arm, the AEs leading to discontinuation were suicidal ideation and insomnia with hyperactivity. None of these AEs were treated with other medications, except the rash that was treated with diphenhydramine and was completely resolved after 4 days. The events of suicidal ideation on arbaclofen and on placebo occurred at different sites but were very similar, with neither participant having method, intent, or plan, and both judged safe to remain in their parents’ custody. Only the event of suicidal ideation on arbaclofen was reported as a ‘serious adverse event’ (SAE). The only other SAE in the study was anaphylaxis, attributed to soy exposure, and occurring 10 days after the participant had completed the study on placebo.
Efficacy
On the primary end point, the ABC-SW/L, participants on arbaclofen and placebo showed no difference in the full intent-to-treat population (change from baseline −5.4±0.78 vs −6.0±0.75, least-squares mean±SEM, p=0.518, Table 3 and Figure 2). Among the five end points identified for the secondary analysis, arbaclofen was associated with a statistically significant advantage on the CGI-S (−0.7±0.10 vs −0.3±0.10, p=0.009, uncorrected) that calls for the treating clinician to integrate all available information on the participants’ disease severity (Guy, 1976) (Table 3 and Figure 3a). No statistically significant differences were observed on the other four secondary end points that focused on specific symptom domains (Table 3). As shown in Figure 3b, the nominally significant difference in change in CGI-S was driven by 10 of 75 participants (13%) that show a shift of two points within the arbaclofen group, such as a change in ratings from ‘severely’ to ‘moderately’ ill. Only three participants in the placebo group showed a change of this magnitude. Exploratory post hoc analyses to examine CGI-S response in subgroups of participants were not statistically significant but showed numerical advantages for younger and higher functioning participants as assessed by IQ or ADOS category (Supplementary Figure S2).

Change in outcome measures over time. The change (mean and SEM) in parent ratings on the Aberrant Behavior Checklist–Community Version (ABC) Social Withdrawal/Lethargy score is shown over the course of the study. The primary outcome measure was assessed at baseline, week 4, and week 12. Uncorrected p=0.477.

Change in the Clinical Global Impression of severity. (a) The change (mean and SEM) in clinician ratings on the Clinical Global Impression of Severity (CGI-S), one of six secondary outcome measures, is shown over the course of the study. The CGI-S was assessed at baseline, week 2, week 4, week 8, and week 12. Uncorrected p=0.009. (b) The number of participants is shown for each degree of change on the CGI-S from baseline to week 12.
In the course of data review, it became evident that a substantial portion of the participants had a rater change on the VABS-II (Sparrow et al, 1984, 2005a), a secondary measure that was intended to be completed by the same clinician and parent at the beginning and end of treatment. An exploratory post hoc analysis of the VABS socialization domain score in the 97 participants with consistent raters on the VABS-II (out of 130 completers) (Supplementary Table S1) revealed greater improvement in those receiving arbaclofen (7.1±1.38 vs 1.8±1.26, p=0.006, uncorrected).
Discussion
The results of this trial of arbaclofen suggest its further study for social function in ASD. No significant difference was seen, however, between arbaclofen and placebo on the study primary outcome measures, the ABC-SW/L, that was chosen based upon FDA acceptance of another ABC subscale for trials targeting Irritability/Agitation symptoms in ASD. Importantly, an expert panel convened by Autism Speaks was unable to recommend without conditions any single measure of social communication as an end point in clinical trials in ASD, but ABC-SW/L was the highest ranked measure on their list based upon potential sensitivity to change in medication treatment trials (Anagnostou et al, 2014). Improvement in ABC-SW/L scores was observed in both the arbaclofen and the placebo groups, similar to previous studies that found substantial placebo effects in ASD (Masi et al, 2015). It is possible that short-term expectancy effects on ABC-SW/L may have been reduced using an alternative study design such as a placebo run-in, although this approach is not without controversy (Emslie et al, 1997; Weimer et al, 2013). Of note, the higher incidence of somnolence adverse events in the arbaclofen group compared with placebo suggests that a subscale that indexes lethargy in addition to social symptoms may not be ideal for assessing the potential benefits of arbaclofen.
The nominally significant difference in change in CGI-S scores suggests a potentially meaningful improvement in a subset of the arbaclofen participants. The CGI measures of Severity and Improvement were originally developed to gauge whether quantitative changes measured using symptom ratings scales were clinically meaningful (Guy, 1976). The CGI-I has also been used as a primary outcome measure in a number of clinical trials where there is no optimal symptom rating scale, including in ASD (King et al, 2009). Changes in CGI-S scores reflect a substantial change in symptoms that is less commonly observed than ratings of improvement on the CGI-I. The CGI-S finding is driven by a subset of participants (13%) who show a shift of two or more points within the arbaclofen group. This result suggests that there was a clinically meaningful change in global ASD symptoms in this subset of children, but planned analyses of other outcome measures did not show statistically significant differences that clarify what specific symptoms are improving in the overall group.
The domains of the VABS have been used to evaluate long-term changes across 1 or 2 years in trials of behavioral interventions in ASD (Dawson et al, 2010), but they were not previously expected to be sensitive to change across shorter medication trials (Anagnostou et al, 2014). Perhaps for this reason, adherence was low to the protocol description that the same raters should be present for all administrations of this secondary measure, despite overall good adherence to other aspects of the protocol across sites. Interrater reliability on the VABS socialization domain is reported to be 0.64 (Sparrow et al, 2005a), suggesting that switching raters is ill advised across a longitudinal treatment study. When restricted to participants with protocol-defined consistent raters, there was a nominally significant change in VABS socialization.
In addition to suggesting further study of arbaclofen in ASD, these results illustrate the central challenges in testing medications for core symptoms in ASD. First, the heterogeneity evident in ASD suggests that no single medication is likely to benefit the full spectrum of affected children. Here, the significant CGI-S finding is driven by a subset of children with ASD who show substantial change of two or more points across the course of the 12-week trial. Subgroup analyses suggest that higher functioning children were more likely to respond based upon CGI-S or VABS-II, but these exploratory analyses were not statistically significant and require replication. Ideally, subgroups in ASD treatment studies would be defined by a biological marker or by a distinct co-occurring diagnosis, such as epilepsy or severe anxiety. Core symptom domains and common co-occurring impairments such as cognition or communication impairment follow a continuum that prevents easy subtyping. Importantly, when a well-defined subgroup cannot be identified a priori, an initial study such as this one could be used to define the population for a replication study.
A second central challenge for ASD trials is the absence of outcome measures that have been shown to be sensitive and specific to change in social symptoms with medication treatment (Anagnostou et al, 2014). Previous work did show change in the ABC-SW/L subscale during open-label treatment with arbaclofen in ASD but without comparison with placebo response (Erickson et al, 2014). The ABC did detect significant improvement in a phase 2, randomized, crossover trial of arbaclofen in FXS (Berry-Kravis et al, 2012), but that study used an FXS-specific Social Avoidance subscale that eliminates some items, including those that index lethargy symptoms, based upon a factor analysis in the FXS population (Sansone et al, 2012). A previous study showed some improvement in ABC-SW/L in a pooled analysis of treatment trials of risperidone for severe irritability/agitation behavior in children with ASD (Scahill et al, 2013), but ABC-SW/L was not a primary outcome measure in those studies, nor was the observed change significant in individual studies. Our results suggest that VABS-II socialization may be a good alternative for assessment of social function in children with ASD, at least when administered by the same raters, even across the relatively short duration of 12 weeks.
There are important limitations to this phase 2, randomized, placebo-controlled trial. First, the primary outcome measure did not show a significant change, falling short of the standard criterion for a positive trial. For this exploratory study, additional measures were analyzed to assess potential outcome measures for future studies, but it is also possible that type I error could account for the nominally significant findings. Second, the CGI-S finding, although nominally significant, is not matched by a significant change in the CGI-I and does not clearly identify the symptoms that improved in participants treated with arbaclofen. Third, the analysis of VABS socialization by consistent raters represents a post hoc analysis that only generates a hypothesis for confirmation in future studies. Similarly, the stronger signal in participants with higher IQ or communication function is difficult to interpret without replication. Finally, it would be more satisfying to connect a subgroup of ASD with the mechanism of action of arbaclofen, either by virtue of a biomarker that defines E/I imbalance or by a cluster of genetic findings.
In conclusion, these results serve two functions. First, they suggest continued study of arbaclofen for core symptoms of ASD, including replication of these exploratory findings and potentially also evaluation of higher doses or earlier time points in development. The post hoc analyses provide guidance regarding choice of primary outcome measure and a potential subgroup where that outcome measure is most likely to demonstrate change. Second, and perhaps most importantly, this study ushers in a new era of ASD clinical trials driven by biological hypotheses based on genetic animal models of ASD and related syndromes. We should not expect to see immediate success in this effort to find new treatments that benefit every child within this heterogeneous spectrum. We can, however, use our results to refine our approach to testing medications that could provide specific benefits to subgroups of children who may share common, underlying neurobiology.
FUNDING AND DISCLOSURE
JV-V received research funding from Seaside Therapeutics to conduct this study. He has consulted or served on advisory boards for Roche, Novartis, and SynapDx; has received research funding from Roche Pharmaceuticals, Novartis, SynapDx, and Forest; and has received stipends for editorial work from Springer and Wiley. EHC consulted with and received research funding from Seaside Therapeutics to conduct this study. BHK consulted with and received research funding from Seaside Therapeutics to conduct this study. He has also consulted with Roche; has served on the Scientific Advisory Board of Confluence Therapeutics; and has received research funding from Roche and Novartis. PZ, MC, KW-B, PPW, and RLC were employees of Seaside Therapeutics at the time of the study. MFB and RLC were co-founders of Seaside Therapeutics.
References
Aman M (1994) Aberrant Behavior Checklist - Community. Slosson Educational Publications: East Aurora, NY.
American Psychiatric Association (2000) Diagnostic and Statistical Manual of Mental Disorders-Text Revision (DSM-IV-TR), 4th edn. American Psychiatric Association Press: Washington, DC.
American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders, 5th edn. DSM-5 American Psychiatric Publishing: Arlington, VA.
Anagnostou E, Jones N, Huerta M, Halladay AK, Wang P, Scahill L et al (2014). Measuring social communication behaviors as a treatment endpoint in individuals with autism spectrum disorder. Autism 19: 622–636.
Anderson GM (2015). Autism biomarkers: challenges, pitfalls and possibilities. J Autism Dev Disord 45: 1103–1113.
Berry-Kravis EM, Hessl D, Rathmell B, Zarevics P, Cherubini M, Walton-Bowen K et al (2012). Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med 4: 152ra127.
Braat S, Kooy RF (2015). The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 86: 1119–1130.
Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P et al (2008). Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol 4: 256–263.
Dawson G, Rogers S, Munson J, Smith M, Winter J, Greenson J et al (2010). Randomized, controlled trial of an intervention for toddlers with autism: the Early Start Denver Model. Pediatrics 125: e17–e23.
De Rubeis S, Buxbaum JD (2015). Genetics and genomics of autism spectrum disorder: embracing complexity. Hum Mol Genet 24 (R1): R24–R31.
DuPaul GJ, Power TJ, Anastopoulos AD, Reid D (1998) ADHD Rating Scale-IV: Checklists, Norms, and Clinical Interpretation. The Guilford Press: New York.
Emslie G, Rush A, Weinberg W, Kowatch R, Hughes C, Carmody T et al (1997). A double-blind, randomized, placebo-controlled trial of fluoxetine in children and adolescents with depression. Arch Gen Psychiatry 54: 1031–1037.
Erickson CA, Veenstra-Vanderweele JM, Melmed RD, McCracken JT, Ginsberg LD, Sikich L et al (2014). STX209 (arbaclofen) for autism spectrum disorders: an 8-week open-label study. J Autism Dev Disord 44: 958–964.
Fung LK, Mahajan R, Nozzolillo A, Bernal P, Krasner A, Jo B et al (2016). Pharmacologic treatment of severe irritability and problem behaviors in autism: a systematic review and meta-analysis. Pediatrics 137 (S2): S124–S135.
Gaetz W, Bloy L, Wang DJ, Port RG, Blaskey L, Levy SE et al (2014). GABA estimation in the brains of children on the autism spectrum: measurement precision and regional cortical variation. Neuroimage 86: 1–9.
Gandal MJ, Sisti J, Klook K, Ortinski PI, Leitman V, Liang Y et al (2012). GABAB-mediated rescue of altered excitatory-inhibitory balance, gamma synchrony and behavioral deficits following constitutive NMDAR-hypofunction. Transl Psychiatry 2: e142.
Guy W (1976) ECDEU Assessment Manual for Psychopharmacology. United States Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health, Psychopharmacology Research Branch. Division of Extramural Research Programs: Rockville, MD.
Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR et al (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABA(B) receptors with arbaclofen. Sci Transl Med 4: 152ra128.
Jeste SS, Geschwind DH (2014). Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol 10: 74–81.
Jeste SS, Tuchman R (2015). Autism spectrum disorder and epilepsy: two sides of the same coin? J Child Neurol 30: 1963–1971.
Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C et al (2014). Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry 19: 872–879.
King BH, Hollander E, Sikich L, McCracken JT, Scahill L, Bregman JD et al (2009). Lack of efficacy of citalopram in children with autism spectrum disorders and high levels of repetitive behavior: citalopram ineffective in children withautism. Arch Gen Psychiatry 66: 583–590.
Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K et al (2015). Excess of rare, inherited truncating mutations in autism. Nat Genet 47: 582–588.
Levin AR, Nelson CA (2015). Inhibition-based biomarkers for autism spectrum disorder. Neurotherapeutics 12: 546–552.
Lord C, Rutter M, DiLavore P, Risi S (2000) Autism Diagnostic Observation Schedule (ADOS). Western Psychological Services: Torrance, CA.
Masi A, Lampit A, Glozier N, Hickie IB, Guastella AJ (2015). Predictors of placebo response in pharmacological and dietary supplement treatment trials in pediatric autism spectrum disorder: a meta-analysis. Transl Psychiatry 5: e640.
McPartland JC (2016). Considerations in biomarker development for neurodevelopmental disorders. Curr Opin Neurol 29: 118–122.
McPheeters ML, Warren Z, Sathe N, Bruzek JL, Krishnaswami S, Jerome RN et al (2011). A systematic review of medical treatments for children with autism spectrum disorders. Pediatrics 127: e1312–e1321.
O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP et al (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246–250.
Qin M, Huang T, Kader M, Krych L, Xia Z, Burlin T et al (2015). R-baclofen reverses a social behavior deficit and elevated protein synthesis in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol 18.
Reichow B, Volkmar FR, Bloch MH (2013). Systematic review and meta-analysis of pharmacological treatment of the symptoms of attention-deficit/hyperactivity disorder in children with pervasive developmental disorders. J Autism Dev Disord 43: 2435–2441.
Robertson CE, Ratai EM, Kanwisher N (2016). Reduced GABAergic action in the autistic brain. Curr Biol 26: 80–85.
Roid GH (2003) Stanrod Binet Intelligence Scales, Fifth Edition. Riverside: Itasca, IL.
Rubenstein JL, Merzenich MM (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2: 255–267.
Ruggeri B, Sarkans U, Schumann G, Persico AM (2014). Biomarkers in autism spectrum disorder: the old and the new. Psychopharmacology (Berl) 231: 1201–1216.
Sahin M, Sur M (2015). Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 350: aab3897.
Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE et al (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87: 1215–1233.
Sansone SM, Widaman KF, Hall SS, Reiss AL, Lightbody A, Kaufmann WE et al (2012). Psychometric study of the Aberrant Behavior Checklist in Fragile X Syndrome and implications for targeted treatment. J Autism Dev Disord 42: 1377–1392.
Scahill L, Hallett V, Aman MG, McDougle CJ, Eugene Arnold L, McCracken JT et al (2013). Brief Report: social disability in autism spectrum disorder: results from Research Units on Pediatric Psychopharmacology (RUPP) Autism Network trials. J Autism Dev Disord 43: 739–746.
Shattuck PT, Narendorf SC, Cooper B, Sterzing PR, Wagner M, Taylor JL (2012). Postsecondary education and employment among youth with an autism spectrum disorder. Pediatrics 129: 1042–1049.
Silverman JL, Pride MC, Hayes JE, Puhger KR, Butler-Struben HM, Baker S et al (2015). GABAB receptor agonist R-baclofen reverses social deficits and reduces repetitive behavior in two mouse models of autism. Neuropsychopharmacology 40: 2228–2239.
Sparrow S, Balla D, Cicchetti D (1984) Vineland Scales of Adaptive Behavior, Survey Form Manual. American Guidance Service: Circle Pines, MN.
Sparrow SS, Cicchetti DV, Balla DA (2005a) Vineland Adaptive Behavior Scales, (Vineland-II). American Guidance Services: Circle Pines, MN.
Sparrow SS, Cicchetti DV, Balla DA (2005b) Vineland Adaptive Behavior Scales, Second Edition. AGS Publishing: Circle Pines.
Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM et al (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318: 71–76.
Weimer K, Gulewitsch MD, Schlarb AA, Schwille-Kiuntke J, Klosterhalfen S, Enck P (2013). Placebo effects in children: a review. Pediatr Res 74: 96–102.
Weitlauf AS, McPheeters ML, Peters B, Sathe N, Travis R, Aiello R et al. Therapies for Children with Autism Spectrum Disorder: Behavioral Interventions Update. AHRQ Comparative Effectiveness Reviews. Agency for Healthcare Research and Quality (US): Rockville, MD, 2014.
Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J et al (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486: 261–265.
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ et al (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477: 171–178.
Acknowledgements
We are grateful to the participants and their families for their dedication to this study. We are also grateful for the efforts of the investigators and staff at the study sites, including Dr Ashraf Attalla and the Institute for Behavioral Medicine team, GA; Dr Amanda Bennett and the Children’s Hospital of Philadelphia team, PA; Dr David Beversdorf and the University of Missouri Columbia Thompson Research Center for Autism & Neurodevelopmental Disorders team, MO; Dr John Duby and the Akron Children’s Hospital team, OH; Dr Jean Frazier and the University of Massachusetts team, MA; Dr Lawrence Ginsberg and the Red Oak Psychiatry team, TX; Dr Randi Hagerman and the University of California Davis Medical Investigation of Neurodevelopmental Disorders Institute team, CA; Dr Shivkumar Hatti and the Suburban Research Associates team, PA; Dr Willis Holloway and the Cutting Edge Research team, OK; the University of Washington and Seattle Children’s Research Institute team, WA; Dr Walter Kaufmann and the Johns Hopkins University and Kennedy Krieger Institute team, MD; Dr Alexander Kolevzon and the Mount Sinai School of Medicine Seaver Autism Center team, NY; Dr Miles Landis and the Lake Mary Pediatrics team, FL; Dr Eric London and the New York State Institute of Basic Research in Developmental Disabilities team, NY; Dr James McCracken and the University of California Los Angeles Neuropsychiatric Institute team, CA; Dr Kathryn McVicar and the University of Tennessee Medical Group team, TN; Dr Fedra Najjar and the University of Illinois at Chicago Institute for Juvenile Research team, IL; Dr Ramzi Nasir and the Children's Hospital Boston team, MA; Dr Raun Melmed and the Southwest Autism Research & Resource Center team, AZ; Dr Roger Rousseau and the Pharmax Research Clinic team, FL; Dr Kevin Sanders and the Vanderbilt University team, TN; Dr Linmarie Sikich and the University of North Carolina Chapel Hill Department of Psychiatry team, NC; Dr Kim Stigler and the University of Indiana Indianapolis, Riley Child and Adolescent Psychiatry Center team, IN; Dr Jerry Tomasovic and the Road Runner Research team, TX; and Dr Alan Yeo and the Summit Research Network team, OR. This study was funded by Seaside Therapeutics that is no longer in business. Seaside Therapeutics was involved in the design of the study and collection, analysis, and interpretation of data. Prior employees of Seaside Therapeutics were involved in writing the manuscript.
Author contributions
JV-V assisted with study design, collected data, participated in data analysis and interpretation of results, and wrote the first draft of the manuscript. EHC and BHK assisted with study design, collected data, participated in interpretation of results, and revised the manuscript for critical content. PZ and MC assisted with study design, oversaw data collection, participated in interpretation of results, and revised the manuscript for critical content. KW-B performed the statistical analysis, participated in interpretation of results, and revised the manuscript for critical content. MFB assisted with study design, participated in analysis and interpretation of results, and revised the manuscript for critical content. PPW assisted in study design, oversaw implementation of the protocol, participated in data analysis and interpretation of results, and revised the manuscript for critical content. RLC assisted in study design, supervised implementation of the study, participated in analysis and interpretation of results, and revised the manuscript for critical content.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article

Cite this article
Veenstra-VanderWeele, J., Cook, E., King, B. et al. Arbaclofen in Children and Adolescents with Autism Spectrum Disorder: A Randomized, Controlled, Phase 2 Trial. Neuropsychopharmacol 42, 1390–1398 (2017). https://doi.org/10.1038/npp.2016.237
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.237

{kind=link}