
Abstract
Autism is a neurodevelopmental disorder characterized by deficits in communication and social skills as well as repetitive and stereotypical behaviors. While much effort has focused on the identification of genes associated with autism, research emerging within the past two decades suggests that immune dysfunction is a viable risk factor contributing to the neurodevelopmental deficits observed in autism spectrum disorders (ASD). Further, it is the heterogeneity within this disorder that has brought to light much of the current thinking regarding the subphenotypes within ASD and how the immune system is associated with these distinctions. This review will focus on the two main axes of immune involvement in ASD, namely dysfunction in the prenatal and postnatal periods. During gestation, prenatal insults including maternal infection and subsequent immunological activation may increase the risk of autism in the child. Similarly, the presence of maternally derived anti-brain autoantibodies found in ~20% of mothers whose children are at risk for developing autism has defined an additional subphenotype of ASD. The postnatal environment, on the other hand, is characterized by related but distinct profiles of immune dysregulation, inflammation, and endogenous autoantibodies that all persist within the affected individual. Further definition of the role of immune dysregulation in ASD thus necessitates a deeper understanding of the interaction between both maternal and child immune systems, and the role they have in diagnosis and treatment.
Similar content being viewed by others
INTRODUCTION
First described by Kanner, 1943, autism is a neurodevelopmental disorder characterized by repetitive, stereotypical behaviors and impaired expressive communication, which has since been folded into the broader classification of autism spectrum disorders (ASD) (American Psychiatric Association, 2013). The most recent (2012) estimates in the United States indicate a rate of 14.6 per 1000 children (one in 68) aged eight (Christensen et al, 2016). With increased prevalence in the population has come increased level of attention to the causes of ASD, which remain elusive yet legion. It is known that autism is strongly influenced by the genome—in contrast to earlier speculation, ranges of heritability estimates generally center ~50 or 55% (Badcock, 2011; Colvert et al, 2015; Hallmayer et al, 2011)—but genes implicated by large genome-wide studies have been shown to account for only a fraction of ASD diagnoses (Abrahams and Geschwind, 2008; Badcock, 2011; Happe et al, 2006).
Nearly 50 years ago, an association of autism with congenital rubella infection was noted, and in the intervening years numerous other infections have been connected to the incidence of ASD. Since that time, mounting evidence for the ability of the immune system and abnormal immune function, including inflammation, cytokine dysregulation, and anti-brain autoantibodies, to act as a significant influence on ASD has prompted researchers to look more closely at the potential role of immune dysregulation and autoimmunity in ASD. This field of research has gained particular traction given that other neurodevelopmental disorders, such as schizophrenia, also present alongside a spectrum of changes in immune function. Here, we review the potential of the immune system to serve as a collective, complex etiology for autism and ASD in at least a subset of cases. A graphic overview of the various neuronal and immune system components involved in the topics under discussion are depicted in Figure 1, indicating the many cells and molecules that may lead from an altered immune system to altered neurodevelopment. We divided this work into two sections, separated into prenatal and postnatal exposures. It begins in the gestational period with an examination of maternal infection, immune activation, and autoantibodies present during pregnancy, followed by the ongoing, postnatal role of dysregulation in the immune system in a child with ASD. We hope that through this comprehensive review, we will provide evidence supporting the notion that immune dysregulation in autism is a viable pathway towards the identification and subsequent treatment of this ASD subpopulation in the future.

Overview of the immune system as a mediator of behavior. Understanding how immune dysfunction in ASD can lead to changes in behavior requires understanding a complex network of interactions between several cell types from both the innate and adaptive arms of the immune system. Several immune factors mediate effects of CNS function. T-cell and NK-cell subpopulations may have altered activity and an impaired reaction to stimulation. Some cytokines can inhibit neurogenesis and promote neuron death, while others can promote the growth and proliferation of neurons and oligodendrocytes. Complement proteins and microglia can participate in synaptic scaling and pruning, while brain-reactive autoantibodies can change the development or function of neurons. When the many components of the immune system are dysregulated, these networks can lead to changes in neurodevelopment and behavior.
THE PRENATAL/GESTATIONAL ENVIRONMENT
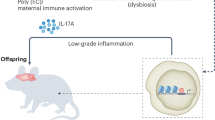
There are a number of ways in which the gestational environment can affect neurodevelopmental outcome in the child. This includes active infection as well as the immune response of the mother to infection as key areas of research when looking for prenatal risk factors. Indeed, it has now been demonstrated through animal models that merely an activated immune response in the mother, absent any infection, is sufficient to lead to changes in the offspring (Shi et al, 2003). Further, autoantibodies to proteins that are a key to healthy neurodevelopment are also thought to be a viable pathogenic mechanism in a subset of cases. Figure 2 summarizes the various prenatal maternal immune factors that can have a role in the development of ASD that will be addressed in the following sections.

Maternal immune dysregulation during gestation is a risk factor for autism and numerous interrelated factors may lead to dysregulation of the maternal immune system. Infection during the pregnancy, such as by rubella or influenza virus, can create an inflammatory immune environment and spur the production of maternal cytokines, which can not only directly affect the placenta but also to a limited degree may cross the placenta and enter the fetal compartment to have lasting effects on the development of the fetus. These effects can also be achieved in the absence of active infection, whether though a generalized inflammatory response or loss of immune regulation. Animal models of MIA have been particularly significant in underscoring the importance of maternal immune regulation. In addition, a subset of women may produce anti-brain autoantibodies that can likewise gain access to the developing fetal brain and bind to fetal proteins, thereby altering the course of neurodevelopment. Activation of the maternal immune system during fetal development is thus an important factor in the etiology of ASD, and may lead to changes in neurodevelopment.
Congenital Infection
In 1964, an epidemic of rubella spread throughout the United States, leading to between 20 000 and 30 000 infants with congenital defects. Following the outbreak, Dr Chess examined 243 preschool children with congenital rubella syndrome (CRS) and found a marked increase in the incidence of autism (Chess, 1971). The study reported 18 children with some degree of autism, implying an incidence of 741 cases per 10 000 among the population of CRS children, considered high even today and an especially high rate for the time. Chess followed-up as the children aged and found even higher rates of autism (905 per 10 000) (Chess, 1977, 1978), thus establishing a link between CRS and autism that has been supported by others (Deykin and MacMahon, 1979). A 2016 reappraisal of rubella-related ASD suggested that the connection persists and offered several reasons for why the rubella–autism connection might be difficult to work out (eg, widespread vaccination, rubella may evade the fetal immune system, children with CRS often lack antibodies to rubella, etc) as well as some ideas for future research, such as using cord blood or prescreening newborns before rubella vaccination (Hutton, 2016).
Rubella is not the only virus, or indeed infection, to show a connection to autism when acquired congenitally (Atladottir et al, 2010; Wilkerson et al, 2009). Varying reports have also implicated measles and mumps (Deykin and MacMahon, 1979), cytomegalovirus (Libbey et al, 2005), polyomaviruses (Lintas et al, 2010), and influenza (Atladottir et al, 2012; Deykin and MacMahon, 1979; Shi et al, 2003; Zhang et al, 2010) in the incidence of ASD. Some studies suggest fever as a correlate (Atladottir et al, 2012), with this risk potentially tempered by anti-fever medications, such as Advil, Tylenol, or Nyquil (Zerbo et al, 2013). A more expansive study by Zerbo et al (2015) found that maternal infection diagnosed at a hospital admission, especially bacterial ones, correlated with increased risk of ASD. A yet-larger 2015 study of over 24 000 Swedish ASD cases observed increased ASD rates among offspring of mothers hospitalized the year before pregnancy; risk for ASD increased under any inpatient diagnosis, including both viral and bacterial (Lee et al, 2015). The authors found no connection with the gestational timing of infection, however, in contrast to previous suggestions of viral or bacterial infection (Atladottir et al, 2010). As research into the role of direct or retrospective infection continues, the evidence presented thus far strongly suggests a connection between maternal infection and altered neurodevelopment. The implication is that the response to the infection, not just a few select pathogens, can lead to ASD in the offspring. This changes how we view the risk of a child developing ASD following a gestational exposure to an infectious agent: we should be looking at the maternal response to the exposure rather than focusing on the specific infectious agent.
Maternal Immune Activation
Some of the strongest support for maternal infection as a contributor to autism has come from the maternal immune activation (MIA) model, which has repeatedly shown consistent results, helping to establish a subset of autism that is supported by observations of autism-relevant atypical behaviors. The MIA model in its most common design involves challenging pregnant rodents via direct infection (eg, influenza, Escherichia coli) or the dsRNA mimic poly(I:C); significant immunological, neurodevelopmental, and behavioral changes are then observed in the offspring (Gilmore et al, 2005; Meyer et al, 2005; Urakubo et al, 2001; Zuckerman and Weiner, 2005). These changes have been linked and employed to model several neurodevelopmental disorders, including schizophrenia (Meyer and Feldon, 2009; Meyer et al, 2005; Zuckerman and Weiner, 2005), cerebral palsy (Boksa, 2010), and autism (Brown et al, 2015).
Shi et al (2003) infected two strains of pregnant mice with human influenza virus and observed abnormal behavior, exploration, and social interaction in their offspring. In support of the congenital infection model, the authors observed similar aberrant phenotypes in the same mice when using only poly(I:C) treatment rather than infection, suggesting that it was the maternal immune response rather than the infection itself that was responsible for the effects of gestational exposure to an infectious agent. The authors followed-up by challenging pregnant dams with either a single injected dose of influenza on embryonic day E9.5 or poly(I:C) on day E12.5; both groups had offspring with altered cerebellar development, akin to that seen in autism (Shi et al, 2009). A more expansive study injected pregnant mice three times with poly(I:C), each with one-fourth the dose of Shi, et al, starting on E10.5 and spaced with 2 days between injections (Malkova et al, 2012). The authors looked at ultrasonic vocalization (USV) responses among male offspring and found lower rates and total length of vocalizations. Sociobehavioral changes were observed using an isolation test and the three-chamber test, a system that presents a test animal with the choice to either explore or avoid novel animals or objects (Kaidanovich-Beilin et al, 2011). The male MIA offspring also showed several autism-relevant changes, such as reduced sociability in the three-chamber apparatus, spending more time in the nonsocial chamber and less time with the social object, as well as significantly higher rates of repetitive behaviors such as self-grooming and marble burying.
A 2014 study by Bauman et al (2014), broadened the poly(I:C) maternal activation model to rhesus macaques, showing many abnormal behaviors, including in vocal communication and social interaction, as well as repetitive behaviors in the offspring of treated females. Pregnant monkeys were injected with poly(I:C) three times in either the late first or late second trimester, and their offspring were subsequently monitored for 2 years. Although there were some differences between the offspring of the two treatment groups, each set of young primates nonetheless presented aberrancies and atypical phenotypes relevant to both ASD and schizophrenia.
Weber-Stadlbauer expanded greatly on this in 2016, finding that offspring of pregnant mice given a single poly(I:C) injection at day E9 showed reduced interaction (via the three-chamber apparatus) and increased fear expression in a cued Pavlovian conditioning test (Weber-Stadlbauer et al, 2016). Significantly, they showed that several autism-relevant traits only persisted through the paternal line, without any further intervention, through the F2 and even the F3 generations, resulting in significant differences in gene expression. A 2015 epigenetic study also using a single poly(I:C) injection found significantly increased rates of 5-methylcytosine and 5-hydroxymethylcytosine on the glutamic acid decarboxylase (GAD1) promoter, which synthesizes the neurotransmitter GABA (Labouesse et al, 2015). These changes correlated with social interaction and working memory impairments and are similar to GABA and GAD1 changes observed in related disorders such as schizophrenia.
Cytokines
Although the alterations in behavior and neurodevelopment of the MIA model have been fairly well established (Gilmore et al, 2005; Meyer et al, 2005; Urakubo et al, 2001; Zuckerman and Weiner, 2005), less well understood is how these changes are mediated through to the offspring. Given the diversity of infections that show a connection to the spectrum of autism disorders (Boksa, 2010; Libbey et al, 2005), as well as evidence from MIA models that reaction to infection rather than infection itself may lead to autism-related symptoms (Shi et al, 2003), a possible common etiology may be the influence of the immune system (Gottfried et al, 2015; Lintas et al, 2010; Zhang et al, 2010). Cytokines, small cell-signaling proteins that act as immunomodulatory and endocrine messengers, have been implicated throughout the process of CNS development (Boulanger, 2009; Jones and Thomsen, 2013). The immune cells that produce cytokines following activation are separated into two populations of cells: innate and adaptive. Both cell populations have the ability to produce cytokines that are linked to inflammation, including monocytes/macrophages and T cells. T cells are divided into subsets based on their function and their cytokine profile. Th or T-helper cells are classified into Th1 cells that produce pro-inflammatory cytokines, Th2 cells that are responsible for driving antibody production and downregulation of the inflammatory response, Th17 cells that also drive inflammation, and Th3 regulatory cells. As we investigate the relationship between the immune system and neurological disorders such as ASD, we look for skewing of the immune response towards an inflammatory or regulatory cytokine profile, whether it is during gestation or in the child.
There is strong evidence that disruption of normal cytokine levels has a significant role as a risk factor for several neurodevelopmental defects, including schizophrenia and autism (Boulanger and Shatz, 2004; Brown et al, 2004b; Deverman and Patterson, 2009; Hsiao et al, 2013; Ponzio et al, 2007). Smith et al (2007) showed in 2007 that a single injection of the pro-inflammatory cytokine IL-6 to pregnant mice at embryonic day 12.5 lead to offspring deficits in prepulse inhibition (PPI) and latent inhibition (LI), both autism-relevant behaviors; these results were not seen following injection of the pro-inflammatory cytokines IL-1α, TNF-α, or IFN-γ. The authors also used a poly(I:C) MIA model, observing basic behavioral changes in agreement with those previously described. When the authors co-administered a neutralizing anti-IL-6 antibody to pregnant dams, they observed an apparent rescue of the aberrant behaviors and reversal of transcriptional changes; these rescue effects were not seen in the context of co-administered anti-IFN-γ or anti-IL-1β antibody, suggesting a specific role for IL-6 in the etiology of ASD.
In further support of IL-6-mediated MIA autism, attempting the poly(I:C) MIA model in IL-6 knockout mice showed no statistically significant differences in PPI or social behavior via the three-chamber test from untreated IL-6 knockouts. Another team also used a poly(I:C) MIA model to show increased IL-6 levels, and found that treatment with Bacteroides fragilis, which can repair tight junction integrity in the gut, corrected and restored IL-6 levels, aberrant behavior, stereotypies, and anxiety-like symptoms (Hsiao et al, 2013).
Although previous work had shown that IL-6 may readily cross the placenta (Zaretsky et al, 2004, Choi et al (2016) extended this pathway further downstream and established that poly(I:C) injections in pregnant dams increased post-induction maternal serum levels of IL-6 3 h later and IL-17 2 days later. These increases did not occur in IL-6 knockout mice; however, increased IL-17 levels were found following injection of IL-6 to wild-type, untreated mothers, indicating that increased IL-6 leads to IL-17 production. MIA pups showed several aberrancies in USVs, sociality, and marble burying, but maternal pretreatment with an anti-IL-17 antibody restored these behaviors to typical measures. Fetal MIA offspring showed increased brain mRNA levels of the IL-17 receptor subunit, effects that were also rescued by maternal anti-IL-17 pretreatment. Injection of IL-17 to E14.5 fetal brain was enough to induce structural changes similar to those seen in MIA offspring. Finally, treatment of the anti-IL-17 antibody 2 days after MIA induction saw rescue of some phenotypes, such as USV calls and marble burying, but not others, including interaction deficits. Put together, this suggests that the Th17 cell/IL-17 pathway may be critical in MIA-induced autism, caused by upregulated IL-17 acting downstream of increased IL-6 levels. Choi et al (2016) also showed that their MIA model induced the production of several other cytokines, finding increased levels of TNF-α, IFN-β, and IL-1β 3 h following MIA induction, but not the anti-inflammatory IL-10. A poly(I:C) MIA model by Meyer et al (2008) suggested that maternal IL-10 may be able to attenuate behavioral and pharmacological dysfunctions.
More recent human studies found increased levels of IFN-γ, IL-4, and IL-5 in sera from mid-gestational mothers with a child that would later be diagnosed (Goines et al, 2011b) and elevated MCP-1 (Abdallah et al, 2012a), IL-4, TNF-α, and TNF-β levels in amniotic fluid (Abdallah et al, 2013). Most recently, studies have demonstrated that elevated mid-gestational levels of inflammatory cytokines and chemokines are more highly associated with the ASD subphentoype that presents with intellectual disability (ID), compared with ASD without ID, developmental delay (DD) without ASD, and typically developing controls (Jones et al, 2016). These included higher levels of GM-CSF, TNF-α, IFN- γ, IL-1α, IL-1β, IL-4, and IL-6. A 2016 study found maternal blood C-reactive protein levels to be lower in weeks 15–19 of pregnancies of a child with ASD than the general population, and although the results appear not to be genetic, the authors lacked reports of disease status and could not say whether infection during pregnancy was a factor (Zerbo et al, 2016). This study was in contrast to an earlier investigation of Finnish children that showed an association between autism and elevated early gestational maternal C-reactive protein (Brown et al, 2014). One proposal suggested three pathways through which a prenatal inflammatory cytokine response may leads to ASD: maternal, in which cytokines from the mother cross the placenta; placental, in which MIA leads to inflammation and cytokine production in the placenta; or fetal, where MIA leads immune and gene dysregulation in the fetus itself (Abdallah et al, 2013). However, it should be considered that there might be a combination of these three pathways. Maternal inflammation history and the pro-inflammatory/anti-inflammatory cytokine balance are thus particularly important avenues for further study, especially at the interface between mother and fetus (Meyer et al, 2008; Jones and Thomsen, 2013; Young et al, 2016).
Transplacental Maternal Autoantibodies to the Fetal Brain
The notion that maternal autoantibodies to elements in the fetal compartment could elicit changes in neurodevelopment began over 25 years ago, in a study that examined serum from 11 mothers of young children and found six with circulating antibodies that targeted lymphocyte antigens of the children (Warren et al, 1990). A decade later, researchers described a mother with serum antibodies that tested positive for reactivity to rat neuronal tissue (Dalton et al, 2003). A subsequent study used samples taken from 11 mothers with autistic children to strengthen these results, finding serum reactivity in those mothers to prenatal, postnatal, and adult rat brain proteins while finding none in control mothers (Zimmerman et al, 2007). Of particular note is that the children in this study ranged in age from 2 to 18 years old, indicating that the presence of autoantibodies in maternal serum is not a transient phenomenon and may persist for many years. Although generally consistent in their findings, the previous studies lacked the large sample sizes that provide convincing evidence.
Braunschweig et al (2008) looked at plasma from 61 mothers of autistic children and 102 controls (62 TD and 40 with other, non-ASD DD) for reactivity to fetal brain proteins. The authors identified two bands, at 37 and 73 kDa, that were significantly more common among mothers with autistic children, in particular children with regression rather than early onset. More importantly, the authors found that 12% of samples from mothers of children with ASD showed both 37 and 73 kDa bands, while none of the mothers of TD or DD children did. Another study published that year looked at serum from 100 mothers of children with autism, again finding higher rates of anti-fetal brain antibodies as compared with 100 control mothers (Singer et al, 2008). These antibodies were positive to adult and fetal human and rat brain proteins, including targets at 73 kDa and 36 kDa, similar to those shown by Braunschweig et al (2008). These authors used specific brain samples rather than a medley, identifying numerous other targets specific to certain brain regions (Singer et al, 2008). In a large study using the Simons Simplex collection, Brimberg et al (2013) examined plasma from 2431 mothers of ASD children and compared it with plasma from 654 GP women, finding that mothers of children with ASD were four times as likely to have circulating anti-brain antibodies. The sample screening process for this study differed from previous research in that the serum samples were tested against mouse tissue sections using immunofluorescence rather than detection of denatured proteins by western blot. This underscores the importance of multiple investigators using different approaches to tackle this critical area of autism research.
Analysis of samples before the birth of the child is a critical next step from the associative studies noted above. In a study utilizing banked mid-pregnancy (prospective) blood samples, the authors noted that maternal autoantibodies reactive to proteins near 37 and 73 kDa were only found in women whose children later received a diagnosis of ASD (Croen and Braunschweig, 2008a). Although the specific maternal anti-fetal brain antibodies were detected more often in mothers of children with ASD, there was still minimal evidence supporting the idea that maternal autoantibodies could alter behavior. Moreover, additional studies are needed determine the predictive ability of the maternal autoantibodies for autism risk; such studies are currently underway utilizing multiple sample populations.
To further explore the relationship between the maternal autoantibodies and behavioral outcome, an expanded study was conducted with a larger cohort of patients to provide additional support for the role of maternal autoantibodies in the pathogenesis of ASD. The authors reported an association between the presence of anti-fetal brain antibodies in the mother and ASD-related deficits in the child, finding that paired brain reactivity (ie, to the 37 and 73 kDa bands) correlated with lower expressive language (Braunschweig et al, 2012a). Furthermore, reactivity to a protein band near 39 kDa was discovered, and paired reactivity to proteins at 39 and 73 kDa correlated with broader diagnosis of ASD as well as increased irritability on the Aberrant Behavior Checklist (ABC) scale, results that have since been supported by subsequent studies (Piras et al, 2014). In addition, it was noted in a follow-up report that children born to mothers with this pattern of autoantibody binding also exhibited higher total cerebral volume when compared both to children with ASD born to autoantibody-negative mothers as well as typically developing control children (Nordahl et al, 2013).
To understand how these anti-brain antibodies could potentially lead to changes in neurodevelopment, it was first necessary to determine the identity of the proteins corresponding to the 37, 39, and 73 kDa bands. Studies by Braunschweig et al (2013) utilized two-dimensional gel electrophoresis followed by tandem mass spectrometry peptide sequencing to identify seven developmentally regulated proteins in the fetal brain that are recognized by maternal autoantibodies. These are lactate dehydrogenase A and B (LDH-A and LDH-B), stress-induced phosphoprotein 1 (STIP1), collapsin response mediator proteins 1 and 2 (CRMP1 and CRMP2), cypin, and Y-box binding protein 1(YBX1). Confirmatory analysis of the identified targets confirmed that 23% of mothers whose children had ASD had reactivity to the highly specific antigen patterns compared with 1% of TD mothers. This preliminary analysis suggests a specificity of 98% with a sensitivity of 23%. Further, when behavioral outcomes were characterized in the children of the 7% of mothers with a combined reactivity to LDH, STIP1, and CRMP1 (corresponding to the original 37/73 kDa band pattern) this pattern of reactivity correlated with an increase in stereotypic behaviors in the child (Braunschweig et al, 2013). In specific combinations, autoantibodies to these proteins were predictive for autism risk, which, although still at the level of laboratory research, suggests the presence of a distinct, maternal autoantibody-driven subset of ASD. Future studies will build upon this work to determine the clinical utility of the maternal autoantibodies as a marker for autism risk.
Animal Models Strongly Support the Maternal Autoantibody Model
As maternal autoantibodies have become increasingly implicated in the behavioral and cognitive deficits that characterize ASD, it has been difficult to determine the pathogenic mechanism by which these antibodies lead to the behaviors observed in children with ASD by relying solely on retrospective clinical studies conducted with human subjects or samples. Toward this goal, animal models have been repeatedly utilized to bolster the findings of maternal anti-brain autoantibodies, suggesting a directly causative effect. As previously mentioned, investigators injected maternal serum containing anti-brain antibodies into pregnant mice daily between E10 and E17, and observed reduced exploration and motor control in their offspring as compared with the offspring of untreated dams (Dalton et al, 2003).
Singer et al (2009) exposed pregnant mice to IgG from mothers of children with autism daily between E13 and E18, observing alterations in sociability and anxiety. Braunschweig et al (2012b) performed a similar study to demonstrate atypical anxiety and sociality alongside impaired motor and sensory abilities following a single intravenous injection. A 2014 study injected pregnant mice only once, intraventricularly, on E14, with IgG from either mothers with the 37 and 73 kDa autoantibodies or control mothers; the authors observed atypical behaviors in the murine offspring, including stereotypical self-grooming and increased marble burying (Camacho et al, 2014). A follow on study using the same technique showed structural changes in the offspring of pregnant mice injected with autoreactive IgG, including higher proliferation of radial glial cells and increased brain and neuron size (Martinez-Cerdeno et al, 2016).
A related study collected sera from both mothers of autistic and TD children, and exposed pregnant macaques to IgG either reactive or nonreactive to monkey brain protein, verified via western blot. The authors found higher levels of motor activity and stereotypies among patient-treated macaque offspring, again supporting a causative agent present in maternal circulation (Martin et al, 2008). Of particular note is that the exogenous IgG was present for only 25% of the pregnancy, given in only three doses at the end of the first trimester, meaning that developing fetuses were likely under-dosed compared with what would be the case during human ASD development.
In a more targeted approach designed to increase gestational exposure, Bauman, et al, purified IgG from mothers who tested positive for antibodies that recognized the 37/73 kDa proteins (LDH, STIP1, and CRMP1) and had children with autism. The authors then treated eight pregnant macaques with that IgG six times over the course of the second and third trimesters, and assessed macaque offspring for 2 years using a battery of developmental and social tests. They observed a number of deviations from typical behavior as compared with animals treated with IgG from mothers of TD children as well as untreated controls, including rebuffed and unreciprocated friendship approaches and inappropriate vocalization (Bauman et al, 2013). These animals also had increased total cerebral volume, further supporting previous work in mice showing that these specific maternal autoantibodies appear to increase brain growth and total cerebral volume (Bauman et al, 2013; Martinez-Cerdeno et al, 2016).
The ideal study would make use of endogenously produced maternal anti-brain auto-antibodies to more readily mimic the theorized pathway. It is also worth keeping in mind that one can have an excellent biomarker of a disease without any direct evidence of pathological significance for that marker. However, while work in this area is currently underway from several lines of investigation and in multiple laboratories, it seems evident that maternal autoantibodies appear to influence, if not define, a specific subset of ASD. Given the existence of autoantibodies associated with other neurological disorders such as schizophrenia (Henneberg et al, 1994) and multiple sclerosis (Ryberg, 1982), there is clearly more to be addressed as to both the genesis and function of these antibodies in neurodevelopmental disorders. It is also critical that this work is validated and replicated by more than one investigator. However, the ultimate payoff for these studies has the potential to be of great benefit to families.
POSTNATAL/ONGOING EFFECTS
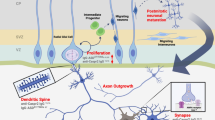
Alterations in immune function have also been noted as factors in children with autism, although this line of research has been somewhat hampered by the heterogeneity that is inherent in ASD. However, as we now recognize that autism would be more accurately labeled as ‘autisms’, the field has begun to look more closely at the relationship between immune dysfunction and the behavioral endophenotypes. To begin with, children may have their own, endogenous anti-brain autoantibodies that are connected with atypical development, distinct from the previously described maternal autoantibodies. Immunogenetic factors are also connected to ASD, including MET and the HLA family of genes. Broad pathways of genetic involvement in immune dysregulation have also been identified, in particular natural killer cells and cytokine regulation. The immune system of children with ASD can also be significantly altered, with a skew toward a pro-inflammatory and elevated Th1 response. Figure 3 is a graphic representation of the various factors found among the pediatric ASD population that will be discussed in more detail in this section.

Ongoing immune dysregulation persists in autism. After birth and at least throughout childhood, an individual with ASD may have endogenous anti-brain autoantibodies, separate from any maternal IgG, which correlate with aberrant behaviors and impaired development. There also exists a broad picture of ASD-related immune dysregulation, including an increased inflammatory cytokine milieu (eg, IL-6, IL-8, and MCP-1), thus leading to an increased, pro-inflammatory Th1/Th2 ratio. T-cell and NK-cell populations may also be skewed, displaying a shift in cell subpopulations. NK cells in particular show an increased baseline activity but a decreased response to activation, rendering the cells unable to properly respond to stimuli. NK cells interact with activating and inhibitory KIRs, many of which are genetically linked to ASD. Other genetic factors include the oncogene MET and members of the diverse family of HLA genes. The broader background of immunogenetic factors of ASD includes multiple networks of the immune system, such as pathways that regulate cytokines and NK cells, which together constitute a broad, endogenous environment of atypical immune regulation and response.
Anti-brain Antibodies in Children with ASD
In addition to the prenatal, maternal autoantibodies previously discussed, numerous studies have reported the presence of circulating anti-brain immunoglobulins in patients with ASD, as well as related conditions such as multiple sclerosis (Ryberg, 1982), schizophrenia (Henneberg et al, 1994), and systemic lupus erythematosus (SLE) (Libbey and Fujinami, 2010; Mackay et al, 2015; Suurmond and Diamond, 2015). Singer et al (2006) found that children with autism were more likely than their non-autistic siblings or controls to have serum that tested positive via western blot to fresh human brain samples (caudate, putamen, and prefrontal cortex). A similar result found that children with autism showed positive serum anti-brain reactivity against prenatal rat proteins by western blot, with a different set of banding patterns relative to their non-autistic siblings and other controls (Zimmerman et al, 2007). A 2009 study of plasma from 63 young children with ASD found that 21% of them had antibodies positive against both human and macaque cerebellum samples via western blot, while only one out of the 63 typical developing (TD) controls (2%) tested positive (Wills et al, 2009). When a subset of subject samples was examined via immunohistochemistry (IHC), 21% of those from children with ASD had ‘intense immunoreactivity’ while none of the TD controls exhibited such reactivity. The western blot and IHC results were strongly correlative, reinforcing the findings of an ASD subset with persistent circulating anti-brain antibodies.
A larger 2011 study expanded on these findings, testing plasma from a cohort of 277 children with ASD and 189 TD controls for reactivity to human and macaque cerebellar protein medleys, with ASD children more likely to test positive for anti-brain autoantibodies via western blotting (Goines et al, 2011a). The authors also found strong correlations between the presence of those antibodies and aberrant behaviors, as well as decreased cognitive and adaptive functioning.
These results were reemphasized by Mostafa and Al-Ayadhi (2012), who found a higher rate of anti-neuronal antibodies in a cohort of 80 Saudi children with ASD vs 80 -GP controls, as tested via immunofluorescence against monkey cerebellum (62.5 vs 5%); the presence of such antibodies was higher in severe diagnoses (87.5%) than mild/moderate diagnoses (25%) as well as in females (90%) compared with males (53.3%). A later study found autoimmunoreactivity to neuronal progenitor cells (NPCs) to be far more common in children with autism than controls, and among positive samples, those with ASD immunostained more strongly than controls (Mazur-Kolecka et al, 2014).
An examination of anti-brain autoantibodies in a cohort of 355 Italian children with autism and their mothers and unaffected siblings found that autoantibodies in autistic individuals correlated with increased severity, stereotypies, and cognitive impairment (Piras et al, 2014). The authors showed that 45 and 62 kDa antibodies in the children correlated with severity of autism, IQ, and impaired motor function and social interaction, as scored on the Vineland Adaptive Behavior Scales. Of particular interest, the authors also examined the 37 and 73 kDa maternal anti-brain autoantibodies mentioned previously, showing that while correlations between the two discrete autoantibody populations do exist, maternal- and child-borne autoantibodies each retain their own distinct and defining characteristics.
Immunogenetic Factors
Numerous studies have shown a connection between the members of the expansive family of HLA alleles and incidence of autism (Torres et al, 2012), in particular the class II DRB1. The hypervariable region 3 of DRB1*0401 has been associated with an increased relative risk for autism in children from Utah (Warren et al, 1996). DRB1 was also associated with autism in Han Chinese (Chien et al, 2012), Egyptian (Mostafa et al, 2013), and Saudi populations (Al-Hakbany et al, 2014). The DRB1*11 (Mostafa et al, 2013) and DRB1*1104 (Al-Hakbany et al, 2014) alleles have specifically been noted to occur at a higher rate in individuals with ASD. Multiple studies have suggested HLA-DR4 as another risk factor, appearing at increased rates in children with autism as well as their mothers (Johnson et al, 2009; Lee et al, 2006). Other positively associated alleles are the class I A*01, A*02, and B*07, while DRB1*03 and some DQB1 alleles have been negatively associated with rates of autism (Al-Hakbany et al, 2014; Mostafa et al, 2013; Torres et al, 2006).
Interestingly, Guerini et al (2015) discovered a 14-basepair insertion in HLA-G that was found more often in individuals with ASD and their mothers. HLA-G, along with HLA-E, -F, and perhaps others, is a non-canonical class I MHC gene that behaves very differently from the classical perception of MHC. These genes, in contrast to the exceptionally polymorphic HLA-A, -B, and -C genes, possess few alleles (Carosella et al, 2003). HLA-G interacts with natural killer cells as part of the innate immune system and is primarily expressed in placental tissues as part of immune tolerance during pregnancy (Carosella et al, 2008). The full meaning of the insertion/deletion is not yet known, but an altered efficacy and faulty tolerance toward the fetus by maternal NK cells during pregnancy, thus compromising the privileged status of the fetus, could provide a mechanism for atypical development.
The MET oncogene, which encodes a receptor tyrosine kinase known as C-Met or hepatocyte growth factor receptor (HGFR), is typically associated with cancer metastasis. However, this pleiotropic receptor is also critical for neuronal migration during development of the cerebellum and cerebral cortex, and functions as a negative immune regulator on antigen presenting cells. Linkage studies by Campbell et al (2006), have found an association between ASD and a single SNP (rs1858830) in the promoter of MET. Recent estimates at this site suggest that the CC and CG alleles represent a 1.76 and 1.59 relative risk, respectively, over the GG allele (Campbell et al, 2008), with an odds ratio of 1.64 for the C variant (Jackson et al, 2009). This MET variant also appears connected to both an increase in 37/73 kDa maternal autoantibodies and a decrease in maternal IL-10, suggesting another mechanism through which MET may associate with ASD as a susceptibility factor (Heuer et al, 2011).
More recently, researchers have utilized the advanced techniques of next-generation sequencing and genome-wide association studies (GWAS) in an attempt to divine the genetic causes of autism, generally finding a complex and varied genomic landscape (Abrahams and Geschwind, 2008; Gaugler et al, 2014; Michel et al, 2012). Voineagu, et al, examined gene expression in ASD and control brains, and as expected found hundreds of genes that were differentially expressed (Voineagu et al, 2011). In the control brains, dozens of genes exhibited different expression levels between the temporal and frontal lobes of the brain; these differences were not mirrored in the ASD samples. The genes significantly upregulated in ASD were heavily enriched for immune and inflammatory response functions, immunoglobulin domains, and other immune-regulatory ontologies.
The authors also defined a module of genes upregulated in ASD that was enriched for astrocytes and activated microglia. They describe a model whereby a significantly altered neuroimmune milieu persists in constant dysregulation. As a complex network of altered expression, the authors suggest that these differences are unlikely to be directly pathological in their own right; rather, the evidence suggests broad immunomodulation as symptomatic of a larger cellular dysregulation (discussed later in more detail).
Several other studies searching for genetic factors of autism and ASD have also found strong connections to the immune system. A 2008 study explored transcriptomic changes in the temporal cortex of children and adults with autism, finding 152 transcripts with varied expression, 130 of which were upregulated (Garbett et al, 2008). When grouped by ontology, the authors found 31 sets of genes that were differentially expressed between autistic and control samples; of these, 19 were connected to the immune system. These immune-relevant gene sets fell into six classes: antigen-specific immune response, inflammation, cell death, autoimmune diseases, migration, and the natural killer T cell pathway. Many of these genes showed increased variability in the autism cohort, which suggests a general dysregulation of the autistic immune system and overall heterogeneous activation response. Another 2008 study identified 11 genes that were differentially expressed in children with autism, as well as within the subsets of early onset and developmental regression, as compared with the general population (overexpression in autism samples were confirmed for seven of 11 via qPCR) (Gregg et al, 2008). These genes are heavily overrepresented in the ontological KEGG natural killer cytotoxicity pathway, and are highly expressed in NK and CD8 T cells.
Ziats and Rennert (2011) performed an in silico analysis using genes from the annotated AutDB portal (Basu et al, 2009) in 2011, and constructed a gene interactome network from highly expressed ASD genes that heavily suggests the immune system has a central role in ASD, specifically via cytokine signaling, as well as in schizophrenia. The authors suggest that immune system genes may function as a fundamental convergence for autistic disorders. A 2014 RNA-seq study built upon this concept with a gene ontology analysis showing that the autism transcriptome was heavily enriched for immune response genes (Gupta et al, 2014).
When Lintas et al (2012) reviewed several genome-wide expression studies in 2012 (including the two just mentioned), they found a strong connection to the immune system; of the 464 genes identified, six out of the top 10 KEGG ontology pathways were functions of the immune systems. Immune dysregulation was a common trend in genomic studies, particularly in the NK cell and cytokine pathways. A 2014 study by Guerini et al (2014) expanded on this trend by looking at the killer-cell immunoglobulin-like receptors (KIRs), the family of receptors responsible for mediating the innate NK cell response. The authors demonstrated that among a cohort of 90 continental Italian children with ASD, rates of activating KIR/HLA complexes (aKIR2DS2) were increased, while inhibitory KIR/HLA complexes (KIR 3DL1/HLABw4-80I) were decreased in the genome relative to controls. Similarly significant differences were seen following genotyping of the mothers, suggesting a broad influence of the genetic background of NK activity, especially given that KIR/HLA complexes are expressed and active in the placenta (Guerini et al, 2014; Parham, 2005).
Lintas et al (2012) proposed a model whereby the dysregulated immune system leads to increased metabolism and reactive oxygen species (ROS) in both the pre- and postnatal environment. They suggest that such alterations would result in prenatal changes in proliferation, migration, and neuronal wiring, while postnatal mitochondrial dysfunction and ROS stress would lead to abnormal synaptic formation.
Taken together, the genetic forces noted in both children and adults with autism are myriad, but insufficiently explanatory. While there appears to be a strong immunogenetic component to many examples of ASD, there is no one single thread. Most studies to date have focused on specific targets, whether they are HLA alleles, inflammation, or NK pathways, but the larger picture is one of a cascading network of genetic interactions that leads to ongoing, persistent immune imbalance. A deeper comprehension of these networks is critical to our understanding of the significant genetic impacts on a life with ASD.
Immune Dysregulation
Skewed and dysfunctioning cell populations
A significant focus of the postnatal immune imbalance has been on specific immune cell subsets, including helper CD4+ and cytotoxic CD8+ T cells (Garbett et al, 2008; Gregg et al, 2008), which may behave differently in individuals with autism (Gupta et al, 1998). A 1986 study found a reduced number of T cells in individuals with autism, as well as an altered helper to suppressor T-cell ratio (Warren et al, 1986), while a supporting 1990 study suggested CD4+ cell levels were decreased in autism (Yonk et al, 1990). A more recent report noted a decrease in CD4+CD25high regulatory T cells in children with autism (Mostafa et al, 2010). Further, Enstrom et al (2010) showed that cultured monocytes from ASD patients responded differently to TLR stimulation, while Ashwood et al (2011b) demonstrated a differential response by PHA-stimulated peripheral blood mononuclear cells (PBMCs), including reduced numbers of CD134- and CD25-activated CD3+, CD4+, and CD8+ T cells. A previously mentioned mouse model also implicated T cell skewing in ASD-relevant behavioral deficits (Ponzio et al, 2007), as did an examination of neuroinflammation in brains from individuals with autism (Vargas et al, 2005).
Natural killer cells have been frequently implicated in autism from a genetic/genomic standpoint (Lintas et al, 2012), and several studies have now examined the broader landscape of NK cell immune dysregulation in ASD. A reduction in NK cell activity was shown in teenage and adult autism patients, with 12 out of 31 patients showing lower activity in PBMCs via K562 chromium-release assays (Warren et al, 1987). Autistic patients showed no decrease in overall NK count, a similar result to schizophrenia (DeLisi et al, 1983), suggesting that it is the cells themselves that are dysfunctional.
A more recent study of NK cells recapitulates the genetic changes previously alluded to, showing that NK cells in children with autism had higher expression of NK cytotoxicity genes (Enstrom et al, 2009b). Among all upregulated genes found in NK cells, annotations were highly enriched for NK activity and immune response and defense, while protein biosynthesis and metabolism were downregulated. The authors also found considerable levels of immune dysregulation, observing significantly more CD56+ NK cells in children with ASD compared with controls. The authors then showed that unstimulated NK cells—specifically the CD56dim subset—stained positive for perforin, granzyme B, and IFN-γ at higher frequencies in ASD patients as compared with general population (GP) controls. Interestingly, upon stimulation, NK cells showed much lower staining to these same targets as compared with GP. In essence, NK cells from individuals with ASD show higher resting but reduced stimulated cytolytic activity as compared with TD controls. NK cells in ASD individuals appear to have a baseline level of activity that is already at peak function, rendering them ill equipped for normal circulation and unable to respond further to stimulation.
A more recent study examined a different subset of natural killer cells, those that are CD57+CD3− (Siniscalco et al, 2016). These NK cells are functionally different than traditional, CD56+ NK cells—they are terminally differentiated and have higher cytolytic capabilities, and generally act as innate immune regulators despite being less responsive to cytokines and connected to autoimmunity and immunodeficiency (Lopez-Verges et al, 2010; Nielsen et al, 2013). Siniscalco et al (2016) examined 104 children with ASD, and found significantly lower, atypical levels of CD57+ NK cells as compared with both age-matched and adult controls. Just 30% of children with ASD had normal CD57+ NK cell levels, despite typical levels of CD56+ NK cells, suggesting that it is the CD57+ cells that are implicated in autism.
Immunoglobulins
Looking at the humoral immune system, a 2008 study of over 250 children found a reduction of plasma IgG and IgM levels circulating in young children with autism, in particular IgG, the levels of which correlated negatively with scores on the ABC, regardless of ASD status (Heuer et al, 2008). A later study with children from the same cohort found increased plasma levels of the weakly active IgG4 subclass among ASD patients (Enstrom et al, 2009a), as did an earlier study comparing ASD children to their siblings (Trajkovski et al, 2004). These findings are further supported by a recent report of lower IgG1 levels in the serum of boys with ASD, along with a similarly negative correlation to communication as scored by the ADI-R (Zaman et al, 2016).
Although there appears to be some discrepancy between studies that show increased (Croonenberghs et al, 2002b; Trajkovski et al, 2004) and decreased (Enstrom et al, 2009a) IgG levels in children with autism, age of the children is most likely a contributing factor, with IgG levels changing significantly and rapidly through the first decade of life (Heuer et al, 2008). A 2012 follow-up study ruled out abnormal B-cell function as the source of aberrant IgG levels (Heuer et al, 2012). Although the contribution of total IgG levels needs to be more thoroughly examined, the apparent shift in subclass to IgG4 means that children with autism are likely to have a higher percentage of circulating IgG with an inherently lower affinity for the antibody receptors on leukocytes (Enstrom et al, 2009a; Zaman et al, 2016).
Cytokines, chemokines, and inflammation
Numerous studies have demonstrated a correlation between cytokine levels and ASD status, and while there is a consensus that cytokines and chemokines are part of the altered immune environment noted in autism (Depino, 2013; Enstrom et al, 2010), there has been some disagreement on the manifestation of such alterations, specifically whether the pro-inflammatory Th1 or anti-inflammatory Th2 cellular response dominates. A 2006 study showed increases in the anti-inflammatory Th2 cytokines IL-4, IL-5, and IL-13 from PHA-stimulated PBMCs (Molloy et al, 2006), which was supported by a later study that indicated a Th2-like response was present at birth and connected increased IL-4 to more severe autism (Krakowiak et al, 2015). On the other hand, a 2002 study demonstrated an increase in the blood cytokines IL-1RA and IFN-γ, suggesting a more Th1-like, pro-inflammatory response (Croonenberghs et al, 2002a), while a mouse model in 2007 showed Th1-skewed T-cell development in amniotic fluid alongside stereotypies in the offspring following treatment of pregnant dams with IL-2 (Ponzio et al, 2007).
The notion of a more pro-inflammatory environment was supported by Li et al (2009) who not only found elevated levels of the chemokine IL-8, pro-inflammatory cytokines IL-6, GM-CSF, and TNF-α, and the Th1 cytokine IFN-γ in autistic brain tissue, but also saw no increase in IL-4 or IL-5; as such, the authors claimed an increase in the Th1/Th2 ratio, suggesting that the Th1 arm is activated in autism. In the context of dietary proteins and food hypersensitivity, PBMCs from children with ASD produce higher levels of TNF-α and IFN-γ but not IL-5, further lending credence to a Th1-dominated response (Jyonouchi et al, 2005; Jyonouchi et al, 2002). Vargas et al (2005) likewise showed the presence of an active neuroinflammatory environment, finding higher levels of MCP-1, TGF-β, and IL-6 in the brains of autistic patients. Of particular note is that many of the above studies found no increase in the Th2 cytokine IL-10 (Croonenberghs et al, 2002a; Krakowiak et al, 2015; Li et al, 2009; Molloy et al, 2006).
A large 2011 study of children with ASD found an increase in plasma levels of a broad range of cytokines, including the Th1-like IL-12p40 and pro-inflammatory cytokines IL-1β, IL-6, IL-8, and GM-CSF (Ashwood et al, 2011a). Examining children from the same cohort, the authors also published on increased levels of the chemokines MCP-1, RANTES, and eotaxin (Ashwood et al, 2011c). In both studies, increased cytokine or chemokine levels were correlated with increased aberrant behavior and impaired development (Ashwood et al, 2011a, 2011c). A related study by the same team supported these findings using PHA-stimulated PBMCs, and correlated a Th1-like response with greater impairments and behavior aberrancies and a Th2-like response to better cognitive and adaptive function (Ashwood et al, 2011b). In contrast to most of the above studies, a 2012 report using dried blood samples suggested that children with ASD had lower levels of both Th1-like and Th2-like cytokines (Abdallah et al, 2012b).
Less clear is the role of pro-inflammatory cytokines IL-17 and IL-23. Enstrom et al (2008) found decreased plasma intracellular IL-23 in children with ASD, but no such change in IL-17, while Al-Ayadhi and Mostafa (2012) and Akintunde et al (2015) found evidence of elevated IL-17 levels among such children in serum and PHA-stimulated PBMCs, respectively. Given that IL-23 induces IL-17 and the importance of IL-17 for inducing the MIA model of autism (Choi et al, 2016), it is clear that these cytokines and the role of Th17 cells in autism warrants further study. This is especially so given the outsized role IL-17 has in other autoimmune conditions such as SLE and rheumatoid arthritis (Konya et al, 2015).
Although an increased pro-inflammatory environment seems well supported (Haroon et al, 2012), there remains a wealth of data to scrutinize and reach agreement on. Further studies should attempt to elucidate the network of interactions leading to systemic immune dysregulation in individuals with autism, in particular the role of IL-17 and the Th1/Th2 balance, with guidance from observations from the MIA model. Given the ability of the immune environment to affect behavior (Filiano et al, 2016), parsing out the different pathways, especially over the rapidly changing immune environment in early childhood as well as the heterogeneity that is inherent in ASD, remains both a challenge and an opportunity for the field.
OVERLAP
Anti-brain Antibodies
The presence of both maternal and patient circulating autoantibodies that target the brain raises the specter of a common provenance. The evidence strongly supports distinct roles for both maternal (Braunschweig et al, 2008; Croen et al, 2008b; Singer et al, 2008; Zimmerman et al, 2007) and patient (Henneberg et al, 1994; Libbey and Fujinami, 2010; Ryberg, 1982) autoantibodies in ASD, including an ability to affect structural brain development (Nordahl et al, 2013; Rossi et al, 2011). Offending antibodies of either origin appear to persist systemically for extended periods of time (Mostafa and Al-Ayadhi, 2012; Piras et al, 2014; Singer et al, 2006; Zimmerman et al, 2007), and although there exists a correlation between the two there does appear to be at least some differences in effect (Piras et al, 2014). Although reports indicate the presence of these antibodies may correlate with adverse conditions in the patients (Braunschweig et al, 2012a; Goines et al, 2011a; Mostafa and Al-Ayadhi, 2012; Piras et al, 2014), the nature of those connections needs to be further investigated, especially as regards different subpopulations of ASD. Doing so could elucidate not only the action but also the cause of these functional autoantibodies, and would allow more accurate models of autoantibody-related autism pathology to be developed.
ASD as an Autoimmune Disorder
The research community has long noticed a correspondence between ASD and autoimmune diseases, with some suggesting autism should itself be considered an autoimmune disorder (Ashwood and Van de Water, 2004). Singh (2009) proposed a theoretical autoimmune mechanism for autism, suggesting that an environmental trigger (eg, a virus) could provoke faulty immune regulation that results in autoimmunity to the brain, leading to the observed neuropathology. Autoantibodies, such as those described above, have been found in patients of other autoimmune disorders, including multiple sclerosis (Ryberg, 1982), schizophrenia (Henneberg et al, 1994), and SLE (Libbey and Fujinami, 2010). Evidence for the association has mounted, including a 2015 study that found a more than double risk of ASD in children of mothers with SLE than those of controls (Vinet et al, 2015). This is especially significant since the authors note that SLE patients present with SLE-related autoantibodies.
Inflammation and immune signaling dysregulation can strongly influence neuropsychiatric behavior beyond just ASD (Haroon et al, 2012; Meyer and Feldon, 2009), with evidence pointing toward roles in bipolar disorder and PTSD (Jones and Thomsen, 2013), and perhaps most emphatically in schizophrenia (Brown et al, 2004b; Meyer and Feldon, 2009; Michel et al, 2012; Patterson, 2009). Schizophrenia and autism share many immune similarities, including a subset of patients with an etiology based in maternal infection and immune activation (Bauman et al, 2014; Boksa, 2010; Brown et al, 2004a; Meyer et al, 2005, 2007; Patterson, 2009; Urakubo et al, 2001). Further lending credence to this association are numerous studies showing that a significant risk factor for autism is a family history of autoimmune disease (Brimberg et al, 2013; Gesundheit et al, 2013; Gottfried et al, 2015; Wu et al, 2015), alongside direct genetic evidence that links ASD and schizophrenia (Cantor and Geschwind, 2008; Ellis et al, 2016; Zhou et al, 2016). Despite the differences between these two disorders, the similarities between them are such that results from animal models like the MIA model may be overlapping and confounded (Bauman et al, 2014; Young et al, 2016).
FUTURE STEPS
The field investigating the immunobiology of autism has grown rapidly in the past decade and is rife with opportunity. In the prenatal environment, a strong line of evidence is beginning to develop in support of maternal immune dysregulation as it affects neurodevelopment based on studies on maternal infection, immune activation, and maternal autoantibodies directed toward the developing fetal brain. Pro-inflammatory cytokines such as IL-6 and IL-17 have been implicated in this process, but the mechanisms remain under investigation. The successful and increasingly prolific MIA model provides an important tool in understanding the pathways involved in maternal immune system-influenced neurodevelopmental disorders.
The postnatal environment echoes those lines of development, with anti-brain antibodies and systemic inflammation providing evidence of anomalies in immune regulation in a subpopulation of children with ASD. Data from various studies regarding immune dysregulation in ASD are beginning to coalesce, but given the heterogeneity of ASD phenotypes, it remains difficult to explain different behavioral outcomes based solely on immune phenotype, and this may contribute to spurious conclusions. However, as more studies in this research area look at both the biological and behavioral phenotype as well as medical co-morbidities such as gastrointestinal symptoms in the same individual, the relationship between immune dysregulation and ASD will likely become clearer. Also exciting and potentially relevant is the recent groundbreaking work by Louveau et al (2015) showing active lymphatic vessels connecting the CNS to the lymphatic system that may possibly act as a conduit for neuroimmunological dysfunction. Further work should be undertaken to understand how this newly revealed lymphatic system in the CNS could function as a pipeline for immunological molecules relevant for the development of ASD.
The application of massive GWAS panels has also greatly informed our understanding of the genetic composition of ASD, and in doing so hundreds of potential avenues of research have opened up, further diversifying patient populations. Regulators of the immune system have long been featured prominently among the genetic components of autism and are likely to continue to do so as we learn more about the myriad interactions taking place between the immune system and the brain. The intersection of genetic underpinnings and neuroimmune dysregulation may provide a door through which both clinical testing and directed therapy may emerge.
FUNDING AND DISCLOSURE
This study was funded by the NIEHS Center for Children’s Environmental Health and Environmental Protection Agency (EPA) grants (2P01ES011269-11, 83543201 respectively), the NIEHS-funded CHARGE study (R01ES015359), and the NICHD funded IDDRC 054 (U54HD079125). There are three patents associated with the work at UCD noted in this review: A provisional ‘Maternal Diagnostic Marker for Autism Risk and Therapeutic Intervention’ UC Case No. 2014-467-1. There are 2 issued patents: The US patent Methods of Diagnosing and Treating Autism, US Patent No. 8,383,360 B2 or Pat. 8,383,360 was filed on 12 August 2010 and issued on 26 February 2013. The client reference number for this patent is 2009-737-2US. This patent is currently in the process of National filing. The first issued patent is Diagnostic Methods for Detection of Autism Spectrum Disorder, US Patent No. US 7,452,681 B2. This patent was filed on 5 May 2006, and was issued 18 November 2008. This patent was filed in the US only. Dr Van de Water received consulting fees from Pediatric Bioscience from 1 Jan 2015 until October 2015. Amory Meltzer reports no biomedical financial interests or potential conflicts of interest.
References
Abdallah MW, Larsen N, Grove J, Norgaard-Pedersen B, Thorsen P, Mortensen EL et al (2012a). Amniotic fluid chemokines and autism spectrum disorders: an exploratory study utilizing a Danish Historic Birth Cohort. Brain Behav Immun 26: 170–176.
Abdallah MW, Larsen N, Grove J, Norgaard-Pedersen B, Thorsen P, Mortensen EL et al (2013). Amniotic fluid inflammatory cytokines: potential markers of immunologic dysfunction in autism spectrum disorders. World J Biol Psychiatry 14: 528–538.
Abdallah MW, Larsen N, Mortensen EL, Atladottir HO, Norgaard-Pedersen B, Bonefeld-Jorgensen EC et al (2012b). Neonatal levels of cytokines and risk of autism spectrum disorders: an exploratory register-based historic birth cohort study utilizing the Danish Newborn Screening Biobank. J Neuroimmunol 252: 75–82.
Abrahams BS, Geschwind DH (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet 9: 341–355.
Akintunde ME, Rose M, Krakowiak P, Heuer L, Ashwood P, Hansen R et al (2015). Increased production of IL-17 in children with autism spectrum disorders and co-morbid asthma. J Neuroimmunol 286: 33–41.
Al-Ayadhi LY, Mostafa GA (2012). Elevated serum levels of interleukin-17A in children with autism. J Neuroinflammation 9: 158.
Al-Hakbany M, Awadallah S, Al-Ayadhi L (2014). The relationship of HLA class I and II alleles and haplotypes with autism: a case control study. Autism Res Treat 2014: 242048.
American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders5th ednAmerican Psychiatric Association: Washington, DC.
Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J (2011a). Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun 25: 40–45.
Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Van de Water J (2011b). Altered T cell responses in children with autism. Brain Behav Immun 25: 840–849.
Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Van de Water J (2011c). Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. J Neuroimmunol 232: 196–199.
Ashwood P, Van de Water J (2004). Is autism an autoimmune disease? Autoimmun Rev 3: 557–562.
Atladottir HO, Henriksen TB, Schendel DE, Parner ET (2012). Autism after infection, febrile episodes, and antibiotic use during pregnancy: an exploratory study. Pediatrics 130: e1447–e1454.
Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M et al (2010). Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 40: 1423–1430.
Badcock C (2011). The imprinted brain: how genes set the balance between autism and psychosis. Epigenomics 3: 345–359.
Basu SN, Kollu R, Banerjee-Basu S (2009). AutDB: a gene reference resource for autism research. Nucleic Acids Res 37 (Database issue): D832–D836.
Bauman MD, Iosif AM, Ashwood P, Braunschweig D, Lee A, Schumann CM et al (2013). Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl Psychiatry 3: e278.
Bauman MD, Iosif AM, Smith SE, Bregere C, Amaral DG, Patterson PH (2014). Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol Psychiatry 75: 332–341.
Boksa P (2010). Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun 24: 881–897.
Boulanger LM (2009). Immune proteins in brain development and synaptic plasticity. Neuron 64: 93–109.
Boulanger LM, Shatz CJ (2004). Immune signalling in neural development, synaptic plasticity and disease. Nat Rev Neurosci 5: 521–531.
Braunschweig D, Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Croen LA et al (2008). Autism: maternally derived antibodies specific for fetal brain proteins. Neurotoxicology 29: 226–231.
Braunschweig D, Duncanson P, Boyce R, Hansen R, Ashwood P, Pessah IN et al (2012a). Behavioral correlates of maternal antibody status among children with autism. J Autism Dev Disord 42: 1435–1445.
Braunschweig D, Golub MS, Koenig CM, Qi L, Pessah IN, Van de Water J et al (2012b). Maternal autism-associated IgG antibodies delay development and produce anxiety in a mouse gestational transfer model. J Neuroimmunol 252: 56–65.
Braunschweig D, Krakowiak P, Duncanson P, Boyce R, Hansen RL, Ashwood P et al (2013). Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry 3: e277.
Brimberg L, Sadiq A, Gregersen PK, Diamond B (2013). Brain-reactive IgG correlates with autoimmunity in mothers of a child with an autism spectrum disorder. Mol Psychiatry 18: 1171–1177.
Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M et al (2004a). Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry 61: 774–780.
Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V et al (2004b). Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry 161: 889–895.
Brown AS, Sourander A, Hinkka-Yli-Salomaki S, McKeague IW, Sundvall J, Surcel HM (2014). Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry 19: 259–264.
Brown AS, Surcel H-M, Hinkka-Yli-Salomäki S, Cheslack-Postava K, Bao Y, Sourander A (2015). Maternal thyroid autoantibody and elevated risk of autism in a national birth cohort. Prog Neuropsychopharmacol Biol Psychiatry 57: 86–92.
Camacho J, Jones K, Miller E, Ariza J, Noctor S, Van de Water J et al (2014). Embryonic intraventricular exposure to autism-specific maternal autoantibodies produces alterations in autistic-like stereotypical behaviors in offspring mice. Behav Brain Res 266: 46–51.
Campbell DB, Li C, Sutcliffe JS, Persico AM, Levitt P (2008). Genetic evidence implicating multiple genes in the MET receptor tyrosine kinase pathway in autism spectrum disorder. Autism Res 1: 159–168.
Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S et al (2006). A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci USA 103: 16834–16839.
Cantor RM, Geschwind DH (2008). Schizophrenia: genome, interrupted. Neuron 58: 165–167.
Carosella ED, Favier B, Rouas-Freiss N, Moreau P, Lemaoult J (2008). Beyond the increasing complexity of the immunomodulatory HLA-G molecule. Blood 111: 4862–4870.
Carosella ED, Moreau P, Le Maoult J, Le Discorde M, Dausset J, Rouas-Freiss N (2003). HLA-G molecules: from maternal-fetal tolerance to tissue acceptance. Adv Immunol 81: 199–252.
Chess S (1971). Autism in children with congenital rubella. J Autism Child Schizophr 1: 33–47.
Chess S (1977). Follow-up report on autism in congenital rubella. J Autism Child Schizophr 7: 69–81.
Chess S, Fernandez P, Korn S (1978). Behavioral consequences of congenital rubella. J Pediatr 93: 699–703.
Chien YL, Wu YY, Chen CH, Gau SS, Huang YS, Chien WH et al (2012). Association of HLA-DRB1 alleles and neuropsychological function in autism. Psychiatr Genet 22: 46–49.
Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV et al (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351: 933–939.
Christensen DL, Baio J, Braun KV, Bilder D, Charles J, Constantino JN et al (2016). Prevalence and characteristics of autism spectrum disorder among children aged 8 years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill Summ 65: 1–23.
Colvert E, Tick B, McEwen F, Stewart C, Curran SR, Woodhouse E et al (2015). Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 72: 415–423.
Croen LA, Braunschweig D (2008a). Maternal mid-pregnancy autoantibodies to fetal brain protein: the early markers for autism study. Biol Psychiatry 64: 583–588.
Croen LA, Braunschweig D, Haapanen L, Yoshida CK, Fireman B, Grether JK et al (2008b). Maternal mid-pregnancy autoantibodies to fetal brain protein: the early markers for autism study. Biol Psychiatry 64: 583–588.
Croonenberghs J, Bosmans E, Deboutte D, Kenis G, Maes M (2002a). Activation of the inflammatory response system in autism. Neuropsychobiology 45: 1–6.
Croonenberghs J, Wauters A, Devreese K, Verkerk R, Scharpe S, Bosmans E et al (2002b). Increased serum albumin, gamma globulin, immunoglobulin IgG, and IgG2 and IgG4 in autism. Psychol Med 32: 1457–1463.
Dalton P, Deacon R, Blamire A, Pike M, McKinlay I, Stein J et al (2003). Maternal neuronal antibodies associated with autism and a language disorder. Ann Neurol 53: 533–537.
DeLisi LE, Ortaldo JR, Maluish AE, Wyatt RJ (1983). Deficient natural killer cell (NK) activity and macrophage functioning in schizophrenic patients. J Neural Transm 58: 99–106.
Depino AM (2013). Peripheral and central inflammation in autism spectrum disorders. Mol Cell Neurosci 53: 69–76.
Deverman BE, Patterson PH (2009). Cytokines and CNS development. Neuron 64: 61–78.
Deykin EY, MacMahon B (1979). Viral exposure and autism. Am J Epidemiol 109: 628–638.
Ellis SE, Panitch R, West AB, Arking DE (2016). Transcriptome analysis of cortical tissue reveals shared sets of downregulated genes in autism and schizophrenia. Transl Psychiatry 6: e817.
Enstrom A, Krakowiak P, Onore C, Pessah IN, Hertz-Picciotto I, Hansen RL et al (2009a). Increased IgG4 levels in children with autism disorder. Brain Behav Immun 23: 389–395.
Enstrom A, Onore C, Hertz-picciotto I, Hansen R, Croen L, Van de Water J et al (2008). Detection of IL-17 and IL-23 in plasma samples of children with autism. Am J Biochem & Biotech 4: 114–120.
Enstrom AM, Lit L, Onore CE, Gregg JP, Hansen RL, Pessah IN et al (2009b). Altered gene expression and function of peripheral blood natural killer cells in children with autism. Brain Behav Immun 23: 124–133.
Enstrom AM, Onore CE, Van de Water JA, Ashwood P (2010). Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav Immun 24: 64–71.
Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I et al (2016). Unexpected role of interferon-gamma in regulating neuronal connectivity and social behaviour. Nature 535: 425–429.
Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K et al (2008). Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis 30: 303–311.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB et al (2014). Most genetic risk for autism resides with common variation. Nat Genet 46: 881–885.
Gesundheit B, Rosenzweig JP, Naor D, Lerer B, Zachor DA, Prochazka V et al (2013). Immunological and autoimmune considerations of autism spectrum disorders. J Autoimmun 44: 1–7.
Gilmore JH, Jarskog LF, Vadlamudi S (2005). Maternal poly I:C exposure during pregnancy regulates TNF alpha, BDNF, and NGF expression in neonatal brain and the maternal-fetal unit of the rat. J Neuroimmunol 159: 106–112.
Goines P, Haapanen L, Boyce R, Duncanson P, Braunschweig D, Delwiche L et al (2011a). Autoantibodies to cerebellum in children with autism associate with behavior. Brain Behav Immun 25: 514–523.
Goines PE, Croen LA, Braunschweig D, Yoshida CK, Grether J, Hansen R et al (2011b). Increased midgestational IFN-gamma, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol Autism 2: 13.
Gottfried C, Bambini-Junior V, Francis F, Riesgo R, Savino W (2015). The impact of neuroimmune alterations in autism spectrum disorder. Front Psychiatry 6: 121.
Gregg JP, Lit L, Baron CA, Hertz-Picciotto I, Walker W, Davis RA et al (2008). Gene expression changes in children with autism. Genomics 91: 22–29.
Guerini FR, Bolognesi E, Chiappedi M, Ghezzo A, Canevini MP, Mensi MM et al (2015). An HLA-G(*)14 bp insertion/deletion polymorphism associates with the development of autistic spectrum disorders. Brain Behav Immun 44: 207–212.
Guerini FR, Bolognesi E, Chiappedi M, Manca S, Ghezzo A, Agliardi C et al (2014). Activating KIR molecules and their cognate ligands prevail in children with a diagnosis of ASD and in their mothers. Brain Behav Immun 36: 54–60.
Gupta S, Aggarwal S, Rashanravan B, Lee T (1998). Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J Neuroimmunol 85: 106–109.
Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J et al (2014). Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 5: 5748.
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T et al (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 68: 1095–1102.
Happe F, Ronald A, Plomin R (2006). Time to give up on a single explanation for autism. Nat Neurosci 9: 1218–1220.
Haroon E, Raison CL, Miller AH (2012). Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 37: 137–162.
Henneberg AE, Horter S, Ruffert S (1994). Increased prevalence of antibrain antibodiesin the sera from schizophrenic patients. Schizophr Res 14: 15–22.
Heuer L, Ashwood P, Schauer J, Goines P, Krakowiak P, Hertz-Picciotto I et al (2008). Reduced levels of immunoglobulin in children with autism correlates with behavioral symptoms. Autism Res 1: 275–283.
Heuer L, Braunschweig D, Ashwood P, Van de Water J, Campbell DB (2011). Association of a MET genetic variant with autism-associated maternal autoantibodies to fetal brain proteins and cytokine expression. Transl Psychiatry 1: e48.
Heuer LS, Rose M, Ashwood P, Van de Water J (2012). Decreased levels of total immunoglobulin in children with autism are not a result of B cell dysfunction. J Neuroimmunol 251: 94–102.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T et al (2013). Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155: 1451–1463.
Hutton J (2016). Does rubella cause autism: a 2015 reappraisal? Front Hum Neurosci 10: 25.
Jackson PB, Boccuto L, Skinner C, Collins JS, Neri G, Gurrieri F et al (2009). Further evidence that the rs1858830 C variant in the promoter region of the MET gene is associated with autistic disorder. Autism Res 2: 232–236.
Johnson WG, Buyske S, Mars AE, Sreenath M, Stenroos ES, Williams TA et al (2009). HLA-DR4 as a risk allele for autism acting in mothers of probands possibly during pregnancy. Arch Pediatr Adolesc Med 163: 542–546.
Jones KA, Thomsen C (2013). The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci 53: 52–62.
Jones KL, Croen LA, Yoshida CK, Heuer L, Hansen R, Zerbo O et al (2016). Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol Psychiatry 1–7 (e-pub ahead of print).
Jyonouchi H, Geng L, Ruby A, Zimmerman-Bier B (2005). Dysregulated innate immune responses in young children with autism spectrum disorders: their relationship to gastrointestinal symptoms and dietary intervention. Neuropsychobiology 51: 77–85.
Jyonouchi H, Sun S, Itokazu N (2002). Innate immunity associated with inflammatory responses and cytokine production against common dietary proteins in patients with autism spectrum disorder. Neuropsychobiology 46: 76–84.
Kaidanovich-Beilin O, Lipina T, Vukobradovic I, Roder J, Woodgett JR (2011). Assessment of social interaction behaviors. J Vis Exp 48: e2473.
Kanner L (1943). Autistic disturbances of affective contact. Nervous Child 2: 217–250.
Konya C, Paz Z, Apostolidis SA, Tsokos GC (2015). Update on the role of Interleukin 17 in rheumatologic autoimmune diseases. Cytokine 75: 207–215.
Krakowiak P, Goines PE, Tancredi DJ, Ashwood P, Hansen RL, Hertz-Picciotto I et al (2015). Neonatal cytokine profiles associated with autism spectrum disorder. Biol Psychiatry 1–10. (available at http://www.biologicalpsychiatryjournal.com/article/S0006-3223(15)00655-1/abstract and http://www.ncbi.nlm.nih.gov/pubmed/26392128).
Labouesse MA, Dong E, Grayson DR, Guidotti A, Meyer U (2015). Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 10: 1143–1155.
Lee BK, Magnusson C, Gardner RM, Blomstrom A, Newschaffer CJ, Burstyn I et al (2015). Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav Immun 44: 100–105.
Lee LC, Zachary AA, Leffell MS, Newschaffer CJ, Matteson KJ, Tyler JD et al (2006). HLA-DR4 in families with autism. Pediatr Neurol 35: 303–307.
Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM et al (2009). Elevated immune response in the brain of autistic patients. J Neuroimmunol 207: 111–116.
Libbey JE, Fujinami RS (2010). Role for antibodies in altering behavior and movement. Autism Res 3: 147–152.
Libbey JE, Sweeten TL, McMahon WM, Fujinami RS (2005). Autistic disorder and viral infections. J Neurovirol 11: 1–10.
Lintas C, Altieri L, Lombardi F, Sacco R, Persico AM (2010). Association of autism with polyomavirus infection in postmortem brains. J Neurovirol 16: 141–149.
Lintas C, Sacco R, Persico AM (2012). Genome-wide expression studies in autism spectrum disorder, Rett syndrome, and Down syndrome. Neurobiol Dis 45: 57–68.
Lopez-Verges S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H et al (2010). CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 116: 3865–3874.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD et al (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523: 337–341.
Mackay M, Tang CC, Volpe BT, Aranow C, Mattis PJ, Korff RA et al (2015). Brain metabolism and autoantibody titres predict functional impairment in systemic lupus erythematosus. Lupus Sci Med 2: e000074.
Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH (2012). Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun 26: 607–616.
Martin LA, Ashwood P, Braunschweig D, Cabanlit M, Van de Water J, Amaral DG (2008). Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain Behav Immun 22: 806–816.
Martinez-Cerdeno V, Camacho J, Fox E, Miller E, Ariza J, Kienzle D et al (2016). Prenatal exposure to autism-specific maternal autoantibodies alters proliferation of cortical neural precursor cells, enlarges brain, and increases neuronal size in adult animals. Cereb Cortex 26: 374–383.
Mazur-Kolecka B, Cohen IL, Gonzalez M, Jenkins EC, Kaczmarski W, Brown WT et al (2014). Autoantibodies against neuronal progenitors in sera from children with autism. Brain Dev 36: 322–329.
Meyer U, Feldon J (2009). Neural basis of psychosis-related behaviour in the infection model of schizophrenia. Behav Brain Res 204: 322–334.
Meyer U, Feldon J, Schedlowski M, Yee BK (2005). Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev 29: 913–947.
Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J (2008). Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry 13: 208–221.
Meyer U, Yee BK, Feldon J (2007). The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist 13: 241–256.
Michel M, Schmidt MJ, Mirnics K (2012). Immune system gene dysregulation in autism and schizophrenia. Dev Neurobiol 72: 1277–1287.
Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, Manning-Courtney P et al (2006). Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol 172: 198–205.
Mostafa GA, Al Shehab A, Fouad NR (2010). Frequency of CD4+CD25 high regulatory T cells in the peripheral blood of Egyptian children with autism. J Child Neurol 25: 328–335.
Mostafa GA, Al-Ayadhi LY (2012). The relationship between the increased frequency of serum antineuronal antibodies and the severity of autism in children. Eur J Paediatr Neurol 16: 464–468.
Mostafa GA, Shehab AA, Al-Ayadhi LY (2013). The link between some alleles on human leukocyte antigen system and autism in children. J Neuroimmunol 255: 70–74.
Nielsen CM, White MJ, Goodier MR, Riley EM (2013). Functional significance of CD57 expression on human NK cells and relevance to disease. Front Immunol 4: 422.
Nordahl CW, Braunschweig D, Iosif AM, Lee A, Rogers S, Ashwood P et al (2013). Maternal autoantibodies are associated with abnormal brain enlargement in a subgroup of children with autism spectrum disorder. Brain Behav Immun 30: 61–65.
Parham P (2005). MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol 5: 201–214.
Patterson PH (2009). Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res 204: 313–321.
Piras IS, Haapanen L, Napolioni V, Sacco R, Van de Water J, Persico AM (2014). Anti-brain antibodies are associated with more severe cognitive and behavioral profiles in Italian children with Autism Spectrum Disorder. Brain Behav Immun 38: 91–99.
Ponzio NM, Servatius R, Beck K, Marzouk A, Kreider T (2007). Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspring. Ann N Y Acad Sci 1107: 118–128.
Rossi CC, Van de Water J, Rogers SJ, Amaral DG (2011). Detection of plasma autoantibodies to brain tissue in young children with and without autism spectrum disorders. Brain Behav Immun 25: 1123–1135.
Ryberg B (1982). Antibrain antibodies in multiple sclerosis. Relation to clinical variables. J Neurol Sci 54: 239–261.
Shi L, Fatemi SH, Sidwell RW, Patterson PH (2003). Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci 23: 297–302.
Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH (2009). Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun 23: 116–123.
Singer HS, Morris C, Gause C, Pollard M, Zimmerman AW, Pletnikov M (2009). Prenatal exposure to antibodies from mothers of children with autism produces neurobehavioral alterations: a pregnant dam mouse model. J Neuroimmunol 211: 39–48.
Singer HS, Morris CM, Gause CD, Gillin PK, Crawford S, Zimmerman AW (2008). Antibodies against fetal brain in sera of mothers with autistic children. J Neuroimmunol 194: 165–172.
Singer HS, Morris CM, Williams PN, Yoon DY, Hong JJ, Zimmerman AW (2006). Antibrain antibodies in children with autism and their unaffected siblings. J Neuroimmunol 178: 149–155.
Singh VK (2009). Phenotypic expression of autoimmune autistic disorder (AAD): a major subset of autism. Ann Clin Psychiatry 21: 148–161.
Siniscalco D, Mijatovic T, Bosmans E, Cirillo A, Kruzliak P, Lombardi VC et al (2016). Decreased numbers of CD57+CD3- cells identify potential innate immune differences in patients with autism spectrum disorder. In Vivo 30: 83–89.
Smith SE, Li J, Garbett K, Mirnics K, Patterson PH (2007). Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 27: 10695–10702.
Suurmond J, Diamond B (2015). Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125: 2194–2202.
Torres AR, Sweeten TL, Cutler A, Bedke BJ, Fillmore M, Stubbs EG et al (2006). The association and linkage of the HLA-A2 class I allele with autism. Hum Immunol 67: 346–351.
Torres AR, Westover JB, Rosenspire AJ (2012). HLA immune function genes in autism. Autism Res Treat 2012: 959073.
Trajkovski V, Ajdinski L, Spiroski M (2004). Plasma concentration of immunoglobulin classes and subclasses in children with autism in the Republic of Macedonia: retrospective study. Croat Med J 45: 746–749.
Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH (2001). Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr Res 47: 27–36.
Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 57: 67–81.
Vinet E, Pineau CA, Clarke AE, Scott S, Fombonne E, Joseph L et al (2015). Increased risk of autism spectrum disorders in children born to women with systemic lupus erythematosus: results from a large population-based cohort. Arthritis Rheumatol 67: 3201–3208.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474: 380–384.
Warren RP, Cole P, Odell JD, Pingree CB, Warren WL, White E et al (1990). Detection of maternal antibodies in infantile autism. J Am Acad Child Adolesc Psychiatry 29: 873–877.
Warren RP, Foster A, Margaretten NC (1987). Reduced natural killer cell activity in autism. J Am Acad Child Adolesc Psychiatry 26: 333–335.
Warren RP, Margaretten NC, Pace NC, Foster A (1986). Immune abnormalities in patients with autism. J Autism Dev Disord 16: 189–197.
Warren RP, Odell JD, Warren WL, Burger RA, Maciulis A, Daniels WW et al (1996). Strong association of the third hypervariable region of HLA-DRβ1 with autism. J Neuroimmunol 67: 97–102.
Weber-Stadlbauer U, Richetto J, Labouesse MA, Bohacek J, Mansuy IM, Meyer U (2016). Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol Psychiatry 1–11 (e-pub ahead of print).
Wilkerson DS, Volpe AG, Dean RS, Titus JB (2009). Perinatal complications as predictors of infantile autism. Int J Neurosci 112: 1085–1098.
Wills S, Cabanlit M, Bennett J, Ashwood P, Amaral DG, Van de Water J (2009). Detection of autoantibodies to neural cells of the cerebellum in the plasma of subjects with autism spectrum disorders. Brain Behav Immun 23: 64–74.
Wu S, Ding Y, Wu F, Li R, Xie G, Hou J et al (2015). Family history of autoimmune diseases is associated with an increased risk of autism in children: A systematic review and meta-analysis. Neurosci Biobehav Rev 55: 322–332.
Yonk LJ, Warren RP, Burger RA, Cole P, Odell JD, Warren WL et al (1990). CD4+ helper T cell depression in autism. Immunol Lett 25: 341–345.
Young AM, Chakrabarti B, Roberts D, Lai MC, Suckling J, Baron-Cohen S (2016). From molecules to neural morphology: understanding neuroinflammation in autism spectrum condition. Mol Autism 7: 9.
Zaman S, Yazdani U, Deng Y, Li W, Gadad BS, Hynan L et al (2016). A search for blood biomarkers for autism: peptoids. Sci Rep 6: 19164.
Zaretsky MV, Alexander JM, Byrd W, Bawdon RE (2004). Transfer of inflammatory cytokines across the placenta. Obstet Gynecol 103: 546–550.
Zerbo O, Iosif AM, Walker C, Ozonoff S, Hansen RL, Hertz-Picciotto I (2013). Is maternal influenza or fever during pregnancy associated with autism or developmental delays? Results from the CHARGE (Childhood Autism Risks from Genetics and Environment) study. J Autism Dev Disord 43: 25–33.
Zerbo O, Qian Y, Yoshida C, Grether JK, Van de Water J, Croen LA (2015). Maternal infection during pregnancy and autism spectrum disorders. J Autism Dev Disord 45: 4015–4025.
Zerbo O, Traglia M, Yoshida C, Heuer LS, Ashwood P, Delorenze GN et al (2016). Maternal mid-pregnancy C-reactive protein and risk of autism spectrum disorders: the early markers for autism study. Transl Psychiatry 6: e783.
Zhang X, Lv CC, Tian J, Miao RJ, Xi W, Hertz-Picciotto I et al (2010). Prenatal and perinatal risk factors for autism in China. J Autism Dev Disord 40: 1311–1321.
Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D et al (2016). Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89: 147–162.
Ziats MN, Rennert OM (2011). Expression profiling of autism candidate genes during human brain development implicates central immune signaling pathways. PLoS One 6: e24691.
Zimmerman AW, Connors SL, Matteson KJ, Lee LC, Singer HS, Castaneda JA et al (2007). Maternal antibrain antibodies in autism. Brain Behav Immun 21: 351–357.
Zuckerman L, Weiner I (2005). Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res 39: 311–323.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Meltzer, A., Van de Water, J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacol 42, 284–298 (2017). https://doi.org/10.1038/npp.2016.158
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.158
This article is cited by
-
Granulocyte macrophage colony-stimulating factor-induced macrophages of individuals with autism spectrum disorder adversely affect neuronal dendrites through the secretion of pro-inflammatory cytokines
Molecular Autism (2024)
-
Understanding immune microenvironment alterations in the brain to improve the diagnosis and treatment of diverse brain diseases
Cell Communication and Signaling (2024)
-
Genetic evidence for causal effects of immune dysfunction in psychiatric disorders: where are we?
Translational Psychiatry (2024)
-
Identification of Immune Infiltration and Iron Metabolism–Related Subgroups in Autism Spectrum Disorder
Journal of Molecular Neuroscience (2024)
-
Untargeted metabolomic, and proteomic analysis identifies metabolic biomarkers and pathway alterations in individuals with 22q11.2 deletion syndrome
Metabolomics (2024)
