
Abstract
Schizophrenia patients exhibit dysfunctional gamma oscillations in response to simple auditory stimuli or more complex cognitive tasks, a phenomenon explained by reduced NMDA transmission within inhibitory/excitatory cortical networks. Indeed, a simple steady-state auditory click stimulation paradigm at gamma frequency (~40 Hz) has been reproducibly shown to reduce entrainment as measured by electroencephalography (EEG) in patients. However, some investigators have reported increased phase locking factor (PLF) and power in response to 40 Hz auditory stimulus in patients. Interestingly, preclinical literature also reflects this contradiction. We investigated whether a graded deficiency in NMDA transmission can account for such disparate findings by administering subanesthetic ketamine (1–30 mg/kg, i.v.) or vehicle to conscious rats (n=12) and testing their EEG entrainment to 40 Hz click stimuli at various time points (~7–62 min after treatment). In separate cohorts, we examined in vivo NMDA channel occupancy and tissue exposure to contextualize ketamine effects. We report a robust inverse relationship between PLF and NMDA occupancy 7 min after dosing. Moreover, ketamine could produce inhibition or disinhibition of the 40 Hz response in a temporally dynamic manner. These results provide for the first time empirical data to understand how cortical NMDA transmission deficit may lead to opposite modulation of the auditory steady-state response (ASSR). Importantly, our findings posit that 40 Hz ASSR is a pharmacodynamic biomarker for cortical NMDA function that is also robustly translatable. Besides schizophrenia, such a functional biomarker may be of value to neuropsychiatric disorders like bipolar and autism spectrum where 40 Hz ASSR deficits have been documented.
Similar content being viewed by others


INTRODUCTION
The sensory cortex exhibits a remarkable ability to faithfully respond to modality-specific stimulus trains (Sannita, 2000). Such entrainment of auditory cortical neurons can be recorded by noninvasive methods such as electroencephalography (EEG) and is termed auditory steady-state response (ASSR). In humans, the scalp-recorded ASSR is maximal when auditory stimuli are delivered at ∼40 Hz (Galambos et al, 1981; Pastor et al, 2002) and appears to reflect resonance frequency of the underlying neural circuits (Llinas, 1988; Llinas et al, 1991; Rosanova et al, 2009). A key part of the circuitry responsible for entrainment to exogenous stimuli appears to involve parvalbumin-positive (PV+), GABAergic basket interneurons and the pyramidal cells of the upper layers of the sensory cortex (Cardin et al, 2009; Carlen et al, 2012; Sohal et al, 2009). Moreover, within this circuitry, NMDA receptor activation on PV+ interneurons appears to be critical for normal 40 Hz entrainment (Carlen et al, 2012; Nakao and Nakazawa, 2014).
Following the original and landmark observation that 40 Hz ASSR is specifically deficient in schizophrenia subjects (Kwon et al, 1999), numerous reports have broadly confirmed the finding (Brenner et al, 2003; Krishnan et al, 2009; Light et al, 2006; Spencer, 2011; Spencer et al, 2008; Vierling-Claassen et al, 2008), and extended it to first-degree relatives of schizophrenia subjects (Hong et al, 2004; Rass et al, 2012), suggesting that such a deficit may be a trait marker. However, it is pertinent to note that 40 Hz ASSR deficit is not specific to schizophrenia and extends to some neuropsychiatric conditions including bipolar disorder (Oda et al, 2012; Rass et al, 2010) and autism spectrum disorders (Rojas et al, 2011; Wilson et al, 2007).
Although a majority of reports from schizophrenia subjects have found a reduction in phase locking factor (PLF) and evoked power (reviewed in O'Donnell et al, 2013), some have found an increase in these measures (Hamm et al, 2012; Hong et al, 2004). Intriguingly, a positive correlation between auditory hallucinations and signal power or phase locking, particularly in the left hemisphere (Mulert et al, 2011, 2012; Spencer et al, 2009; Ying et al, 2013), has been observed (Mulert et al, 2011, 2012; Spencer et al, 2009; Ying et al, 2013) in the presence of concurrent increases in spontaneous or induced gamma power (Baldeweg et al, 1998; Hirano et al, 2015; Spencer, 2011). Thus, there appears to be a positive or negative modulation of the gamma oscillatory signal including the 40 Hz ASSR in schizophrenia.
Acute NMDA antagonism by drugs like phencyclidine and ketamine is known to mimic a brief schizophrenia-like state in healthy individuals (Javitt and Zukin, 1991; Krystal et al, 1994; Luby et al, 1962). Taking advantage of this important validation, preclinical studies often employ a short-term disruption in NMDA neurotransmission to mimic a psychosis-like state (Adell et al, 2012; Frohlich and Van Horn, 2014; Gunduz-Bruce, 2009). Several groups have used this approach to understand the role of NMDA receptors in the 40 Hz ASSR (Sivarao et al, 2013; Sullivan et al, 2015; Vohs et al, 2012). Unfortunately, these studies reported opposite modulation of the ASSR signal with NMDA antagonists while employing divergent methodology. Sivarao et al (2013) showed that a low dose of the potent NMDA antagonist MK-801 attenuated ASSR at 40 Hz in anesthetized rats, Sullivan et al (2015) reported that a single dose of MK-801 increased phase locking at 40 Hz in awake behaving rats, whereas Vohs et al (2012) reported an increase in phase locking at multiple frequencies after a single dose of ketamine in conscious rats. Thus, anesthetized and awake rats produced opposite effects on 40 Hz ASSR following acute administration of an NMDA antagonist. Interestingly, in the only known clinical study that tested the effect of a relatively large dose of intravenous ketamine on the 40 Hz ASSR, the authors reported a robust increase in evoked power (Plourde et al, 1997).
In the current study, we tested the hypothesis that intravenous ketamine can increase or decrease the 40 Hz ASSR depending on dose and interval between dosing and measurement. Moreover, to fully contextualize the early effects of ketamine on the 40 Hz ASSR, we conducted in vivo NMDA channel occupancy and exposure studies in separate cohorts of rats.
MATERIALS AND METHODS
Surgery
All experimental procedures were approved by the Bristol Myers Squibb institutional animal care and use committee. Twelve adult male Sprague-Dawley (SD) rats were anesthetized with isoflurane and implanted with epidural screw electrodes at the following coordinates: frontal, 6 mm anterior to bregma and 1 mm lateral to midline; vertex, 5.5 mm caudal to bregma and 1 mm lateral to midline. Access to the electrodes was through a multi-channel pedestal (Plastics One, Roanoke, VA) fixed on the skull at the time of the surgery. After implantation, the rats were housed singly and had unrestricted access to food and water. Rats were thoroughly acclimated to handling and brief restraint procedure described below as well as the recording environment in the presence of auditory stimuli. These rats were previously used for another pharmacological study and were used in the current study after an interval of at least 3 weeks. The approximate age of the rats was 8 months at the time of the study.
Pharmacological Treatments
Rats were individually restrained in a plexiglas restrainer and the tail was secured and swabbed and prepared for i.v. dosing. Percutaneous intravenous bolus injection was given in a 0.5 ml/kg volume and the rat was returned to home cage. The drug doses included 1, 3, 10, and 30 mg/kg of racemic ketamine hydrochloride (Ketaset, Fort Dodge, IA), chosen to obtain a wide range of NMDA channel occupancy (see below) and were split into two panels: in the first panel, vehicle, ketamine 1 and 30 mg/kg were used in a crossover study with at least 4 days in between treatments. Following the completion of the study, after an interval of several weeks, doses 3 and 10 mg/kg were tested along with a vehicle control in the same subjects in a crossover design as above. Drug response was compared with the within-group vehicle response and when comparisons were made across dose panels, it was done after normalization to within-group vehicle response. Thus, any order effects if present were randomized across the three treatment groups. Separate cohorts of adult rats were dosed and killed for characterizing in vivo NMDA channel occupancy as well as for tissue exposure (see below). We did not specifically test for loss of consciousness in these studies. However, rats in the EEG study were video monitored throughout the experiment and it was noted that rats receiving the 30 mpk (mg per kg body weight) dose showed ataxic gait and stereotypic head movements by the first ASSR recording (~7 min), suggesting that loss of consciousness if any was short-lived. Gait abnormalities at this dose lasted till the end of the recording period (64 min). Lower doses of ketamine (3 and 10 mpk, i.v.) showed hyperactivity, circular ambulation along with stereotypic head movements but recovered substantially by the end of the recording period. No behavioral changes were noted at the 1 mpk dose.
EEG Recording
Rats in their home cages were placed individually in sound-attenuated recording boxes equipped with a video camera, a house speaker, and a shielded light weight cable attached to a low-torque commutator (Plastics One). Using the plastic head mounts, the cables were attached for continuous EEG recording while permitting the animals free movement within the cage. For pharmacological studies, rats were first treated and then placed inside the recording boxes. Thus, a pretreatment baseline was not recorded as part of this protocol. Immediately after connecting the recording cable, spontaneous EEG was recorded for a few minutes to confirm signal quality followed by recording in the presence of auditory clicks (see below) that continued up to 64 min after treatment. A total of 75 epochs of 3 s EEG were digitized in succession at a sampling frequency of 2 kHz by the acquisition system (CED Power 1401; Cambridge Electronics Design, Cambridge, UK). Signal 4.10 or higher software was used to generate auditory clicks by feeding a 1 V monophasic square wave (1 ms long with a 0.5 ms raise and fall times) into the house speaker through an audio amplifier (Insignia IS-HC040918; Richfield, MN). The intensity of the 40 Hz click train was adjusted to be 70±1.0 dB when measured from the center of the recording chamber. During each 3 s EEG epoch recording, a single auditory click was presented 1 s after epoch onset, whereas at 2 s, a train of 20 clicks was presented in a half a second period (ie, 40 Hz). Single-click data are not discussed in this report. Four sets of EEG, each lasting for a 3.75-min sample period, were collected after intravenous dosing (approximated to 7, 12, 32, and 62 min after i.v. dosing). The experimental protocol is graphically summarized in Figure 1a and b.

Graphical representation of the experimental paradigm. (a) A schematic of the procedure: following intravenous dosing at time zero, EEG was collected at four times (duration of collection indicated by gray rectangular boxes), each approximated to a discrete time point shown within the gray boxes. For the 7 min time point, using a separate cohort, in vivo NMDA blockade was quantified. Plasma and brain exposure to ketamine was also quantified at indicated times. (b) Schematic representation of auditory stimuli presented in a 3-s epoch; a single click was presented at 1 s from epoch onset, whereas a 40-Hz click train was presented between 2 and 2.5 s. Only 40 Hz data are discussed in this report. (c) For illustration, an averaged and band-pass (35–45 Hz) filtered evoked response from a vehicle-treated subject is shown. Baseline total gamma power was computed using the first 0.5 s of an epoch, and mean ASSR measures were computed through the time of ASSR train presentation (2–2.5 s).
Data Analysis
EEG epochs were visually evaluated and epochs with movement-related artifact were removed from further analysis using an automated script (Signal 4.10). Typically, <10% of the trials were rejected in such analysis. Artifact-free EEG epochs were saved as text and imported into MS Windows-based proprietary software for pre-processing and time-frequency analysis (EMSE Data editor v5.5.2 or higher, Cortech Solutions, Wilmington, NC). Briefly, a second-order Butterworth IIR zero phase-shift band-pass (35–45 Hz) digital filter was applied to the individual EEG epochs and baseline corrected using the time period of 0.5 s preceding the stimulus onset. EEG segments corresponding to the baseline (0–0.5 s) and ASSR stimulus duration (2.0–2.5 s) (Figure 1c) were subjected to time-frequency decomposition using a Morlet wavelet algorithm. Total power (0–0.5 s segment only), mean PLF, and evoked and induced powers (2.0–2.5 s segment only) were computed as below.
Time-varying wavelet transform coefficients (complex valued) are obtained by convolving a Morlet wavelet of a given frequency (complex valued) with an EEG epoch (real valued). Convolution is calculated by multiplying the discrete Fourier transform (DFT) of an EEG epoch with the DFT of a Morlet wavelet followed by inverse DFT (Torrence and Compo, 1998). Total power for a given time-frequency combination is obtained as the average of the modulus squared of the associated wavelet coefficients across epochs. Evoked power is obtained as the modulus squared of the average of the associated wavelet coefficients across epochs. Because of linearity of both the wavelet transform and averaging, evoked power is equivalent to the modulus squared of the time-frequency coefficient obtained from the wavelet transform of the average ERP epoch. Induced power is defined as total power minus evoked power (Roach and Mathalon, 2008). To a first approximation, evoked power measures the EEG signal that is phase locked to the stimulus. This approximation is valid if the phase-locked signal is homogeneous across epochs. Second-order phase-locked variations around the average ERP (such as variations of amplitude scaling) will be allocated to the induced power component (Truccolo et al, 2002).
Data are expressed as group mean along with the SEM. Statistical comparisons were made using one- or two-way analysis of variance (ANOVA) with Dunnett’s test or t-tests as appropriate for significance evaluation. A P<0.05 was deemed to be statistically significant. GraphPadPrism v6.0 (GraphPad Corporation, San Diego, CA) was used for statistical analysis and graphing of the data.
Drugs
A racemic form of the marketed drug ketamine hydrochloride (Ketaset, Fort Dodge, NY) was used for intravenous dosing, and sterile saline for injection was used as a control (Teknova, Hollister, CA). Intravenous dosing was done as a slow bolus over a period of ~1 min (0.5 ml/kg). [3H]MK-801 was synthesized by the Radio synthesis Group at Bristol Myers Squibb, and cold MK-801 (as hydrogen malate) was obtained from Sigma-Aldrich (St Louis, MO).
In Vivo NMDA Occupancy
Ketamine’s occupancy of the NMDA channel was determined using an in vivo [3H]MK-801 binding method modified from that reported previously (Murray et al, 2000). Adult male SD rats equipped with jugular vein catheters (Hilltop Lab Animals, Scottsdale, PA) were dosed with [3H]-MK-801 (200 mCi/2 ml/kg, i.v.) at time zero, and at 3 min, ketamine (1, 3, 10, or 30 mg/kg, i.v.) was administered. At 7 min after ketamine dosing, the rats were killed by decapitation and the forebrain (minus the cerebellum and brainstem) was collected and split into hemispheres. One hemisphere was weighed and homogenized in 30 volumes of ice-cold 5 mM Tris-acetate buffer (pH 7.0). A 600 μl aliquot of the homogenate was filtered through GF/B Whatman filters. Filters were washed with ice-cold Tris-acetate buffer, and then placed in a scintillation vial with 10 ml of scintillation fluid. Radioactivity was counted using a Wallac Microbeta counter (Perkin Elmer, Waltham, MA). Total [3H]MK-801 binding was determined by measuring radioactivity in the forebrain of animals treated with vehicle, whereas the nonspecific binding was determined by measuring the radioactivity in animals administered with MK-801 at 5 mg/kg i.v. Specific [3H]MK-801 binding was calculated by subtracting the value of the nonspecific binding from that of the total binding in each sample.
Quantification of Ketamine in Plasma and Brain
Groups of male SD rats (n=3) were dosed with ketamine (1, 3, 10, or 30 mg/kg, i.v.) at time zero and killed at designated times (7, 32, or 62 min) and tissues were collected and stored at −20 °C until analysis. Brains were thawed and homogenized in four volumes (v/v) of tissue homogenate buffer (133 mM sodium phosphate-buffered saline, pH 7.4). Ketamine plasma and brain concentrations were determined using an LC-MS/MS assay.
RESULTS
Phase Locking Factor
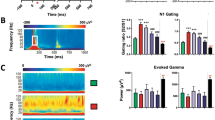
The PLF is a measure of trial-to-trial consistency in phase of the 40 Hz ASSR signal with reference to the time of stimulus onset (also referred to in literature as phase synchrony or intertrial coherence; Roach and Mathalon, 2008). Analysis of the PLF data from dose panel 1 using a two-way ANOVA with repeated measures showed a significant time (F(3, 33)=2.94; P=0.0475) and treatment (F(2, 22)=4.135; P=0.0299) effect and a strong interaction between the two (F(6, 66)=9.552; P<0.0001). Dunnett’s multiple comparison tests were performed to further clarify the dose levels and time points that differentiated from the vehicle group. The grand average heat maps of the PLF response for the 7 min time point are shown in Figure 2. In comparison with vehicle (Figure 2a), ketamine (1 mg/kg; Figure 2b) significantly augmented PLF, whereas at the 30 mg/kg dose (Figure 2c), there was a marked reduction.

Heat map representation of mean PLF measure at the 7 min point following vehicle (a) or ketamine [1 (b) or 30 (c) mg/kg] treatments. Dashed boxes indicate computed activity within the gamma band (35–45Hz) for the duration of the stimulus train (0.5 s). In comparison to the vehicle group, note a clear increase after 1mg/kg ketamine treatment and a reduction after 30 mg/kg treatment. Statistical significance indicated by *P<0.05; Dunnett’s test.
The time course of PLF after vehicle or ketamine (1 or 30 mg/kg) is summarized in Figure 3a. Whereas the augmentation produced by the low dose of ketamine (1 mg/kg) disappeared by 12 min, the suppression produced by the high dose of ketamine (30 mg/kg) lasted up to 32 min (Figure 3a) followed by a reversal at 62 min. The two intermediate doses (3 and 10 mg/kg) showed a significant time effect (F(3, 33)=3.025; P=0.0433), a trend-level treatment effect (F(2, 22)=0.0695), and a strong interaction between the two (F(6, 66)=5.699; P<0.001). Dunnett’s posttests confirmed that the 10 mg/kg dose showed a significant augmentation in PLF at 32 and 62 min time points (Supplementary Figure S1A). Moreover, linear regression analysis between dose and normalized PLF at the 7 min time point showed a robust inverse relationship (R2, 0.43; P<0.0001).

Line graphs summarizing the time course of PLF (a), evoked power (b), and induced power (c) following vehicle or ketamine (1 or 30 mg/kg) treatment. Statistical significance: *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 (Dunnett’s test using vehicle response as the comparator).
Evoked Power
The time- and phase-locked signal power through the period of stimulus presentation at 1 and 30 mg/kg across four time points is compared with that of the vehicle treatment in Figure 3b. Two-way ANOVA revealed significant time (F(3, 33)=3.422; P=0.0284), treatment (F(2, 22)=3.479; P=0.0487), as well as interaction (F(6, 66)=8.804; P<0.0001) effects. The post hoc comparisons unveiled robust effects at multiple time points. Whereas the 1 mg/kg dose increased evoked power at 7 min only (nearly three times the vehicle mean), no change in evoked power was noted at 30 mg/kg dose at the first three time points. However, 62 min after ketamine (30 mg/kg) injection, a robust increase in evoked power, a little over four times the vehicle mean, was observed.
The effect of ketamine (3 and 10 mg/kg, i.v.) on evoked 40 Hz ASSR power showed a significant treatment (F(2, 22)8.727; P=0.0016) and treatment × time interaction (F(6, 66)=4.572; P=0.0006). Dunnett’s multiple comparisons showed significant increase over vehicle treatment for all time points at the 10 mg/kg dose and for the first two time points only for the 3 mg/kg dose. These data are summarized in Supplementary Figure S1B. Unlike for PLF, regression analysis between dose and normalized evoked power at the 7 min time point yielded no significant relationship (R2, 0.06; P>0.05).
Induced Power
Induced power is considered as the phase-variant power instigated by the stimulus and was defined as the residual gamma band power through the duration of ASSR presentation, after the (phase-locked) evoked power has been subtracted from the total power (Roach and Mathalon, 2008). Two-way ANOVA revealed significant time (F(3, 33)=10.66; P<0.0001) and treatment (F(2, 22)=38.37; P<0.0001) effects but no interaction. Dunnett’s multiple comparisons showed robust increase at all time points for the 30 mg/kg dose relative to vehicle and 1 mg/kg dose had significant increase at the two early time points only (Figure 3c).
Robust time (F(3, 33)=7.917; P=0.0004), treatment (F(2, 22)=35.83; P<0.0001), and time × treatment (F(6, 66)=3.258; P=0.0072) effects were noted in the study with the two intermediate doses of ketamine (3 and10 mg/kg). Dunnett’s multiple comparisons revealed that all four time points at both doses were significantly different from that of the vehicle group (Supplementary Figure S1C).
To understand whether the induced gamma is a mere reflection of the elevation in baseline gamma activity that NMDA antagonists are well known to engender (Hakami et al, 2009; Kocsis, 2012; Sebban et al, 2002), we compared induced power through duration of train stimulation with the total gamma band power at the beginning of each epoch when there was no auditory stimulation. We found a highly significant positive correlation (Pearson’s r) and no statistical difference between the two measures (paired t-test; data not shown) under all treatment conditions, suggesting that induced power in our paradigm was merely reflecting the gamma power present in the signal. For example, Pearson’s r for baseline gamma power vs induced gamma power at the 7 min point were as follows (vehicle, 0.95 (P<0.0001); ketamine 1 mpk, 0.81 (P=0.0012); ketamine 30 mpk, 0.95 (P<0.0001).
In Vivo NMDA Channel Occupancy and Ketamine Exposure
In order to contextualize the disparate effects of ketamine on ASSR within the context of target engagement and exposure, in vivo NMDA occupancy and ketamine exposure analysis were undertaken. The linear and dose-dependent NMDA channel occupancy at the 7 min period are summarized and contrasted with the observed PLF at this time in Figure 4. In addition, time course of systemic plasma and brain exposures following ketamine dosing was also investigated as summarized in Table 1 and shows a dose-dependent exposure that exhibited a rapid decay over time.

Mean PLF (% vehicle; filled circles) at the 7 min time point for each ketamine dose is contrasted with NMDA channel occupancy (triangles) derived from separate cohorts. Note the inverse relationship between the two measures.
DISCUSSION
To date, there have been over a dozen reports on 40 Hz ASSR in schizophrenia patients (reviewed in O'Donnell et al, 2013; Roach et al, 2013). In a majority of these, a deficit in PLF and/or evoked response has been found relative to healthy controls. On the other hand, some investigators reported an increase in PLF and evoked power in patients when challenged with 40 Hz auditory stimuli (Hamm et al, 2012). The main contribution of the current report is that a framework to reconcile these contradicting sets of observations is provided by using a well-established experimental model of acute NMDA hypofunction with relevance to schizophrenia. Moreover, our findings argue for a need to carefully explore dose–response relationships when testing NMDA blocker effects on the 40 Hz ASSR. Finally, we posit 40 Hz ASSR as an easily accessible translational biomarker for in vivo cortical NMDA activity that may find application in a number of neuropsychiatric disorders.
Approximately 7 min after i.v. ketamine, a period when exposure is likely to be closest to Cmax, we found that the 40 Hz ASSR was increased, unaffected, or reduced in a dose-dependent manner. The crucial determinant of this full-spectrum modulation appears to be the extent of NMDA channel engagement. If ASSR PLF data at 7 min are considered together with the corresponding NMDA occupancy data, the reciprocal relationship between the two is striking (Figure 4). Whereas at a dose that produced a relatively small fraction of occupancy of the available channel pool, an increase in PLF and evoked power was observed, at a dose that produced a high degree of blockade, a robust reduction in PLF was noted. In addition, the pharmacological modulation of the 40 Hz ASSR at a given dose was temporally highly dynamic and required careful monitoring to capture the full range of effects. In contrast to the effects on ASSR, the increase in induced gamma power across a 30-fold dose range was qualitatively unremarkable while being highly robust (Figure 3c and Supplementary Figure S1C). These two contrasting indicators may help us understand the on-going dynamics at the cortical microcircuit level and their impact on ASSR as discussed below.
It has been suggested that NMDA antagonists tilt the excitatory/inhibitory balance at the cortical microcircuit toward an increased excitability (Gandal et al, 2012; Murray et al, 2000; Yizhar et al, 2011). Functional evidence for this hypothesis can be found in increased pyramidal cell firing and reduced putative GABAergic interneuron activity within the same cortical region following systemic, subanesthetic NMDA antagonist treatment (Homayoun and Moghaddam, 2007). A robust disinhibition at the thalamic nuclei by systemic NMDA antagonists may also increase activity in thalamocortical afferents projecting onto cortical layers (Ferrarelli and Tononi, 2011). A consequence of these events is increased cortical synaptic glutamate (Lopez-Gil et al, 2007; Moghaddam et al, 1997). The resulting hyperglutamatergic state is believed to mediate the enhanced field gamma activity by stimulating kainate and AMPA receptor-mediated activation of the cortical interneuron–pyramidal cell network (McNally et al, 2011; Roopun et al, 2008). Indeed, a robust increase in field gamma oscillations is one of the most consistent findings in response to systemic treatment with NMDA antagonists (Hakami et al, 2009; Kocsis, 2012; Sebban et al, 2002).
Based on the foregoing discussion, and the empirical finding of a robust elevation of gamma band oscillations in our study, it is reasonable to infer an excess of synaptic glutamate after ketamine. At the lowest dose, we speculate that the enhanced synaptic glutamate allows many hitherto unoccupied NMDA sites to be engaged (despite ~14% block of the total available pool), leading to enhanced NMDA transmission and an increase in 40 Hz PLF. Interestingly, both PLF and induced gamma return to baseline levels relatively quickly, suggesting rapid normalization in synaptic glutamate levels at this low dose. On the other hand, at the two intermediate doses, despite a robust and persistent increase in induced gamma (and therefore, increased synaptic glutamate), we speculate that there are not enough available NMDA sites to mediate ASSR enhancement. Finally, at the highest dose of ketamine, as an estimated 83% of the cortical NMDA channels are unavailable, a significant reduction in PLF occurs, despite a very strong induced gamma activity, and therefore we speculate high levels of synaptic glutamate. This line of reasoning also presupposes that in the baseline condition, the evoked ASSR PLF is likely to be submaximal and that it can be increased by exogenous factors such as augmenting synaptic glutamate. This assumption seems reasonable as the NMDA receptors on PV+ neurons have been shown to be critical for entrainment at 40 Hz in empirical as well as modeling studies (Carlen et al, 2012; Kirli et al, 2014; Nakao and Nakazawa, 2014; Spencer, 2009), and because baseline 40 Hz ASSR can be improved by exogenous modulators like attention (Albrecht et al, 2013; Skosnik et al, 2007; Ross et al, 2004) or by pharmacological intervention that affect key neurotransmitters (Albrecht et al, 2013; Sivarao et al, 2013; Sullivan et al, 2015; Vohs et al, 2012). As the two intermediate doses of ketamine produced 34 and 62% of NMDA block while not significantly affecting ASSR PLF, we speculate that a baseline PLF response involves engagement of ∼38–66% of the available cortical NMDA receptors. The dissociation between spontaneous gamma power and evoked gamma power at the top dose of ketamine also alerts us to the fact that despite the shared nomenclature, these measures reflect different mechanisms. Whereas NMDA-mediated inhibitory interneuron activity is required for evoked gamma, the spontaneous gamma may be sustained by AMPA/kainate-mediated inhibitory mechanisms. It is however unclear at this time whether AMPA/kainate receptors also contribute to 40 Hz ASSR in a significant manner.
To date, the only clinical study that examined the effect of intravenous ketamine on 40 Hz ASSR in healthy subjects reported an increase in evoked power after ketamine (Plourde et al, 1997). A close examination of the time-course data from Plourde et al (1997) indicates that ketamine’s effects were more nuanced than their conclusion. Immediately after intravenous dosing, there was a reduction in 40 Hz ASSR followed by normalization followed by an increase in power (see Table 1 in Plourde et al, 1997). Thus, a temporally dynamic response was evident; a reflection perhaps of the rapid pharmacokinetic properties of the drug. In our experiment as well, at each dose, a temporally evolving modulation of the ASSR was seen. The delayed increase at the high dose suggests significant unblocking of NMDA channels over time concomitant with high synaptic glutamate.
Clinical studies showed that PLF of the 40 Hz ASSR showed excellent test–retest reliability across sessions separated by several days (McFadden et al, 2014; Tan et al, 2015). Although we did not study this aspect specifically, the consistency we observed at the group level (mean and %CV) in the vehicle cohort, both within study and across studies, appears to reinforce the reliability of this measure. Moreover, PLF showed an inverse relationship to predicted NMDA occupancy as well as in vivo ketamine exposure, whereas evoked activity indicated only a unidirectional change (increase only). This highlights a frequent observation from relevant literature on ASSR that PLF and evoked power do not necessarily covary (Roach and Mathalon, 2008). An increase in phase synchrony in the absence of a change in evoked power has been cited as evidence for phase resetting (Shah et al, 2004). However, this has been recently challenged (Ding and Simon, 2013). Although we do not completely understand the dynamics behind these measures, PLF may be a more sensitive index of neuropsychiatric dysfunction associated with NMDA transmission (Hirano et al, 2015; Kirihara et al, 2012; Spencer et al, 2008; Sullivan et al, 2015).
Mechanistically, ketamine-induced disinhibition is not limited to the glutamatergic system alone and affects other neurotransmitters as well (Giovannini et al, 1994; Yan et al, 1997). It is therefore possible for acetylcholine, dopamine, or serotonin to play a mediatory role in ASSR PLF effects. Indeed, nicotinic cholinergic stimulation has been previously shown to augment 40 Hz ASSR power and PLF (Sivarao et al, 2013). Similarly, dopamine release-promoting agents like dexamphetamine have been shown to augment 40 Hz ASSR evoked power but not PLF (Albrecht et al, 2013). However, to our knowledge there are no data to suggest that increases in these neurotransmitter levels would result in a reduction in ASSR PLF. In contrast, the causal role played by NMDA receptor activation on PV+ neurons in 40 Hz ASSR generation has been recently demonstrated using molecular biology and optogenetic techniques (Carlen et al, 2012; Nakao and Nakazawa, 2014). Thus, a loss of NMDA function on PV+ neurons following a high dose of an NMDA antagonist is fully consistent with a reduction of response to a 40 Hz driving stimulus.
It is unclear whether a complete block of NMDA receptors would result in a complete loss of PLF. We were not able to directly test this in our study as we could only achieve ∼83% occupancy. As ketamine displays rapid kinetics following a bolus dosing paradigm (Table 1), it may not be suitable to obtain and maintain high levels of NMDA receptor blockade for durations sufficient to ask this question. In future studies, this issue can be addressed by switching to a bolus+infusion paradigm to maintain steady-state saturation blockade and/or by using a high-affinity antagonist like MK-801 that exhibits a more persistent block.
In summary, we show here that acute ketamine can augment or curtail the 40 Hz ASSR in the awake rat depending on the dose and, likely, the degree of NMDA channel blockade. Our findings reveal for the first time a robust reciprocal relationship between 40 Hz ASSR PLF and NMDA channel blockade, suggesting that 40 Hz ASSR could be an easy-to-access translational biomarker that closely reflects cortical NMDA receptor function that is of significant relevance in a number of neuropsychiatric conditions.
Funding and disclosure
All the authors are full-time employees of the Bristol Myers Squibb Company. The authors declare no conflict of interest.
References
Adell A, Jimenez-Sanchez L, Lopez-Gil X, Romon T (2012). Is the acute NMDA receptor hypofunction a valid model of schizophrenia? Schizophr Bull 38: 9–14.
Albrecht MA, Price G, Lee J, Iyyalol R, Martin-Iverson MT (2013). Dexamphetamine selectively increases 40 Hz auditory steady state response power to target and nontarget stimuli in healthy humans. J Psychiatry Neurosci 38: 24–32.
Baldeweg T, Spence S, Hirsch SR, Gruzelier J (1998). Gamma-band electroencephalographic oscillations in a patient with somatic hallucinations. Lancet 352: 620–621.
Brenner CA, Sporns O, Lysaker PH, O'Donnell BF (2003). EEG synchronization to modulated auditory tones in schizophrenia, schizoaffective disorder, and schizotypal personality disorder. Am J Psychiatry 160: 2238–2240.
Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K et al (2009). Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459: 663–667.
Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D et al (2012). A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry 17: 537–548.
Ding N, Simon JZ (2013). Power and phase properties of oscillatory neural responses in the presence of background activity. J Comput Neurosci 34: 337–343.
Ferrarelli F, Tononi G (2011). The thalamic reticular nucleus and schizophrenia. Schizophr Bull 37: 306–315.
Frohlich J, Van Horn JD (2014). Reviewing the ketamine model for schizophrenia. J Psychopharmacol 28: 287–302.
Galambos R, Makeig S, Talmachoff PJ (1981). A 40-Hz auditory potential recorded from the human scalp. Proc Natl Acad Sci USA 78: 2643–2647.
Gandal MJ, Sisti J, Klook K, Ortinski PI, Leitman V, Liang Y et al (2012). GABAB-mediated rescue of altered excitatory-inhibitory balance, gamma synchrony and behavioral deficits following constitutive NMDAR-hypofunction. Transl Psychiatry 2: e142.
Giovannini MG, Mutolo D, Bianchi L, Michelassi A, Pepeu G (1994). NMDA receptor antagonists decrease GABA outflow from the septum and increase acetylcholine outflow from the hippocampus: a microdialysis study. J Neurosci 14 (3 Pt 1): 1358–1365.
Gunduz-Bruce H (2009). The acute effects of NMDA antagonism: from the rodent to the human brain. Brain Res Rev 60: 279–286.
Hakami T, Jones NC, Tolmacheva EA, Gaudias J, Chaumont J, Salzberg M et al (2009). NMDA receptor hypofunction leads to generalized and persistent aberrant gamma oscillations independent of hyperlocomotion and the state of consciousness. PLoS One 4: e6755.
Hamm JP, Gilmore CS, Clementz BA (2012). Augmented gamma band auditory steady-state responses: support for NMDA hypofunction in schizophrenia. Schizophr Res 138: 1–7.
Hirano Y, Oribe N, Kanba S, Onitsuka T, Nestor PG, Spencer KM (2015). Spontaneous gamma activity in schizophrenia. JAMA Psychiatry 72: 813–821.
Homayoun H, Moghaddam B (2007). NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27: 11496–11500.
Hong LE, Summerfelt A, McMahon R, Adami H, Francis G, Elliott A et al (2004). Evoked gamma band synchronization and the liability for schizophrenia. Schizophr Res 70: 293–302.
Javitt DC, Zukin SR (1991). Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148: 1301–1308.
Kirihara K, Rissling AJ, Swerdlow NR, Braff DL, Light GA (2012). Hierarchical organization of gamma and theta oscillatory dynamics in schizophrenia. Biol Psychiatry 71: 873–880.
Kirli KK, Ermentrout GB, Cho RY (2014). Computational study of NMDA conductance and cortical oscillations in schizophrenia. Front Comput Neurosci 8: 133.
Kocsis B (2012). Differential role of NR2A and NR2B subunits in N-methyl-D-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biol Psychiatry 71: 987–995.
Krishnan GP, Hetrick WP, Brenner CA, Shekhar A, Steffen AN, O'Donnell BF (2009). Steady state and induced auditory gamma deficits in schizophrenia. Neuroimage 47: 1711–1719.
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD et al (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: 199–214.
Kwon JS, O'Donnell BF, Wallenstein GV, Greene RW, Hirayasu Y, Nestor PG et al (1999). Gamma frequency-range abnormalities to auditory stimulation in schizophrenia. Arch Gen Psychiatry 56: 1001–1005.
Light GA, Hsu JL, Hsieh MH, Meyer-Gomes K, Sprock J, Swerdlow NR et al (2006). Gamma band oscillations reveal neural network cortical coherence dysfunction in schizophrenia patients. Biol Psychiatry 60: 1231–1240.
Llinas RR (1988). The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science 242: 1654–1664.
Llinas RR, Grace AA, Yarom Y (1991). In vitro neurons in mammalian cortical layer 4 exhibit intrinsic oscillatory activity in the 10- to 50-Hz frequency range. Proc Natl Acad Sci USA 88: 897–901.
Lopez-Gil X, Babot Z, Amargos-Bosch M, Sunol C, Artigas F, Adell A (2007). Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology 32: 2087–2097.
Luby ED, Gottlieb JS, Cohen BD, Rosenbaum G, Domino EF (1962). Model psychoses and schizophrenia. Am J Psychiatry 119: 61–67.
McFadden KL, Steinmetz SE, Carroll AM, Simon ST, Wallace A, Rojas DC (2014). Test-retest reliability of the 40 Hz EEG auditory steady-state response. PLoS One 9: e85748.
McNally JM, McCarley RW, McKenna JT, Yanagawa Y, Brown RE (2011). Complex receptor mediation of acute ketamine application on in vitro gamma oscillations in mouse prefrontal cortex: modeling gamma band oscillation abnormalities in schizophrenia. Neuroscience 199: 51–63.
Moghaddam B, Adams B, Verma A, Daly D (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17: 2921–2927.
Mulert C, Kirsch V, Pascual-Marqui R, McCarley RW, Spencer KM (2011). Long-range synchrony of gamma oscillations and auditory hallucination symptoms in schizophrenia. Int J Psychophysiol 79: 55–63.
Mulert C, Kirsch V, Whitford TJ, Alvarado J, Pelavin P, McCarley RW et al (2012). Hearing voices: a role of interhemispheric auditory connectivity? World J Biol Psychiatry 13: 153–158.
Murray F, Kennedy J, Hutson PH, Elliot J, Huscroft I, Mohnen K et al (2000). Modulation of [3H]MK-801 binding to NMDA receptors. in vivo and in vitro. Eur J Pharmacol 397: 263–270.
Nakao K, Nakazawa K (2014). Brain state-dependent abnormal LFP activity in the auditory cortex of a schizophrenia mouse model. Front Neurosci 8: 168.
O'Donnell BF, Vohs JL, Krishnan GP, Rass O, Hetrick WP, Morzorati SL (2013). The auditory steady-state response (ASSR): a translational biomarker for schizophrenia. Suppl Clin Neurophysiol 62: 101–112.
Oda Y, Onitsuka T, Tsuchimoto R, Hirano S, Oribe N, Ueno T et al (2012). Gamma band neural synchronization deficits for auditory steady state responses in bipolar disorder patients. PLoS One 7: e39955.
Pastor MA, Artieda J, Arbizu J, Marti-Climent JM, Penuelas I, Masdeu JC (2002). Activation of human cerebral and cerebellar cortex by auditory stimulation at 40 Hz. J Neurosci 22: 10501–10506.
Plourde G, Baribeau J, Bonhomme V (1997). Ketamine increases the amplitude of the 40-Hz auditory steady-state response in humans. Br J Anaesth 78: 524–529.
Rass O, Forsyth JK, Krishnan GP, Hetrick WP, Klaunig MJ, Breier A et al (2012). Auditory steady state response in the schizophrenia, first-degree relatives, and schizotypal personality disorder. Schizophr Res 136: 143–149.
Rass O, Krishnan G, Brenner CA, Hetrick WP, Merrill CC, Shekhar A et al (2010). Auditory steady state response in bipolar disorder: relation to clinical state, cognitive performance, medication status, and substance disorders. Bipolar Disord 12: 793–803.
Roach BJ, Ford JM, Hoffman RE, Mathalon DH (2013). Converging evidence for gamma synchrony deficits in schizophrenia. Suppl Clin Neurophysiol 62: 163–180.
Roach BJ, Mathalon DH (2008). Event-related EEG time-frequency analysis: an overview of measures and an analysis of early gamma band phase locking in schizophrenia. Schizophr Bull 34: 907–926.
Rojas DC, Teale PD, Maharajh K, Kronberg E, Youngpeter K, Wilson LB et al (2011). Transient and steady-state auditory gamma-band responses in first-degree relatives of people with autism spectrum disorder. Mol Autism 2: 11.
Roopun AK, Cunningham MO, Racca C, Alter K, Traub RD, Whittington MA (2008). Region-specific changes in gamma and beta2 rhythms in NMDA receptor dysfunction models of schizophrenia. Schizophr Bull 34: 962–973.
Rosanova M, Casali A, Bellina V, Resta F, Mariotti M, Massimini M (2009). Natural frequencies of human corticothalamic circuits. J Neurosci 29: 7679–7685.
Ross B, Herdman AT, Pantev C (2004). Stimulus induced reset of 40-Hz auditory steady-state responses. Neurol Clin Neurophysiol 2004: 21.
Sannita WG (2000). Stimulus-specific oscillatory responses of the brain: a time/frequency-related coding process. Clin Neurophysiol 111: 565–583.
Sebban C, Tesolin-Decros B, Ciprian-Ollivier J, Perret L, Spedding M (2002). Effects of phencyclidine (PCP) and MK 801 on the EEGq in the prefrontal cortex of conscious rats; antagonism by clozapine, and antagonists of AMPA-, alpha(1)- and 5-HT(2A)-receptors. Br J Pharmacol 135: 65–78.
Shah AS, Bressler SL, Knuth KH, Ding M, Mehta AD, Ulbert I et al (2004). Neural dynamics and the fundamental mechanisms of event-related brain potentials. Cereb Cortex 14: 476–483.
Sivarao DV, Frenkel M, Chen P, Healy FL, Lodge NJ, Zaczek R (2013). MK-801 disrupts and nicotine augments 40 Hz auditory steady state responses in the auditory cortex of the urethane-anesthetized rat. Neuropharmacology 73: 1–9.
Skosnik PD, Krishnan GP, O'Donnell BF (2007). The effect of selective attention on the gamma-band auditory steady-state response. Neurosci Lett 420: 223–228.
Sohal VS, Zhang F, Yizhar O, Deisseroth K (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459: 698–702.
Spencer KM (2009). The functional consequences of cortical circuit abnormalities on gamma oscillations in schizophrenia: insights from computational modeling. Front Hum Neurosci 3: 33.
Spencer KM (2011). Baseline gamma power during auditory steady-state stimulation in schizophrenia. Front Hum Neurosci 5: 190.
Spencer KM, Niznikiewicz MA, Nestor PG, Shenton ME, McCarley RW (2009). Left auditory cortex gamma synchronization and auditory hallucination symptoms in schizophrenia. BMC Neurosci 10: 85.
Spencer KM, Salisbury DF, Shenton ME, McCarley RW (2008). Gamma-band auditory steady-state responses are impaired in first episode psychosis. Biol Psychiatry 64: 369–375.
Sullivan EM, Timi P, Hong LE, O'Donnell P (2015). Effects of NMDA and GABA-A receptor antagonism on auditory steady-state synchronization in awake behaving rats. Int J Neuropsychopharmacol 18: pyu118.
Tan HR, Gross J, Uhlhaas PJ (2015). MEG-measured auditory steady-state oscillations show high test-retest reliability: a sensor and source-space analysis. Neuroimage 122: 417–426.
Torrence C, Compo GP (1998). A practical guide to wavelet analysis. B Am Meteorol Soc 79: 61–78.
Truccolo WA, Ding M, Knuth KH, Nakamura R, Bressler SL (2002). Trial-to-trial variability of cortical evoked responses: implications for the analysis of functional connectivity. Clin Neurophysiol 113: 206–226.
Vierling-Claassen D, Siekmeier P, Stufflebeam S, Kopell N (2008). Modeling GABA alterations in schizophrenia: a link between impaired inhibition and altered gamma and beta range auditory entrainment. J Neurophysiol 99: 2656–2671.
Vohs JL, Chambers RA, O'Donnell BF, Krishnan GP, Morzorati SL (2012). Auditory steady state responses in a schizophrenia rat model probed by excitatory/inhibitory receptor manipulation. Int J Psychophysiol 86: 136–142.
Wilson TW, Rojas DC, Reite ML, Teale PD, Rogers SJ (2007). Children and adolescents with autism exhibit reduced MEG steady-state gamma responses. Biol Psychiatry 62: 192–197.
Yan QS, Reith ME, Jobe PC, Dailey JW (1997). Dizocilpine (MK-801) increases not only dopamine but also serotonin and norepinephrine transmissions in the nucleus accumbens as measured by microdialysis in freely moving rats. Brain Res 765: 149–158.
Ying J, Yan Z, Gao XR (2013). 40Hz auditory steady state response to linguistic features of stimuli during auditory hallucinations. J Huazhong Univ Sci Technolog Med Sci 33: 748–753.
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ et al (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477: 171–178.
Acknowledgements
We thank Dr Robert McCarley (Harvard Medical School, Boston, MA) for helpful comments on an early draft of this manuscript. We also thank Dr Mark Pflieger (Cortech Solutions, La Mesa, CA) for helpful discussion on the time-frequency analysis as implemented in the EMSE Data editor software. Bristol Myers Squibb Company has funded this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article

Cite this article
Sivarao, D., Chen, P., Senapati, A. et al. 40 Hz Auditory Steady-State Response Is a Pharmacodynamic Biomarker for Cortical NMDA Receptors. Neuropsychopharmacol 41, 2232–2240 (2016). https://doi.org/10.1038/npp.2016.17
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.17
This article is cited by
-
Developmental delays in cortical auditory temporal processing in a mouse model of Fragile X syndrome
Journal of Neurodevelopmental Disorders (2023)
-
An IGFBP2-derived peptide promotes neuroplasticity and rescues deficits in a mouse model of Phelan-McDermid syndrome
Molecular Psychiatry (2023)
-
Alterations of auditory-evoked gamma oscillations are more pronounced than alterations of spontaneous power of gamma oscillation in early stages of schizophrenia
Translational Psychiatry (2023)
-
Detection of the 40 Hz auditory steady-state response with optically pumped magnetometers
Scientific Reports (2022)
-
Altered neural oscillations and behavior in a genetic mouse model of NMDA receptor hypofunction
Scientific Reports (2021)
