
Abstract
The mechanisms by which stressful life events increase the risk of relapse in recovering cocaine addicts are not well understood. We previously reported that stress, via elevated corticosterone, potentiates cocaine-primed reinstatement of cocaine seeking following self-administration in rats and that this potentiation appears to involve corticosterone-induced blockade of dopamine clearance via the organic cation transporter 3 (OCT3). In the present study, we use a conditioned place preference/reinstatement paradigm in mice to directly test the hypothesis that corticosterone potentiates cocaine-primed reinstatement by blockade of OCT3. Consistent with our findings following self-administration in rats, pretreatment of male C57/BL6 mice with corticosterone (using a dose that reproduced stress-level plasma concentrations) potentiated cocaine-primed reinstatement of extinguished cocaine-induced conditioned place preference. Corticosterone failed to re-establish extinguished preference alone but produced a leftward shift in the dose–response curve for cocaine-primed reinstatement. A similar potentiating effect was observed upon pretreatment of mice with the non-glucocorticoid OCT3 blocker, normetanephrine. To determine the role of OCT3 blockade in these effects, we examined the abilities of corticosterone and normetanephrine to potentiate cocaine-primed reinstatement in OCT3-deficient and wild-type mice. Conditioned place preference, extinction and reinstatement of extinguished preference in response to low-dose cocaine administration did not differ between genotypes. However, corticosterone and normetanephrine failed to potentiate cocaine-primed reinstatement in OCT3-deficient mice. Together, these data provide the first direct evidence that the interaction of corticosterone with OCT3 mediates corticosterone effects on drug-seeking behavior and establish OCT3 function as an important determinant of susceptibility to cocaine use.
Similar content being viewed by others


INTRODUCTION
Glucocorticoids exert profound effects on behavior and are critical mediators of adaptive stress responses. Although glucocorticoid-mediated stress responses are often attributed to transcriptional regulation resulting from activation of the cytosolic glucocorticoid receptor (GR), a growing body of evidence suggests that many glucocorticoid effects, particularly those that involve acute regulation of behavior, are mediated by non-canonical, GR-independent mechanisms. One such mechanism appears to involve impairment of monoamine clearance via a direct inhibition of the low-affinity, high-capacity monoamine transporter, organic cation transporter 3 (OCT3).
It has long been known that corticosteroids impair monoamine clearance mediated by what historically has been termed uptake2, a high-capacity transport system originally characterized in peripheral tissues (Iversen and Salt, 1970). More recently, uptake2-mediated monoamine clearance has been attributed to a group of transporters that includes the organic cation transporter (OCT) family (OCTs 1, 2, and 3) and the plasma membrane monoamine transporter (PMAT) (Gründemann et al, 1998a; Shang et al, 2003; Gorboulev et al, 2005; Schomig et al, 1998; Duan and Wang, 2010). All of these transporters have been reported to be expressed in the brain, although levels of OCT1 expression are likely very low (Schmitt et al, 2003; Engel et al, 2004; Vialou et al, 2004; Amphoux et al, 2006; Baganz et al, 2008; Gasser et al, 2009). Although each of these transporters is sensitive to inhibition by corticosterone, there is significant variation in sensitivity. Among these transporters, PMAT is the least sensitive to corticosterone (Ki=450 μm), followed by OCT1 (IC50=150 μm), OCT2 (IC50=5 μm), and OCT3 (IC50=0.04–0.2 μm) (Gründemann et al, 1998b; Shang et al, 2003; Gorboulev et al, 2005; Schomig et al, 1998; Duan and Wang, 2010). Thus it appears that OCT3 is the only uptake2 transporter sensitive to inhibition by corticosterone at concentrations observed during stress. Corticosterone-induced inhibition of OCT3-mediated transport is GR-independent, non-genomic, and likely involves direct binding to OCT3 (Gründemann et al, 1999; Horvath et al, 2003; Gorboulev et al, 2005). Moreover, it appears to elevate monoamine levels in vivo (Gasser et al., 2006; Baganz et al., 2010; Graf et al, 2013), posing it as a potential GR-independent mechanism through which glucocorticoids regulate behavior. However, to date, there is no direct evidence that glucocorticoids exert actions on behavior via inhibition of OCT3.
Glucocorticoid-mediated stress-adaptive responses include altered processing of motivational and rewarding stimuli. A consequence of such alterations appears to be a heightened susceptibility to drug-seeking behavior. We recently established a rat self-administration/reinstatement model in which acute stress potentiates the reinstatement of cocaine seeking in response to an otherwise subthreshold priming dose of cocaine but does not by itself directly evoke cocaine seeking (Graf et al, 2013; McReynolds et al., 2016). This stage-setting effect of stress appears to be mediated by glucocorticoids, in that the ability of footshock to potentiate reinstatement in response to a subthreshold low dose of cocaine is eliminated by adrenalectomy, and mimicked in the absence of stress by administration of corticosterone at a dose that results in plasma levels comparable to those observed during stress. The effect also appears to be mediated by a GR-independent mechanism, as it is insensitive to pretreatment with the GR antagonist RU38486 (Graf et al, 2013). A role for inhibition of OCT3 in these actions of corticosterone is suggested by the fact that corticosterone potentiates low-dose cocaine-induced increases in nucleus accumbens (NAc) dopamine, that corticosterone treatment decreases dopamine clearance in the NAc via a DAT-independent mechanism, and that corticosterone-induced potentiation of reinstatement is mimicked by treatment with normetanephrine, a non-glucocorticoid blocker of OCT3-mediated transport (Graf et al, 2013).
Although the above findings suggest that the effects of corticosterone on cocaine seeking may be attributable to its ability to inhibit OCT3-mediated dopamine clearance, definitive evidence that these effects are mediated by inhibition of OCT3 is lacking. To address this, we sought to examine the effects of corticosterone on cocaine-seeking behavior in mutant mice deficient in OCT3 function (Zwart et al, 2001; Vialou et al, 2008; Cui et al, 2009). Here we establish a mouse model wherein corticosterone potentiates the reinstatement of extinguished cocaine-induced conditioned place preference (CPP) resulting from priming injections of cocaine. Using this model, we report that the ability of stress-level corticosterone or normetanephrine to potentiate cocaine-induced reinstatement following CPP requires functional expression of OCT3, thereby providing the first direct evidence that acute behavioral effects of corticosterone can result from its interaction with OCT3.
MATERIALS AND METHODS
Subjects
Male C57BL/6 mice were purchased from Harlan Laboratories and used for Experiment #1. Male OCT3-deficient and wild-type control mice (Zwart et al, 2001; see Supplementary Materials and Methods) were used for Experiment #2. Mice were housed individually in a humidity- and temperature-controlled, AAALAC-accredited animal facility under a 12 h/12 h light/dark cycle (lights on at 0700 hours) with food and water available ad libitum, except when in the experimental chambers, and were tested starting at an age of 8–10 weeks. All procedures were carried out in compliance with the NIH guidelines and the Guide for Care and Use of Laboratory Animals.
CPP, Extinction, and Reinstatement
CPP was conducted as previously described (Mantsch et al, 2010; Vranjkovic et al, 2012; see Supplementary Materials and Methods). Briefly, following determination of initial preference, compartments in a three-compartment apparatus were paired with cocaine (15 mg/kg, ip) or saline during alternating 30-min conditioning sessions over an 8-day period using a biased approach. CPP was defined according to the time spent in the cocaine-paired compartment relative to preconditioning. Mice that displayed CPP underwent daily extinction training during the 30-min sessions until the extinction criterion was met, at which point reinstatement testing began the next day (see Supplementary Materials and Methods). Reinstatement testing conditions were identical to extinction except that the sessions were immediately preceded by cocaine priming injections that followed pretreatment with corticosterone, normetanephrine, or saline. Most mice underwent repeated reinstatement testing, in which case reinstatement sessions were separated by additional extinction training with a requirement that the extinction criterion was met prior to the next reinstatement test.
Experiment 1: Effects of Corticosterone and the Non-Glucocorticoid OCT3 Blocker, Normetanephrine, on Cocaine-Primed Reinstatement Following CPP in C57 BL/6 mice
We initially tested C57BL/6 mice for the effects of pretreatment with corticosterone; the non-glucocorticoid OCT3 inhibitor, normetanephrine; or vehicle on cocaine-primed reinstatement. Soluble HBC-complexed corticosterone (2 mg/kg corticosterone; Sigma-Aldrich) and normetanephrine (2.5 mg/kg; Sigma-Aldrich) were dissolved in saline. Doses were selected based on our findings that they potentiated cocaine-primed reinstatement following self-administration in rats (Graf et al, 2013). Following CPP and extinction, mice were tested for reinstatement in response to a range of cocaine doses (0, 0.93, 1.875, 3.75, 7.5, 15 mg/kg, ip), 40 min after corticosterone, normetanephrine, or saline pretreatment. Each mouse was tested multiple times and received no more than six combinations of saline, normetanephrine, or corticosterone pretreatment/cocaine doses in randomized sequence (average of four tests). Pretreatment effects were assessed using two-way ANOVAs with cocaine dose and pretreatment as between-subject measures. Reinstatement was defined as the change in the time spent in the cocaine-paired compartment relative to the prior extinction session.
To determine plasma corticosterone levels resulting from HBC-corticosterone administration, mice were decapitated 40 min after ip corticosterone or saline administration and trunk blood was collected and plasma isolated for measurement of corticosterone using a commercial RIA Kit (MP Biomedicals). To allow comparison with stressor-induced corticosterone levels, a separate group of mice underwent a 6-min forced swim in 22 °C water (Mantsch et al, 2010) prior to decapitation and trunk blood collection, 40 min later. Plasma corticosterone levels following ip vehicle or corticosterone administration were also measured in wild-type and OCT3-deficient mice (see Experiment 2).
Experiment 2: Contribution of OCT3 to Corticosterone- and Normetanephrine-Potentiated Reinstatement
To examine the contribution of OCT3 to corticosterone- and normetanephrine-potentiated reinstatement, separate groups of OCT3-deficient and wild-type mice underwent CPP followed by extinction and were tested for reinstatement in response to ip saline or low-dose (0.93 mg/kg) cocaine after pretreatment with corticosterone, normetanephrine, or vehicle. The 0.93 mg/kg cocaine dose was selected because, in C57BL/6 mice, (1) it was subthreshold in terms of reinstatement and (2) robust corticosterone- and normetanephrine-induced potentiation was observed at this cocaine dose. Each mouse was tested for reinstatement at least twice (once with corticosterone, normetanephrine, or vehicle administered prior to injection with 0.93 mg/kg cocaine and once for the same treatment prior to a saline injection) and as many as four times. Each reinstatement test was separated by additional extinction sessions with the requirement that extinction-level preference was re-established prior to retesting. Separate two-way pretreatment × cocaine treatment ANOVAs were conducted in each genotype with planned comparisons for pretreatment effects at the 0. 93 mg/kg dose.
To determine whether OCT3 deficiency alone promoted low-dose cocaine-induced reinstatement, we compared pooled reinstatement data from all vehicle-pretreated wild-type mice with pooled data from all vehicle-pretreated OCT-deficient mice and conducted a two-way (genotype × cocaine/saline treatment) ANOVA. Additionally, to more closely examine differences in corticosterone and normetanephrine effects across genotypes, three-way genotype × pretreatment × cocaine treatment ANOVAs were also conducted.
RESULTS
Experiment #1: Effects of Corticosterone and the Non-Glucocorticoid OCT3 Inhibitor, Normetanephrine, on Cocaine-Primed Reinstatement of Extinguished CPP in C57BL/6 Mice
Overall, C57BL/6 mice displayed robust CPP and this preference was attenuated as a result of extinction (Figure 1a). One-way ANOVA showed a significant overall effect of test condition (preconditioning, postconditioning, last extinction session prior to reinstatement testing) on preference for the cocaine compartment (one-way ANOVA; F2,118=187.72; P<0.001). Post-hoc testing revealed that mice spent more time in the cocaine compartment after conditioning (P<0.05, postconditioning vs preconditioning) and that time spent in the cocaine compartment was reduced as a result of extinction training (P<0.05, last extinction vs postconditioning). There were no differences in CPP or extinction between groups of mice that subsequently received vehicle, corticosterone, or normetanephrine pretreatments (Supplementary Tables S1 and S2). A one-way ANOVA showed that cocaine dose-dependently reinstated extinguished CPP (F5,89=7.551; P<0.001; Figure 1b) with significant reinstatement observed at the 3.75, 7.5, and 15.0 mg/kg cocaine-priming doses (P<0.05 vs 0 mg/kg; Bonferroni-corrected t-tests). No significant differences in the total time spent in the cocaine and saline compartments combined were observed during reinstatement testing. To examine the effects of corticosterone or normetanephrine pretreatment on reinstatement, separate cohorts of C57BL/6 mice were tested for cocaine-primed reinstatement following pretreatment with 2.0 mg/kg corticosterone, 2.5 mg/kg normetanephrine, or vehicle. Separate analyses were conducted to assess the effects of corticosterone (two-way corticosterone treatment × cocaine dose ANOVA) and normetanephrine (two-way normetanephrine treatment × cocaine dose ANOVA).

Cocaine-induced conditioned place preference (CPP), extinction, and cocaine-primed reinstatement in C57BL/6 mice. Mice underwent CPP at a conditioning dose of 15 mg/kg, ip prior to extinction and testing for reinstatement in response to ip cocaine priming doses (0–15 mg/kg, ip). Data in panel (a) represent time spent in the cocaine-paired compartment (seconds; mean±SE) prior to (Pre-Cond) and after (Post-Cond) conditioning and on the final day of extinction (prior to reinstatement testing). Data from all mice used for reinstatement testing were combined for analyses (n=66). Mice showed significant cocaine-induced CPP (*P<0.05 Pre- vs Post-Cond) and preference was diminished as a result of extinction training (#P<0.05 Extinction vs Post-Cond). Data in panel (b) represent a dose–response curve for cocaine-primed reinstatement of extinguished preference (change in time spent in the cocaine-paired compartment relative to the prior extinction session; seconds, mean±SE). Cocaine dose-dependently reinstated preference for the cocaine compartment (*P<0.05 vs 0 mg/kg dose; n=15–26 per cocaine dose).
Effects of Corticosterone on Cocaine-Primed Reinstatement
A two-way ANOVA revealed a significant main effect of cocaine dose (F5,201=6.255; P<0.001; Figure 2a) but not of corticosterone pretreatment on reinstatement and a significant corticosterone pretreatment × cocaine dose interaction (F5,201=3.537; P<0.01). Corticosterone did not induce reinstatement on its own but produced a leftward shift in the dose–response curve for cocaine-primed reinstatement. In vehicle-pretreated mice, cocaine dose-dependently reinstated cocaine seeking with significant increases relative to saline observed at the 3.75, 7.5, and 15 mg/kg cocaine-priming doses (P<0.05). Corticosterone-pretreated mice were more sensitive to cocaine-induced reinstatement and displayed significant reinstatement in response to 0.93 and 1.88 mg/kg cocaine, in addition to the 3.75 mg/kg cocaine dose. Moreover, post-hoc testing showed that, while there were no differences between corticosterone- and vehicle-pretreated mice at the 0 mg/kg cocaine dose, corticosterone-pretreated mice displayed significant increases in reinstatement relative to vehicle-pretreated mice that received either 0.93 or 1.88 mg/kg cocaine (P<0.05; Bonferroni-corrected t-tests). At higher cocaine doses, the magnitude of reinstatement declined in corticosterone-pretreated mice, resulting in a descending limb in the reinstatement dose–response curve. The times spent in the cocaine-paired compartment on the reinstatement test days and preceding extinction sessions are shown in Supplementary Table S1.

Cocaine-primed reinstatement following CPP is potentiated by pretreatment with corticosterone or normetanephrine. Neither pretreatment with corticosterone (2.0 mg/kg, ip; 40 min; (a)) nor normetanephrine (2.5 mg/kg, ip; 40 min; (b)) reinstated extinguished preference alone, but each potentiated cocaine-primed reinstatement as indicated by a leftward shift in the dose–response curve for cocaine-primed reinstatement. Data represent the change in the time spent in the cocaine-paired compartment (sec; mean±SE) upon reinstatement testing relative to the prior extinction session. Separate cohorts of mice were pretreated with vehicle (n=15–26 per cocaine dose), corticosterone (Cort; n=12–20 per cocaine dose), and normetanephrine (Normet; n=13–18 per cocaine dose). Separate analyses were conducted comparing the effects of vehicle and corticosterone (a) and vehicle and normetanephrine (b) on cocaine-primed reinstatement. Cocaine dose-dependently increased preference (#P<0.05), and reinstatement of CPP in response to lower doses of cocaine was augmented following pretreatment with corticosterone or normetanephrine (*P<0.05 vs Vehicle).
Corticosterone administration significantly increased blood levels relative to vehicle injection (t17=11.50; P<0.001; two-tailed t-test; Supplementary Table S3). Although plasma corticosterone concentrations resulting from corticosterone administration were significantly higher than those resulting from forced swim (t23=5.70; P<0.001; two-tailed t-test), they were within previously reported stress-induced ranges (Aliczki et al, 2013; Gong et al, 2015; Malisch et al, 2007).
Effects of Normetanephrine on Cocaine-Primed Reinstatement
A two-way ANOVA showed a significant main effect of cocaine dose (F5,186=6.823; P<0.001) but not of normetanephrine pretreatment on reinstatement and a significant normetanephrine pretreatment × cocaine dose interaction (F5,186=2.967; P<0. 05; Figure 2b). Normetanephrine did not induce reinstatement on its own, but, similar to corticosterone, it shifted the dose–response curve for cocaine-primed reinstatement to the left. In contrast to vehicle-pretreated mice, in which reinstatement was only observed at and above the 3.75 mg/kg cocaine priming dose, in normetanephrine-pretreated mice, reinstatement was observed at 0.93 and 1.88 mg/kg cocaine. Moreover, while the effects of normetanephrine did not differ from vehicle at the 0 mg/kg dose, normetanephrine-induced increases in reinstatement relative to vehicle were also observed at 0.93 and 1.88 mg/kg cocaine. With the exception of the 15 mg/kg cocaine dose, as the cocaine dose increased, the magnitude of reinstatement also declined in normetanephrine-pretreated mice, again suggestive of a descending limb in the reinstatement dose–response curve. The times spent in the cocaine-paired compartment on the reinstatement test days and preceding extinction sessions are shown in Supplementary Table S1.
Experiment #2: Role of OCT3 in Corticosterone-Potentiated Cocaine-Primed Reinstatement of CPP, Extinction, and Cocaine-Primed Reinstatement in Wild-Type and OCT3-Deficient Mice
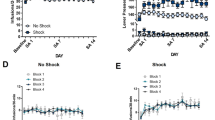
No differences in cocaine-induced CPP or extinction were observed between wild-type and OCT3-deficient mice (Figure 3a; Supplementary Table S4). A two-way conditioning session × genotype ANOVA showed a significant overall effect of conditioning (F1,184=203.84; P<0.0001) but no significant effect of genotype or genotype × conditioning session interaction.

Cocaine-induced conditioned place preference (CPP), extinction, and cocaine-primed reinstatement in OCT3-deficient and wild-type mice. Cocaine-induced CPP and extinction (a) and reinstatement in response to a low dose of cocaine (0.93 mg/kg, ip; (b)) did not significantly differ between OCT3-deficient (n=101) and wild-type mice (n=85). Data in panel (a) represent time spent in the cocaine-paired compartment (seconds; mean±SE) prior to and after conditioning and on the final day of extinction prior to reinstatement testing in wild-type and OCT3 knockout (KO) mice. Overall, cocaine-induced CPP (*P<0.05 Post-Cond vs Pre-Cond) and extinction (#P<0.05 Last Ext vs Post-Cond) were observed in both genotypes and did not differ between wild-type and OCT3 KO mice. Data in panel (b) represent reinstatement of extinguished preference (change in time spent in the cocaine-paired compartment relative to the prior extinction session; seconds; mean±SE) following saline (0 mg/kg, ip) or low-dose cocaine (0.93 mg/kg, ip) administration in wild-type (n=26 saline; n=32 cocaine) and OCT3 KO mice (n=49 saline; n=57 cocaine). Overall, 0.93 mg/kg cocaine failed to reinstate preference and preference levels during reinstatement testing did not significantly differ between wild-type and OCT3 KO mice.
As the primary objective of the experiment was to determine whether the absence of OCT3 prevents corticosterone- and normetanephrine-potentiated reinstatement, our primary statistical analyses consisted of separate two-way ANOVAs within genotypes (see below). However, to determine whether there were any complex interactions between pretreatment effects, cocaine-primed reinstatement, and OCT3 deficiency, we also conducted three-way genotype × corticosterone/normetanephrine × low-dose cocaine-induced reinstatement ANOVAs. These analyses failed to demonstrate significant (a) genotype × pretreatment interactions or (b) genotype × pretreatment × cocaine treatment interactions. We also tested for basal differences in reinstatement in response to 0.93 mg/kg cocaine, a dose at which both corticosterone and normetanephrine potentiated reinstatement in C57BL/6 mice, by comparing pooled data from all vehicle-pretreated wild-type and OCT3-deficient mice. A two-way cocaine treatment (0.93 mg/kg cocaine vs saline) × genotype ANOVA revealed no significant main effects of cocaine or genotype or a cocaine × genotype interaction (Figure 3b).
Contribution of OCT3 to Corticosterone-Potentiated Cocaine-Induced Reinstatement
Pretreatment with corticosterone potentiated cocaine-primed reinstatement in wild-type but not in OCT3-deficient mice (Figure 4a). A two-way cocaine treatment (0.93 mg/kg cocaine vs saline) × corticosterone pretreatment (2.0 mg/kg corticosterone vs vehicle) ANOVA revealed significant main effects of both cocaine treatment (F1,67=5.66; P<0.05) and corticosterone pretreatment (F1,67=4.82; P<0.05), as well as a significant cocaine treatment × corticosterone pretreatment interaction (F1,67=4.21; P<0.05) in wild-type mice. Post-hoc testing using Bonferroni-corrected t-tests, as well as planned comparisons, revealed that, compared with vehicle-pretreated mice, wild-type mice pretreated with corticosterone showed a significantly greater increase in time spent in the cocaine-paired compared compartment in response to low-dose cocaine (P<0.05). Likewise, corticosterone- but not vehicle-pretreated wild-type mice displayed significant low-dose cocaine-induced reinstatement, as defined by a greater increase in time spent in the cocaine-paired compartment following 0.93 mg/kg cocaine relative to saline (P<0.05). In contrast, in OCT3-deficient mice, neither main effects of cocaine or corticosterone nor a cocaine × corticosterone interaction were observed (Figure 4b), suggesting that the effects of corticosterone on reinstatement were indeed mediated through its interaction with OCT3. The times spent in the cocaine-paired compartment on the reinstatement test days and preceding extinction sessions are shown in Supplementary Table S6. Plasma corticosterone levels did not differ between wild-type and OCT3-deficient mice following ip administration of corticosterone (Supplementary Table S5).

Corticosterone potentiates cocaine-primed reinstatement in wild-type but not OCT3-deficient mice. Data represent reinstatement of extinguished preference (change in time spent in the cocaine-paired compartment relative to the prior extinction session; seconds; mean±SE) following saline (0 mg/kg, ip) or low-dose cocaine (0.93 mg/kg, ip) administration in wild-type (a) and OCT3 KO (b) mice after pretreatment with corticosterone (Cort; 2.0 mg/kg, ip; 40 min; wild-type n=22; KO n=23) or vehicle (Veh; wild-type n=16; KO n=22). In wild-type mice, neither 0.093 mg/kg cocaine following vehicle pretreatment nor saline injection following corticosterone pretreatment reinstated preference. However, injection of 0.093 mg/kg cocaine did reinstate preference when it was administered following corticosterone pretreatment (*P<0.05 vs Cort/0 mg/kg cocaine and Veh/0.093 mg/kg cocaine; (a)). By contrast, no significant effects of cocaine or corticosterone pretreatment were observed in OCT3 KO mice (b).
Contribution of OCT3 to Normetanephrine-Potentiated Cocaine-Induced Reinstatement
Normetanephrine pretreatment also potentiated low-dose cocaine-primed reinstatement in wild-type but not in OCT3-deficient mice (Figure 5a). A two-way cocaine treatment (0.93 mg/kg cocaine vs saline) × normetanephrine pretreatment (2.5 mg/kg normetanephrine vs veh) ANOVA revealed main effects of both cocaine treatment (F1,97=11.15; P<0.05) and normetanephrine pretreatment (F1,97=6.68; P<0.05) in wild-type mice. However, despite a trend (P=0.10), a significant cocaine × normetanephrine interaction was not observed. Planned comparisons revealed that, compared with vehicle-pretreated mice, wild-type mice pretreated with normetanephrine displayed a significantly greater increase in preference in response to low-dose cocaine (t53=3.38; P=0.001; unpaired t-test) and that low-dose cocaine administration resulted in significant reinstatement in normetanephrine- but not in vehicle-pretreated wild-type mice (0.93 mg/kg cocaine vs saline; t41=3.35; P<0.01; unpaired t-test). By contrast, in OCT3-deficient mice, no main effects of cocaine or normetanephrine were observed, and there was no cocaine × normetanephrine interaction (Figure 5b). The times spent in the cocaine-paired compartment on the reinstatement test days and preceding extinction sessions are shown in Supplementary Table S7.

Normetanephrine potentiates cocaine-primed reinstatement in wild-type but not OCT3-deficient mice. Data represent reinstatement of extinguished preference (change in time spent in the cocaine-paired compartment relative to the prior extinction session; seconds; mean±SE) following saline (0 mg/kg, ip) or low-dose cocaine (0.93 mg/kg, ip) administration in wild-type (a) and OCT3 KO (b) mice following pretreatment with normetanephrine (Normet; 2.5 mg/kg, ip; 40 min; wild-type n=32; KO n=40) or vehicle (wild-type n=23; KO n=17). In wild-type mice, neither 0.093 mg/kg cocaine following vehicle pretreatment nor saline injection following normetanephrine pretreatment reinstated preference. However, injection of 0.093 mg/kg cocaine did reinstate preference when it was administered following normetanephrine pretreatment (*P<0.05 vs Normet/0 mg/kg cocaine and Veh/0.093 mg/kg cocaine; Figure 4a). No significant effects cocaine or normetanephrine pretreatment were observed in OCT3 KO mice (Figure 4b).
DISCUSSION
We previously reported that, under conditions in which stressors do not directly evoke cocaine seeking in rats, they can ‘set the stage’ for relapse, enhancing the reinstatement of cocaine seeking in response to an otherwise subthreshold priming dose of cocaine (Graf et al, 2013; McReynolds et al, 2016). These stage-setting effects of stress are mediated by glucocorticoids (Graf et al, 2013). The ability of electric footshock stress to potentiate reinstatement is eliminated by surgical adrenalectomy and is reproduced in the absence of shock by administration of corticosterone at a dose that reproduces plasma levels observed during stress. Here we demonstrate that stress-level corticosterone administration also potentiates cocaine-induced reinstatement of extinguished CPP in mice. Following CPP, cocaine dose-dependently reinstated preference for the cocaine-paired compartment. As reported in rats, pretreatment of mice with corticosterone, at a dose that alone did not produce reinstatement, increased the sensitivity to cocaine-induced reinstatement as indicated by a leftward shift in the cocaine dose–response curve. The ability of circulating glucocorticoids to influence relapse susceptibility is significant in that elevated cortisol levels in cocaine-dependent human subjects are associated with stress- and cue-induced increases in subjective craving measures (Sinha et al, 1999) and that craving measures can be predicted by individual cortisol responses (Sinha et al, 2006). Thus identifying mechanisms through which glucocorticoids promote cocaine seeking may guide the development of interventions aimed at relapse prevention.
The establishment of a protocol for assessing corticosterone-potentiated cocaine seeking in mice is significant, as it permits the utilization of available transgenic mouse models to study the glucocorticoid-dependent mechanisms through which stressors promote cocaine use. One mechanism of interest is the OCT (OCT3), a low-affinity, high-capacity monoamine transporter that is inhibited by glucocorticoids. Using our rat model, we previously provided indirect evidence that glucocorticoid treatment potentiates subthreshold cocaine-induced reinstatement by inhibiting OCT3-mediated dopamine clearance in the NAc (Graf et al, 2013). Specifically, we found that the effect of corticosterone on cocaine-primed reinstatement (1) was not blocked by the GR antagonist RU486, (2) was reproduced by intra-NAc corticosterone injections, (3) was dependent on NAc dopamine receptor activation and (4) was associated with potentiation of the NAc dopamine response to cocaine. Importantly, we demonstrated that acute treatment of rats with stress-level corticosterone decreased dopamine clearance via a DAT-independent mechanism and that stress- and corticosterone-induced potentiation of cocaine-primed reinstatement were reproduced by administration of the non-glucocorticoid OCT3 blocker, normetanephrine. In the present study, we demonstrate that pretreatment with either corticosterone or normetanephrine potentiates low-dose cocaine-induced reinstatement of extinguished CPP in mice. As with corticosterone, normetanephrine, at a dose that fails to re-establish preference for the cocaine-paired compartment, produces a leftward shift in the dose–response curve for cocaine-primed reinstatement.
Although collectively these findings strongly suggest that corticosterone enhances cocaine seeking by inhibiting OCT3-mediated transport, they are not conclusive. To more rigorously test the hypothesis that corticosterone acts by blocking OCT3, we examined the effects of OCT3 inhibitors on low-dose cocaine-induced reinstatement in wild-type and OCT3-deficient mice (Zwart et al, 2001). Prior work using these mice has implicated OCT3 in anxiety-related behavioral responses (Vialou et al, 2008) and in salt appetite (Vialou et al, 2004). However, there are, to our knowledge, no reports on the effects of corticosterone on behavior in OCT3-deficient mice. Here, in the first studies to examine the role of this corticosterone-sensitive transporter in behavioral actions of the hormone, we demonstrate that the ability of corticosterone to potentiate cocaine-primed reinstatement is eliminated in OCT3-deficient mice. Pretreatment with corticosterone potentiated low-dose cocaine-primed reinstatement of extinguished CPP in wild-type, but not in OCT3-deficient, mice. Thus these are the first studies to directly implicate OCT3, not only in corticosterone’s effects on cocaine seeking but also in any behavioral effect of corticosterone.
OCT3-deficient mice did not display differences in the acquisition or extinction of cocaine-induced CPP and, despite hints of increased sensitivity to cocaine-primed reinstatement, did not display significant increases in CPP reinstatement in the absence of corticosterone or normetanephrine. This finding is somewhat surprising, given the well-established contribution of monoamines to the induction, retrieval, extinction, and reinstatement of cocaine-induced CPP. However, it is consistent with a previous study examining the effects of amphetamine and cocaine in OCT3-deficient mice. In that study, OCT3-deficient mice displayed no differences in either amphetamine-induced CPP or behavioral sensitization to amphetamine compared with wild-type mice (Vialou et al, 2008). In the same study, OCT3-deficient mice did display enhanced locomotor responses to acute treatment with cocaine and amphetamine but only at very high doses of the drugs (Vialou et al, 2008). Whereas no differences in basal or low-to-moderate dose (10 or 20 mg/kg, ip) cocaine-induced increases in locomotor activity were observed, the locomotor response to a very high cocaine dose (40 mg/kg, ip) was augmented in knockout mice. Definitive conclusions regarding the contribution of OCT3 to acquisition and reinstatement of CPP in the absence of corticosterone/normetanephrine will require testing across a broader range of cocaine doses during conditioning and reinstatement and is beyond the scope of the present study, which was aimed solely at confirming the contribution of OCT3 to the potentiating effects of corticosterone.
Interestingly, although reductions in the total tissue content of dopamine and its metabolites have been found in OCT3 knockout mice (Vialou et al, 2008), basal extracellular dopamine, as measured using in vivo microdialysis, is not altered (Cui et al, 2009), suggesting that deletion of OCT3 may result in compensatory alterations in the expression of other transporters that limit effects on neurotransmission. Consistent with this possibility, it has been reported that constitutive deletion of one monoamine transporter often leads to changes in the expression and/or function of others (Shen et al, 2004; Baganz et al, 2008; Daws, 2009). Thus, while dopamine clearance at sites in which OCT3 represents the primary transport mechanism will be impaired in OCT3-deficient mice, increased expression of DAT and other dopamine-clearing transporters may compensate for the loss of OCT3-mediated dopamine clearance. For this reason, it is likely that the loss of OCT3-mediated clearance would be most evident under extreme conditions, when the capacity for other transporters to clear dopamine is limiting. This is consistent with a report that enhanced behavioral responses to cocaine and amphetamine were only observed in OCT3-deficient mice at very high doses of the drugs (Vialou et al, 2008). Under our hypothesis that the potentiating effects of corticosterone on reinstatement are attributable solely to its ability to inhibit OCT3, compensatory changes in the expression of other transporters would not be predicted to restore corticosterone sensitivity.
The results of our previous studies in rats strongly suggest that corticosterone acts directly in the NAc to potentiate the effects of low-dose cocaine on behavior and dopamine signaling (Graf et al, 2013). Specifically: (a) direct delivery of corticosterone into this OCT3-expressing brain region potentiated low-dose cocaine effects on behavior (reinstatement); (b) the ability of ip corticosterone to potentiate reinstatement is blocked by intra-NAc injections of dopamine receptor antagonists; and (c) acute corticosterone treatment potentiates the effects of low-dose cocaine on extracellular dopamine concentrations measured using microdialysis and decreases DAT-independent dopamine clearance measured using fast-scan cyclic voltammetry. However, while these findings provide strong evidence for NAc dopamine in the effects of corticosterone, other sites of action and cellular mechanisms cannot be ruled out. OCT3 is expressed throughout the brain, including in other regions that contribute to cocaine-primed reinstatement, such as the prefrontal cortex (Gasser et al, 2009). Moreover, OCT3 mediates corticosterone-sensitive transport of other monoamine substrates, including serotonin and norepinephrine (Gründemann et al, 1998a, 1998b). Both of these monoamines have also been implicated in the acute regulation of cocaine-seeking behavior (see, eg, Baker et al, 2001; Fletcher et al, 2002; Burmeister et al, 2004; Nic Dhonnchadha et al, 2009) (Erb et al, 2000; Zhang and Kosten, 2005; Platt et al, 2007; Mantsch et al, 2010).
The present studies demonstrate that OCT3 expression is required for potentiation of low-dose cocaine-induced reinstatement of cocaine seeking not only by corticosterone but also by the non-glucocorticoid OCT3 inhibitor normetanephrine. This is important, because, while normetanephrine is a potent inhibitor of OCT3-mediated transport (Martel et al, 1999), it has been reported to have other pharmacological effects, including weak agonist actions at alpha-1 adrenergic receptors (Langer and Rubio 1973) and low-affinity antagonism of alpha-2 adrenergic receptors (Lenz et al, 1991). The present findings indicate that, to the extent that these secondary pharmacological effects occur, the regulation of cocaine-induced reinstatement by normetanephrine is likely the result of OCT3 blockade, thereby validating the use of this compound as a pharmacological tool for studying OCT3-mediated processes.
To summarize, using a CPP/reinstatement approach in mice, we provide converging lines of evidence that OCT3 is necessary for the potentiating effects of corticosterone on the reinstatement of cocaine-seeking behavior. These results build on our prior findings in rats (Graf et al, 2013) to demonstrate, for the first time, that inhibition of OCT3 by corticosterone, likely via non-genomic effects on monoamine clearance, has behavioral consequences. Moreover, they pose OCT3 blockade as a potential mechanism through which stressful stimuli influence drug craving and relapse susceptibility and as a potential contributor to a wide range of stress-related behavioral responses.
FUNDING AND DISCLOSURE
This work was supported by NIH grants R01 DA015758 and R01 DA032895. All drugs used are commercially available. Homozygotic OCT3-deficient and wild-type mice were provided by Dr Kim Tieu, previously at the University of Rochester and now at Plymouth University, UK, with permission from Dr Bruno Giros and Dr Sophie Gautron (INSERM) and were bred at the Marquette University Animal Research Center. The authors declare no conflict of interest.
References
Aliczki M, Zelena D, Mikics E, Varga ZK, Pinter O, Bakos NV et al (2013). Monoacylglycerol lipase inhibition-induced changes in plasma corticosterone levels, anxiety and locomotor activity in male CD1 mice. Horm Behav 63: 752–758.
Amphoux A, Vialou V, Drescher E, Bruss M, La Cour CM, Rochat C et al (2006). Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology 50: 941–952.
Baganz NL, Horton RE, Calderon AS, Owens WA, Munn JL, Watts LT et al (2008). Organic cation transporter 3: Keeping the brake on extracellular serotonin in serotonin-transporter-deficient mice. Proc Natl Acad Sci USA 105: 18976–18981.
Baganz N, Horton R, Martin K, Holmes A, Daws LC (2010). Repeated swim impairs serotonin clearance via a corticosterone-sensitive mechanism: organic cation transporter 3, the smoking gun. J Neurosci 30: 15185–15195.
Baker DA, Tran-Nguyen TL, Fuchs RA, Neisewander JL (2001). Influence of individual differences and chronic fluoxetine treatment on cocaine-seeking behavior in rats. Psychopharmacology 155: 18–26.
Burmeister JJ, Lungren EM, Kirschner KF, Neisewander JL (2004). Differential roles of 5-HT receptor subtypes in cue and cocaine reinstatement of cocaine-seeking behavior in rats. Neuropsychopharmacology 29: 660–668.
Cui M, Aras R, Christian WV, Rappold PM, Hatwar M, Panza J, Jackson-Lewis V et al (2009). The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc Natl Acad Sci USA 106: 8043–8048.
Daws LC (2009). Unfaithful neurotransmitter transporters: focus on serotonin uptake and implications for antidepressant efficacy. Pharmacol Ther 121: 89–99.
Duan H, Wang J (2010). Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. J Pharmacol Exp Ther 335: 743–753.
Engel K, Zhou M, Wang J (2004). Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem 279: 50042–50049.
Erb S, Hitchcott PK, Rajabi H, Mueller D, Shaham Y, Stewart J (2000). Alpha-2 adrenergic receptor agonists block stress-induced reinstatement of cocaine seeking. Neuropsychopharmacology 23: 138–150.
Fletcher PJ, Grottick AJ, Higgins GA (2002). Differential effects of the 5-HT(2A) receptor antagonist M100907 and the 5-HT(2C) receptor antagonist SB242084 on cocaine-induced locomotor activity, cocaine self-administration and cocaine-induced reinstatement of responding. Neuropsychopharmacology 27: 576–586.
Gasser PJ, Lowry CA, Orchinik M (2006). Corticosterone-sensitive monoamine transport in the rat dorsomedial hypothalamus: potential role for organic cation transporter 3 in stress-induced modulation of monoaminergic neurotransmission. J Neurosci 26: 8758–8766.
Gasser PJ, Orchinik M, Raju I, Lowry CA (2009). Distribution of organic cation transporter 3, a corticosterone-sensitive monoamine transporter, in the rat brain. J Comp Neurol 512: 529–555.
Gong S, Miao YL, Jiao GZ, Sun MJ, Li H, Lin J et al (2015). Dynamics and correlation of serum cortisol and corticosterone under different physiological or stressful conditions in mice. PLoS One 10: e0117503.
Gorboulev V, Shatskaya N, Volk C, Koepsell H (2005). Subtype-specific affinity for corticosterone of rat organic cation transporters rOCT1 and rOCT2 depends on three amino acids within the substrate binding region. Mol Pharmacol 67: 1612–1619.
Graf EN, Wheeler RA, Baker DA, Ebben AL, Hill JE, McReynolds JR et al (2013). Corticosterone acts in the nucleus accumbens to enhance dopamine signaling and potentiate reinstatement of cocaine seeking. J Neurosci 33: 11800–11810.
Gründemann D, Köster S, Kiefer N, Breidert T, Engelhardt M, Spitzenberger F et al (1998a). Transport of monoamine transmitters by the organic cation transporter type 2, OCT2. J Biol Chem 273: 30915–30920.
Gründemann D, Schechinger B, Rappold GA, Schömig E (1998b). Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nat Neurosci 1: 349–351.
Gründemann D, Liebich G, Kiefer N, Köster S, Schömig E (1999). Selective substrates for non-neuronal monoamine transporters. Mol Pharmacol 56: 1–10.
Horvath G, Sutto Z, Torbati A, Conner GE, Salathe M, Wanner A (2003). Norepinephrine transport by the extraneuronal monoamine transporter in human bronchial arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 285: L829–L837.
Iversen LL, Salt PJ (1970). Inhibition of catecholamine uptake-2 by steroids in the isolated rat heart. Br J Pharmacol 40: 528–530.
Langer SZ, Rubio MC (1973). Effects of the noradrenaline metabolites on the adrenergic receptors. Naunyn Schmiedebergs Arch Pharmacol 276: 71–88.
Lenz T, Werle E, Strobel G, Weicker H (1991). O-Methylated and sulfoconjugated catecholamines: differential activities at human platelet alpha 2-adrenoceptors. Can J Physiol Pharmacol 69: 929–937.
Mantsch JR, Weyer A, Vranjkovic O, Beyer CE, Baker DA, Caretta H (2010). Involvement of noradrenergic neurotransmission in the stress- but not cocaine-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: role for β-2 adrenergic receptors. Neuropsychopharmacology 35: 2165–2178.
Malisch JL, Saltzman W, Gomes FR, Rezende EL, Jeske DR, Garland T Jr. (2007). Baseline and stress-induced plasma corticosterone concentrations of mice selectively bred for high voluntary wheel running. Physiol Biochem Zool 80: 146–156.
Martel F, Ribeiro L, Calhau C, Azevedo I (1999). Comparison between uptake2 and rOCT1: effects of catecholamines, metanephrines and corticosterone. Naunyn Schmiedebergs Arch Pharmacol 359: 303–309.
McReynolds JR, Doncheck EM, Vranjkovic O, Ganzman GS, Baker DA, Hillard CJ, Mantsch JR (2016). CB1 receptor antagonism blocks stress-potentiated reinstatement of cocaine seeking in rats. Psychopharmacology 233: 99–109.
Nic Dhonnchadha BA, Fox RG, Stutz SJ, Rice KC, Cunningham KA (2009). Blockade of the serotonin 5-HT2A receptor suppresses cue-evoked reinstatement of cocaine-seeking behavior in a rat self-administration model. Behav Neurosci 123: 382–396.
Platt DM, Rowlett JK, Spealman RD (2007). Noradrenergic mechanisms in cocaine-induced reinstatement of drug seeking in squirrel monkeys. J Pharmacol Exp Ther 322: 894–902.
Schomig E, Russ H, Staudt K, Martel F, Gliese M, Grundemann D (1998). The extraneuronal monoamine transporter exists in human central nervous system glia. Adv Pharmacol 42: 356–359.
Schmitt A, Mössner R, Gossmann A, Fischer IG, Gorboulev V, Murphy DL et al (2003). Organic cation transporter capable of transporting serotonin is up-regulated in serotonin transporter-deficient mice. J Neurosci Res 71: 701–709.
Shang T, Uihlein AV, Van Asten J, Kalyanaraman B, Hillard CJ (2003). 1-Methyl-4-phenylpyridinium accumulates in cerebellar granule neurons via organic cation transporter 3. J Neurochem 85: 358–367.
Shen HW, Hagino Y, Kobayashi H, Shinohara-Tanaka K, Ikeda K, Yamamoto H et al (2004). Regional differences in extracellular dopamine and serotonin assessed by in vivo microdialysis in mice lacking dopamine and/or serotonin transporters. Neuropsychopharmacology 29: 1790–1799.
Sinha R, Catapano D, O'Malley S (1999). Stress-induced craving and stress response in cocaine dependent individuals. Psychopharmacology 142: 343–351.
Sinha R, Garcia M, Paliwal P, Kreek MJ, Rounsaville BJ (2006). Stress-induced cocaine craving and hypothalamic-pituitary-adrenal responses are predictive of cocaine relapse outcomes. Arch Gen Psychiatry 63: 324–331.
Vialou V, Amphoux A, Zwart R, Giros B, Gautron S (2004). Organic cation transporter 3 (Slc22a3) is implicated in salt-intake regulation. J Neurosci 24: 2846–2851.
Vialou V, Balasse L, Callebert J, Launay JM, Giros B, Gautron S (2008). Altered aminergic neurotransmission in the brain of organic cation transporter 3-deficient mice. J Neurochem 106: 1471–1482.
Vranjkovic O, Hang S, Baker DA, Mantsch JR (2012). Beta adrenergic receptor mediation of stress-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: roles for beta-1 and beta-2 adrenergic receptors. J Pharmacol Exp Ther 342: 541–551.
Zhang XY, Kosten TA (2005). Prazosin, an alpha-1 adrenergic antagonist, reduces cocaine-induced reinstatement of drug seeking. Biol Psychiatry 57: 1202–1204.
Zwart R, Verhaagh S, Buitelaar M, Popp-Snijders C, Barlow DP (2001). Impaired activity of the extraneuronal monoamine transporter system known as uptake-2 in Orct3/Slc22a3-deficient mice. Mol Cell Biol 21: 4188–4196.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article

Cite this article
McReynolds, J., Taylor, A., Vranjkovic, O. et al. Corticosterone Potentiation of Cocaine-Induced Reinstatement of Conditioned Place Preference in Mice is Mediated by Blockade of the Organic Cation Transporter 3. Neuropsychopharmacol 42, 757–765 (2017). https://doi.org/10.1038/npp.2016.187
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.187
This article is cited by
-
Ethanol inhibits dopamine uptake via organic cation transporter 3: Implications for ethanol and cocaine co-abuse
Molecular Psychiatry (2023)
-
Chronically dysregulated corticosterone impairs dopaminergic transmission in the dorsomedial striatum by sex-divergent mechanisms
Neuropsychopharmacology (2023)
-
Neuroinflammatory Response in Reward-Associated Psychostimulants and Opioids: A Review
Cellular and Molecular Neurobiology (2023)
-
Increased cocaine reward in offspring of females exposed to morphine during adolescence
Psychopharmacology (2019)
