
Abstract
Mounting evidence indicates that proinflammatory signaling in the brain affects mood, cognition, and behavior and is linked with the etiology of psychiatric disorders, including anxiety and depression. The purpose of this review is to focus on stress-induced bidirectional communication pathways between the central nervous system (CNS) and peripheral immune system that converge to promote a heightened neuroinflammatory environment. These communication pathways involve sympathetic outflow from the brain to the peripheral immune system that biases hematopoietic stem cells to differentiate into a glucocorticoid-resistant and primed myeloid lineage immune cell. In conjunction, microglia-dependent neuroinflammatory events promote myeloid cell trafficking to the brain that reinforces stress-related behavior, and is argued to play a role in stress-related psychiatric disorders. We will discuss evidence implicating a key role for endothelial cells that comprise the blood–brain barrier in propagating peripheral-to-central immune communication. We will also discuss novel neuron-to-glia communication pathways involving endogenous danger signals that have recently been argued to facilitate neuroinflammation under various conditions, including stress. These findings help elucidate the complex communication that occurs in response to stress and highlight novel therapeutic targets against the development of stress-related psychiatric disorders.
Similar content being viewed by others

Introduction
Exposure to chronic stress or traumatic events increases the risk of developing mood or anxiety disorders and can severely impact quality of life (Kessler, 1997; Gilman et al, 2013). However, not everyone who experiences an adverse or stressful event succumbs to negative outcomes and enters a pathological state. As such, the biological mechanisms underlying the deleterious effects of stress on psychiatric health are of clinical importance in order to identify factors that promote resistance vs pathology. Advancement in our understanding of how neuroimmune processes mediates behavioral, affective, and neurochemical functions offers a new perspective for understanding the etiology of psychiatric disorders.
The central nervous system (CNS) and peripheral immune system are in constant communication and influence how each other respond to a challenge. Prolonged or severe stress exposure disrupts homeostatic or ‘healthy’ communication between the CNS and peripheral immune system, shifting immune signaling toward a proinflammatory state. Part of this response includes elevated and prolonged proinflammatory signaling in the CNS that is argued to be linked with stress-related psychiatric disorders (Miller and Raison, 2015). This argument comes from the observation that activation of proinflammatory signaling molecules engage neural mechanisms that mediate changes to mood, cognition, and physiology that are adaptive for the host defense against infection or injury (ie, the ‘sickness’ response) (Dantzer et al, 1998; Maier and Watkins, 1998). However, this response closely resembles several characteristics observed in mood disorders, including withdrawal from social and physical environments, anhedonia, and sleep disturbances (Dantzer et al, 2008). Dysregulated or enhanced neuroinflammation is argued to facilitate the etiology of mood disorders, particularly in treatment-resistant depressed patients (Hodes et al, 2015). In support of this, elevated proinflammatory cytokine has been reported in the cerebrospinal fluid of clinically depressed patients (Levine et al, 1999; Miller et al, 2009). Perhaps the most compelling evidence to date comes from a recent neuroimaging study demonstrating significant association between activated microglia and major depression in several brain regions linked with depression (Setiawan et al, 2015). In the early 1990s, Ronald Smith proposed that immune dysregulation contributes to depression based on the observation of increased myeloid cells in circulation of patients with MDD (Smith, 1991) that communicates with the CNS via humoral and neural pathways (Maier and Watkins, 2003). To implicate a causal role of immune-to-brain communication and depression, investigators have long noted that a high percentage of cancer patients treated with the proinflammatory cytokine interferon-α (IFN-α) develop depressive-like behaviors (Raison et al, 2006). INF-α is a potent inducer of proinflammatory cytokines, thereby supporting the contention that peripheral inflammation induces a ‘depressive-like’ state.
These studies led investigators to the hypothesis that chronic stress induces vulnerability to psychiatric disorders by activating neuroimmune circuitry. Rodent stress models provide means to empirically study communication between stress-reactive neurocircuitry and immunity. Repeated social defeat (RSD) is an ethologically relevant social stressor that recapitulates many of the stress-induced immune and behavioral outcomes of stress (Reader et al, 2015). RSD disrupts established social hierarchy by introducing a larger aggressor into the residents’ cage for 2 h. After six consecutive cycles, the residents display submissive behavior to the aggressor. As illustrated in Figure 1, RSD is accompanied by activation of stress-reactive neurocircuitry, prolonged anxiety-like behavior, and neuroinflammatory events including microglial activation and increased cytokine production. Furthermore, sympathetic outflow increases production, egress, and trafficking of inflammatory myeloid cells, typically monocytes, that are insensitive to glucocorticoids (GCs). In this review, we outline recent advances using the RSD stress model, highlighting a monocyte-dependent signaling pathway that promotes neuroinflammation and affects stress-induced mood and behavior. We discuss CNS-mediated events that recruit monocytes via blood–brain barrier (BBB) endothelial cells followed by clinical significance of brain-immune interactions. Finally, we discuss specific neuron–glia communication pathways that are recently implicated in stress-induced neuroinflammation. This includes investigating endogenous danger signals that could provide novel therapeutic targets against the development of stress-related mood disorders.

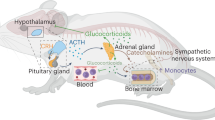
Overview of stress-induced myelopoiesis and monocyte trafficking to the CNS. Social defeat stress activates fear/threat appraisal circuitry. In corresponding regions, microglia are activated and increase proinflammatory cytokine and chemokine expression that interacts with BBB-derived endothelial cells to recruit circulating monocytes. Social defeat also initiates brain-to-immune outflow via SNS and HPA axis activation. SNS and HPA activation shifts peripheral immune responses through production of primed and GC-insensitive monocytes from the bone marrow into circulation. These monocytes have an increased capacity to traffic throughout the body and interact with BBB endothelial cells to traffic into the CNS.
Rsd-responsive neuroendocrine pathways alter homeostatic hematopoiesis: enhanced myelopoiesis leads to production of primed and GC-resistant myeloid cells
A hallmark of the stress response is activation of sympathetic nervous system (SNS) and hypothalamic–pituitary–adrenal (HPA) axis. The paraventricular nucleus (PVN) of the hypothalamus is a key common efferent structure that integrates stress signals and regulates HPA and autonomic responses to stressors. The PVN houses distinct neuron populations projecting to autonomic targets in the brain stem and spinal cord (Ulrich-Lai and Herman, 2009). Descending sympathetic nerve fibers innervate target organs throughout the body with the purpose of activating physiological changes that occur during the fight or flight response. This includes increased blood flow to the muscles, increased muscle tension for increase in speed and strength, and increased blood pressure, heart rate, and glucose to supply the body with extra energy. In addition, sympathetic nerve fibers innervate peripheral immune lymphoid tissue including the thymus, spleen, lymph nodes, and bone marrow (BM), thereby providing a direct signaling pathway to regulate peripheral immune processes (Felten et al, 1985). Brief or acute stressors activate sympathetic signaling that increases norepinephrine (NE) to mobilize ‘housed’ leukocytes into circulation from reservoirs including the spleen, BM, and lymph nodes (Dhabhar et al, 2012). This is to provide a quick and nonspecialized immune response to the acute stressor. However, rodent models of repeated or chronic stressors indicate that stress duration can affect proliferation, release, and function of leukocytes to promote the production of more specialized immune cells (Engler et al, 2004; Dhabhar et al, 2012; Heidt et al, 2014). The BM is of particular interest here, as it is the site of proliferation and maturation of multipotent stem cells into immune cells. Chronic stressors or repeated exposure to social defeat shifts hematopoietic stem cell differentiation toward the myeloid cell lineage. For example, RSD increases monocyte and granulocyte progenitor cells in the BM that traffic into circulation and are redistributed throughout the body (Engler et al, 2004; Powell et al, 2013). The myelopoietic response to RSD requires repeated bouts with the aggressor, as monocytes begin to accumulate in circulation after three cycles of social defeat and are robustly increased after six cycles (Wohleb et al, 2013). This effect is not specific to RSD as chronic variable stress also increases the most primitive self-renewal Lin−Sca-1+c-Kit+ hematopoietic stem cells in the BM (Heidt et al, 2014). Again, the progenitor cells were biased toward the myeloid cell lineage as neutrophils and monocytes were increased in circulation. However, a similar accumulation is not observed by lymphocyte production. In fact, RSD significantly decreases B-cell and T-cell lymphocytes in the BM and circulation (Engler et al, 2004; McKim et al, 2015), thereby shifting hematopoiesis to produce myeloid lineage cells.
Further investigation revealed that the myelopoietic response of RSD is mediated by NE (Wohleb et al, 2011; Hanke et al, 2012; Heidt et al, 2014). NE is synthesized downstream of the rate-limiting tyrosine hydroxylase. RSD increases tyrosine hydroxylase and NE in the BM stroma (Heidt et al, 2014). Furthermore, pharmacological blockage of the β-adrenergic receptor before RSD prevented BM-derived myeloid cell differentiation and redistribution in circulation (Wohleb et al, 2011; Hanke et al, 2012; Powell et al, 2013). One such way that leukocytes are stimulated by NE is by decreasing the inhibitory chemokine CXCL12 (Katayama et al, 2006; Heidt et al, 2014). In the hematopoietic niche, CXCL12 functions to inhibit hematopoietic stem and progenitor cell proliferation (Nie et al, 2008; Eash et al, 2009). For example, CXCL12-deficient mice and mice lacking the target receptor, CXCR4, display increased hematopoietic stem cell cycling, progenitor pool expansion, and increased trafficking of neutrophils in circulation (Eash et al, 2009). As such, stress-induced hematopoietic stem cell differentiation was associated with decreased CXCL12 expression in the BM. A direct link between NE and CXCL12 comes from studies in which NE signaling is blocked, either through genetic deletion or pharmacological inhibition. Here, either genetic deletion or pharmacological inhibition of β3 adrenergic receptor restored CXCL12 mRNA and protein that reduced hematopoietic stem cell progenitors in the BM and lowered circulating neutrophils and monocytes (Heidt et al, 2014). In opposition to CXCL12, stress can stimulate myeloid differentiation by increasing the myelopoietic growth factor, granulocyte-macrophage colony-stimulating factor (GM-CSF). Specifically, GM-CSF signaling mediates differentiation of granulocytes and monocytes from their common progenitor, granulocyte-macrophage colony-forming unit (GM-CFU) (Hamilton and Achuthan, 2013). Indeed, exposure to RSD increased expression of GM-CSF and its receptor (CD116) (Powell et al, 2013), consistent with increased expansion of granulocytes and monocytes after RSD. Furthermore, treatment with a GM-CSF neutralizing antibody prevented RSD-induced expansion of granulocytes and monocytes. As such, repeated or chronic stressors activate sympathetic signaling that reduces CXCL12 and increases GM-CSF, thereby enhancing immune cell production toward the myeloid cell lineage.
An interesting characteristic of RSD is that the newly generated monocytes are less mature and more inflammatory than ‘homeostatic’ monocytes. Newly differentiated monocytes express high levels of the monocyte marker Ly6C (Ly6Chi) (Rose et al, 2012). Ly6Chi monocytes have a high capacity for pro-inflammatory processes, including potent phagocytic capacity and increased secretion of reactive oxygen species (ROS), nitric oxides, and pro-inflammatory cytokines (Bailey et al, 2007). Critically, Ly6Chi monocytes express a chemokine receptor profile that allows for trafficking to inflamed tissue. Ly6Chi monocytes are positive for C-C chemokine receptor type 2 (CCR2) that detects chemotactic CCL2 to mediate cell trafficking (Audoy-Remus et al, 2008). Monocytes begin to reduce Ly6C and CCR2 expression as they differentiate toward an anti-inflammatory phenotype, contributing to tissue repair. This includes increased expression of the fractalkine receptor (CX3CR1) and secretion of anti-inflammatory molecules such as IL-10 (Auffray et al, 2007). Based on data from flow cytometry scatter properties, RSD preferentially increases the amount of circulating Ly6Chi monocytes (Powell et al, 2013; Wohleb et al, 2013; Ramirez et al, 2015a) that is prevented by blockage of the β-adrenergic receptor (Hanke et al, 2012; Powell et al, 2013). In addition, newly differentiated Ly6chi monocytes express surface activation markers that enhance immune function. For example, surface activation markers including Toll-like receptors (TLRs), chemotactic receptors including CCR2, and co-stimulator receptors are upregulated by RSD (Avitsur et al, 2003; Engler et al, 2004; Bailey et al, 2007; Powell et al, 2009; Hanke et al, 2012). These cells are termed ‘primed’ because they produce enhanced proinflammatory signaling molecules and bactericidal activity when stimulated with an immune challenge (Bailey et al, 2007; Powell et al, 2009). As such, newly differentiated monocytes produced by chronic stress have increased trafficking capabilities and a primed proinflammatory phenotype compared with that of the typical monocyte during ‘healthy’ or homeostatic conditions (Figure 2).

Stress-responsive neuroendocrine pathways alter homeostatic hematopoiesis: enhanced myelopoiesis leads to production of primed and GC-resistant myeloid cells. (a) Hematopoietic stem cells (HPSCs) begin with the capacity to differentiate into myeloid or lymphocyte progenitor cells. With the focus on myeloid lineage, myeloid progenitor cells differentiate into erythroid–megakaryocyte (MK) progenitor cells or granulocyte-macrophage colony-forming unit (GM-CFU). GM-CFU differentiates into basophils, neutrophils, eosinophils, and monocytes. Monocytes further differentiate into Ly6Chi expression that is associated with elevated expression of proinflammatory molecules and increased ability to traffic to tissue (ie, inflammatory). Ly6Clo monocytes are associated with increased expression of anti-inflammatory molecules and tissue repair (ie, patrolling). (b) Stress exposure can influence myelopoiesis via descending sympathetic nerve fibers that innervate lymphoid tissue. Increased NE alters growth factors (GM-CSF) and chemokines (CXCL12) that promote GM-CFU differentiation to primed/GC-resistant Ly6Chi monocytes.
RSD promotes differentiation of monocytes that are insensitive to GCs. Elevated GCs are a hallmark of stress and potently inhibit inflammatory processes and reduce immune cell viability (Coutinho and Chapman, 2011). With regard to RSD, the aggressor is introduced to the subject mouse at the beginning of the dark cycle. This produces a robust increase in circulating GCs but, importantly, GC levels return to baseline within a few hours after the stress cycle and circadian rhythmicity is not affected. However, RSD promotes the development of myeloid cells that are resistant to the inhibitory effects of elevated GCs, with the outcome being enhanced immune function. For example, GCs potently reduce LPS-induced cytokine production and cell viability ex vivo. However, this immunosuppressive effect does not occur in splenocytes isolated from mice exposed to RSD (Stark et al, 2001). It should be noted that the repeated and chronic nature of RSD is a critical factor for immune cells to develop GC resistance, as it requires 6 days of social defeat to develop, and persists for at least 10 days after stress termination (Avitsur et al, 2002). GC resistance is likely mediated by loss of anti-inflammatory feedback from the GC receptor. Typically, when GCs bind to GC receptors, GC receptors translocate into the nucleus to activate glucocorticoid-responsive elements (GREs). Activated GREs promote anti-inflammatory effects by inhibiting the proinflammatory transcription factor NF-κB and transcribing anti-inflammatory signaling molecules (Haegeman et al, 2003). However, GC receptor translocation is impaired in splenocytes after RSD (Quan et al, 2003), thereby reducing the ability of GCs to inhibit NF-κB-dependent transcription. Indeed, splenocytes isolated from RSD-exposed mice have enhanced NF-κB activity following LPS stimulation that is likely the cause of exaggerated cytokine production (Quan et al, 2003). Recent evidence suggests RSD may interfere with the ability of GC receptors to traffic via immunophilin FK506 binding proteins (FKBPs) (Galigniana et al, 2012). FKBPs include either FKBP51 or FKBP52. Binding of the GC receptor by FKBP51 prevents nuclear translocation, whereas binding by FKBP52 facilitates nuclear translocation (Davies et al, 2002). Indeed, impaired nuclear trafficking of GC receptors in splenocytes was associated with decreased FKBP52, thereby increasing the ratio of FKBP51/FKBP2 in order to reduce GC receptor nuclear translocation (Jung et al, 2015). These data indicate that RSD promotes accumulation of innately GC-insensitive monocytes, underlying the stress-induced proinflammatory phenotype.
Thus far, the focus has been on the ability of RSD to selectively increase the production of immunologically primed and GC-insensitive monocytes. It is tempting to generalize the myelopoietic response of RSD to all rodent models of chronic stress. However, it is important to keep in mind that ‘stress’ does not produce a homologous response via common mechanisms between all stress paradigms. In fact, there are very few studies demonstrating myelopoiesis in chronic stress models outside of RSD, whereas certain models reduce microbial activity. For example, several studies investigated the immune response to chronic restraint stress and reported diminished natural killer (NK) cell function, cytokine secretion, and enhanced viral replication (Hunzeker et al, 2004; Tseng et al, 2005; Jin et al, 2013). The opposing immune effects on myeloid cells between RSD and chronic restraint can be accounted for by differences in circulating GCs. Elevated GCs are potently immunosuppressive (Coutinho and Chapman, 2011). Chronic restraint stress is a potent activator of the HPA axis and is associated with elevated resting levels of circulating GCs (Bauer et al, 2001). As noted above, RSD increases circulating GCs during the stressor but, importantly, GCs return to baseline levels shortly after the end of the cycle, and GC levels during the diurnal nadir are not affected (Hanke et al, 2012). Therefore, elevated resting levels of GCs may be particularly important for the immunosuppressive effects of chronic restraint.
If the myeloid cell response to chronic stress is mediated by the circadian rhythm of GCs, then other models of chronic stress that maintain the diurnal nadir of GCs would be expected to increase myeloid cell production. For example, a recent study reported that 8 weeks of chronic unpredictable stress increased GCs during the diurnal zenith but GC levels during the diurnal nadir were normal (Monteiro et al, 2015). This was accompanied by increased population of monocytes and neutrophils, but not eosinophils, in the spleen. Similar to RSD, chronic unpredictable stress alters sympathetic innervation in the BM that strengthens adrenergic signaling (Heidt et al, 2014). Enhanced NE in the BM decreased CXCL12 via the β3-adrnergic receptor and shifted hematopoiesis to increase production of neutrophils and Ly6Chi inflammatory monocytes (Heidt et al, 2014). The consequence of chronic stress is dependent on HPA and sympathetic outflow from the CNS, and stress models differentially effect myeloid cell production depending on how stressors affect these pathways.
Another factor that makes RSD unique is the physical nature of the stressor. RSD is accompanied by biting and increased pain associated with the bite that is necessary to generate primed and GC-insensitive monocytes. Within a socially established cage, there are dominant and submissive residents. The dominant mouse receives significantly reduced number of bites compared with submissive mice in the cage and is protected against myelopoiesis of primed and GC-insensitive monocytes (Avitsur et al, 2007). However, myelopoiesis is not solely dependent on biting and attacks from the aggressor. Clonazepam is a common anxiolytic that acts by enhancing GABAergic activity in the brain. Daily treatment of clonazepam during RSD prevents myelopoiesis, although the same amount of biting from the aggressor persists, indicating that biting wounds are not sufficient to induce myelopoiesis of primed and GC-resistant monocytes (Ramirez et al, 2015a). The protective effects of clonazepam are attributed to attenuated sympathetic and HPA axis outflow from the CNS. In support of this, daily administration of the β-adrenergic antagonist propranolol before social defeat reduced primed and GC-resistant splenetic CD11b+ macrophages and significantly decreased surface expression of TLR2, TLR4, and CD86 on these cells (Hanke et al, 2012). The current opinion is that interactions between sympathetic and HPA axis outflow from fear/threat neurocircuitry and biting from the aggressor are required in the RSD model to induce myelopoiesis of primed and GC-resistant monocytes.
RSD activates stress circuitry, promotes neuroinflammation, and affects behavior
Exposure to potent or chronic stressors is known to produce a variety of changes in subsequent behavior often summarized as either anxiety like or depressive like. For example, RSD increases anxiety-like behaviors on standard measures including reduced exploration on open field and elevated plus maze, and increases dark preference in the light/dark test (Wohleb et al, 2011, 2013, 2014b; Hanke et al, 2012). RSD also increases depressive-like behaviors, including social avoidance (Wohleb et al, 2012; Ramirez et al, 2015b). It is important to note that the effects of RSD can be long lasting, as many of these behavioral changes persist at least 8 days after stressor termination with some (social avoidance) persisting up to 24 days later (Ramirez et al, 2015b).
The behavioral effects of RSD are indicative of enhanced fear/anxiety circuitry that is linked with anxiety and depression (Calhoon and Tye, 2015). The amygdala (AMYG) is the regulatory structure that integrates relevant sensory and cortical information and projects to hypothalamic and brainstem structures to produce fear responses (LeDoux, 2003). RSD robustly activates the AMYG and associated projection regions. For example, RSD increased the amount of c-fos-positive neurons in the bed nucleus of the stria terminalis (BNST), lateral septum (LS), hypothalamus, hippocampus, and frontal cortex (Wohleb et al, 2011). Importantly, increased c-fos expression occurred after a single cycle of social defeat that precedes any neuroinflammatory or anxiety-like behavior that is associated with 6 days of RSD. It is therefore likely that activation of fear/anxiety circuitry is a critical early mediator that initiates subsequent neuroinflammatory processes. In support of this, pharmacological treatment with common anti-anxiety (clonazepam) or antidepressants (imipramine) that inhibit neuronal activation prevents RSD-induced neuroinflammation and associated changes to behavior (Ramirez et al, 2015a, b). Therefore, neuronal activation is likely upstream of the events discussed below.
In addition to activating neuronal fear/anxiety circuitry, neuroinflammatory signaling is increased by RSD (Kinsey et al, 2007; Wohleb et al, 2012) that is argued to facilitate vulnerability toward mood disorders (Haroon et al, 2012). Microglia are the predominate innate immune cell in the brain parenchyma, and are a key cell population that propagates neuroinflammatory signaling in the CNS. Microglia serve many functions including immunosurveillance for inflammatory molecules, pathogens, cellular debris, apoptotic cells, and alterations in neuronal phenotype (Ransohoff and Cardona, 2010). Any disturbance or ‘danger’-associated change in brain homeostasis evokes rapid changes to microglia morphology, gene expression, and production of inflammatory signaling molecules (Gehrmann et al, 1995; Aloisi, 2001; Bianchi, 2007; Kettenmann et al, 2011). Microglia are sensitive to the physiological response of stress as they express β-adrenergic and GC receptors (Kettenmann et al, 2011). There is a growing literature indicating that both stress-induced NE and GCs activate and/or prime microglia to future immune challenges. This has been demonstrated particularly in stress-reactive brain regions like the hypothalamus, pituitary, and hippocampus (Blandino et al, 2006; Frank et al, 2012; Johnson et al, 2013). Blocking either the β-adrenergic (Wohleb et al, 2011, 2013; Hanke et al, 2012) or GC receptor (Frank et al, 2012) during stress prevents stress-induced microglial activation/priming. Direct priming effects of adrenergic signaling (Johnson et al, 2013) or GC signaling (Frank et al, 2012) have been attributed to microglia. Therefore, microglia are sensitive to homeostatic changes evoked during the stress response.
Numerous studies have revealed microglial activation and enhanced neuroinflammatory signaling in a variety of stress models (O’Connor et al, 2003b; Nair and Bonneau, 2006; Frank et al, 2007; Kopp et al, 2013; Iwata et al, 2015). Moreover, recent studies indicate that microglial activation and neuroinflammatory signaling occurs particularly within stress-reactive brain regions including the frontal cortex, hypothalamus, amygdala, and hippocampus (Wohleb et al, 2011; Hinwood et al, 2012). RSD is associated with an increased neuroinflammatory profile characterized by increased cytokines (IL-1β, IL-6, TNF-α) and the chemotactic CCL2, and reduced anti-inflammatory regulation of neuronal-derived fractalkine ligand (CX3CL1) and receptor (CX3CR1) that are expressed on microglia (Wohleb et al, 2013, 2014b; Ramirez et al, 2015a). A hallmark of activated microglia is a change in morphology that includes repressed processes and enlarged soma, giving them an ‘amoeboid’ appearance (Kettenmann et al, 2011). A similar morphological profile occurs after RSD in stress-reactive regions (Wohleb et al, 2011, 2013). In addition, key surface adhesion molecules are upregulated to increase the sensitivity of microglia, including CD14, TLRs, and CD86 (Wohleb et al, 2011). Functionally, microglia are ‘primed’ to later activation, as they produce an exaggerated proinflammatory response to subsequent immunological challenges (Wohleb et al, 2012). This microglia profile has also been demonstrated in other stress models including tail shock (Johnson et al, 2003; Frank et al, 2007) and chronic unpredictable stress (Nair and Bonneau, 2006; Tynan et al, 2010; Bian et al, 2012). Therefore, microglia are activated in a variety of stress models, and is argued to contribute to the maladaptive effects of stress.
Although microglial activation, and the resulting increase in neuroinflammatory mediators, regionally correlates with neuronal activation, it does not imply that microglial activation is a causative link to produce the behavioral sequelae associated with chronic stress. In support of this notion, microglia activation was inhibited by pharmacological treatment with minocycline in a variety of stress models. In each case, inhibiting microglia during stress exposure attenuated the increase of proinflammatory cytokines normally produced by stress (Hinwood et al, 2012, 2013; Kreisel et al, 2014; McKim et al, 2016). Furthermore, minocycline protected against the development of stress-associated deficits in cognitive memory tasks in Morris water maze and Barnes maze (McKim et al, 2016) and depressive-like and anxiety-like behaviors such as reduced social interaction, sucrose preference, and open field exploration (Hinwood et al, 2012; Kreisel et al, 2014; Liu et al, 2015). In addition, stimulating stress-exposed microglia with an immune challenge reduces social interaction and open field exploration (Wohleb et al, 2012; Frank et al, 2015a) that is prevented by minocycline treatment (Chijiwa et al, 2015). Therefore, microglia are a key cell population that responds to stress exposure by increasing neuroinflammatory signaling to influence the behavioral response of stress. However, recent evidence across several laboratories indicates that microglia are not the sole immune cell in the CNS that influences the neuroinflammatory and behavioral response of stress.
RSD-induced primed and GC-resistant monocytes traffic to the brain to elevate proinflammatory signaling that is associated with changes to behavior
Monocytes function to mediate host antimicrobial defense (Serbina et al, 2008) and do so by differentiating and trafficking to site-specific tissue where they assist with microbial clearance. The ability of monocytes to mobilize and traffic is a critical and seemingly complex process that is necessary for promoting immune defense during infection or injury. In the past, the CNS was considered to be devoid of peripheral immune cells because of the tight regulation of BBB. However, it has now been well demonstrated in models of severe trauma or advanced disease states that BM-derived monocytes traffic to and influence inflammatory signaling in the brain (Zhou et al, 2009; Grozdanov et al, 2014). Typically, monocyte trafficking into the CNS can be attributed to an opening of the BBB that occurs during severe trauma or pathology, and suggests a more passive recruitment (Stamatovic et al, 2008). However, recent evidence indicates that stress-induced circulating monocytes are actively recruited to the brain in the absence of a leaky BBB.
The first evidence that chronic stress promotes trafficking of BM-derived monocytes was by Brevet et al (2010). In this study, host mice were irradiated and transplanted with cells from the BM of GFP+ donor mice. At 1 month after the transplant, mice were exposed to 1 h of intermittent footshock for 5 consecutive days. After day 5 of footshock, there was a significant increase of GFP+ cells (ie, BM-derived cells) in the hippocampus (Brevet et al, 2010). Furthermore, GFP+ cells were also found in mice that were placed adjacent to mice receiving footshock (Ataka et al, 2013). That is to say, merely being placed in an environment in which other mice received footshocks was sufficient to traffic BM-derived immune cells to the brain. It is important to note that irradiation-induced BM transplants compromises BBB permeability (Diserbo et al, 2002), and hence GFP+ cells in the parenchyma may be an artifact of a leaky BBB. This potential confound led to the development of BM ablation using low doses of busulfan that is far less toxic than irradiation-induced BM ablation (Kierdorf et al, 2013).
Under the context of RSD, Wohleb et al (2013) used a low-dose busulfan protocol that partially ablated the BM. Although BBB permeability was unaffected as indicated by the Evan’s blue dye test, exposure to RSD increased the presence GFP+ cells specifically in brain regions associated with fear, anxiety, and threat appraisal, including the hypothalamus, amygdala, and hippocampus (Wohleb et al, 2013). Notably, GFP+ monocytes accumulated in the same threat appraisal areas that had increased c-Fos and IBA-1 activation after RSD, but not in other brain regions like the motor cortex or cerebellum, suggesting site-specific recruitment to stress-responsive brain regions (Wohleb et al, 2011, 2013). These findings are corroborated by flow cytometry assessment of microglia/macrophage cell populations in the absence of busulfan. Here, RSD increased the presence of CD11b+/CD45high monocyte population in CNS (Wohleb et al, 2011). In addition, these cells highly expressed Ly6C, similar to the expression pattern of circulating monocytes induced by RSD (Powell et al, 2013). It is important to note that the increase of CD11b+/CD45high persisted despite vascular perfusion, and hence the CD11b+/CD45high monocyte/macrophages adhered to the vasculature or entered the parenchyma (Wohleb et al, 2012).
With RSD, temporal relationships exist between the development and resolution of immune cell trafficking and neuroinflammatory effects of RSD. For example, accumulation of Ly6Chi monocytes in circulation begins after 3 cycles and peaks after 6 cycles of social defeat, indicating that repeated neuroendocrine activation is required to generate this response (Wohleb et al, 2013). Furthermore, peripherally derived Ly6Chi monocytes selectively traffic to CNS threat appraisal centers including the frontal cortex, hypothalamus, amygdala, and hippocampus after 3 cycles and continue to accumulate after 6 cycles. Peripheral monocytes/macrophages highly express IL-1β and retain high expression levels after entering the CNS parenchyma (Ataka et al, 2013). Indeed, proinflammatory gene expression, including IL-1β, is increased after 3 cycles and peaks after 6 cycles in areas of monocyte trafficking that temporally correlates with expression of anxiety-like behavior (Wohleb et al, 2013). Similarly, the resolution between brain monocyte trafficking, neuroinflammation, and anxiety-like behavior are correlated. Peripherally derived monocytes, elevated proinflammatory gene expression, and anxiety-like behavior persist for at least 8 days after the last cycle of social defeat but are no longer apparent after 24 days (Wohleb et al, 2014a), suggesting that these parameters are temporally connected. Taken together, trafficked monocytes/macrophages represent a second immune cell population that influences immune processes/behaviors in the brain.
Support for this view comes from studies in which blocking monocyte trafficking to the brain attenuates stress-induced proinflammatory cytokine response, and related changes to behavior (Wohleb et al, 2011, 2013, 2014b; Ramirez et al, 2015a, b). For example, the CCL2–CCR2 and CX3CL1–CX3CR1 chemotactic axis are two ligand–receptor signaling pathways that recruit monocytes to sites of inflammation (Auffray et al, 2007; Tsou et al, 2007), including the brain (D’Mello et al, 2009). This notion is particularly appreciated in varying CNS disease/injury models associated with monocyte trafficking, including EAE (Saederup et al, 2010), intracerebral hemorrhage (Hammond et al, 2014), prion disease (Gomez-Nicola et al, 2014), and spinal cord injury (Donnelly et al, 2011). Under the context of RSD, genetic knockout of CCR2 or CX3CR1 prevented monocyte recruitment to the brain and blocked development of anxiety-like behaviors (Wohleb et al, 2013). However, RSD still caused the release of Ly6Chi monocytes into circulation, independent of CCR2 or CX3CR1. This suggests that the release of monocytes into circulation is not sufficient for monocytes to traffic to the brain. Instead, signaling from the CNS must occur to recruit monocytes to the brain. Under this view, circulating monocytes do not passively diffuse to the brain, but instead are actively recruited via communication from the CNS.
Reactive endothelium actively recruits peripheral monocytes to the CNS
Monocyte recruitment to the CNS involves dynamic interactions with vascular endothelial cells that comprise the BBB. Leukocyte recruitment to the CNS is a multistep process that is regulated through expression of adherent surface molecules on leukocytes and endothelial cells (Ransohoff et al, 2003). Typically, the first step involves rolling and capture of leukocytes onto the surface of endothelial cells. This occurs via enhanced expression of the capture molecules, L-selectin and P-selectin glycoprotein ligand 1 (PSGL1) on leukocytes and E- and P-selectin on the luminal surface of endothelial cells. Engagement between leukocytes and endothelial cells allows for crosstalk that assists leukocyte trafficking. Here, chemokine activation of leukocytes initiates relocation and conformational changes to the integrins, lymphocyte function-associated antigen 1 (LFA-1) and very late antigen 4 (VLA4), that form high-affinity binding sites for firm adhesion onto endothelial cells (Kim et al, 2003). The CCL2–CCR2 chemotactic axis, in particular, is implicated in mediating leukocyte infiltration into the CNS under a variety of conditions including HIV encephalitis (Eugenin et al, 2006), ischemia (Dimitrijevic et al, 2006), prion disease (Gomez-Nicola et al, 2014), and stress (Wohleb et al, 2013). Next, dynamic interactions between integrins and the cell adhesion molecules (CAMs), intercellular CAM (ICAM) and vascular CAM (VCAM), arrest the leukocyte onto the endothelial cell surface. Once the leukocyte has arrested on the endothelial surface, it may then polarize and migrate through tightly regulated integrin/CAM-mediated events (eg, VCAM/ICAM) to enter into the perivascular space (Greenwood et al, 2011). Therefore, if exposure to stressors, such as RSD, were to increase expression of adhesion molecules, this would promote trafficking of circulating immune cells to the CNS.
Indeed, RSD increased expression of E-selectin selectively in stress-reactive brain regions where monocyte trafficking occurs (Sawicki et al, 2015). Furthermore, RSD increased ICAM/VCAM protein expression on BBB endothelial cells in similar regions. This increase of adhesion molecules was dependent on the number of social defeat cycles the animal was exposed to. Both ICAM and VCAM were increased after 6 days of social defeat that temporally correlated with peripheral monocyte trafficking (Sawicki et al, 2015). Importantly, endothelial cells are responsive to proinflammatory cytokine and chemokine signaling. For example, activation of the endothelium by IL-1β and TNF-α increases the expression of E-selectin and P-selectin and ICAM/VCAM (O’Carroll et al, 2015). As discussed above, stress exposure activates microglia and increases proinflammatory cytokine (IL-1β, TNF-α, IL-6) and chemokine (CCL2) signaling (Wohleb et al, 2012), thereby providing a direct pathway to upregulate adhesion molecules.
Once macrophages traffic into the vasculature, the astrocytic foot comprises an additional barrier that prevents monocytes from entering the parenchyma. Macrophages express degrading enzymes like matrix metalloproteinases (MMPs) that break down the astrocytic barrier that exacerbates infiltration from the vasculature into the parenchyma (Bechmann et al, 2007). This notion is predominately characterized under gross pathological conditions such as cerebral ischemia/reperfusion (Asahi et al, 2000) and neurodegenerative disorders including multiple sclerosis (Avolio et al, 2003), Alzheimer’s disease (Duits et al, 2015), and Parkinson’s disease (He et al, 2013) where peripherally derived immune cells contribute to pathology. It is known within the cancer literature that stress levels of NE increase MMP-9 and MMP-2 that increase invasive potential of the tumor cells (Sood et al, 2006; Yang et al, 2006). Furthermore, MMP upregulation and invasiveness of the tumor cells were reversed by propranolol treatment, highlighting the link between stress hormones and MMPs (Sood et al, 2006). Therefore, infiltrating monocytes may upregulate MMPs in response to increased adrenergic signaling to facilitate diapedisis and entry into the brain parenchyma. In support of this, genome-wide transcriptional profiling indicates that splenetic monocytes upregulate MMP 8/9/13/14 in response to RSD (Powell et al, 2013). However, further studies are needed to determine whether MMP-mediated mechanisms regulate monocyte entrance into the brain parenchyma.
In addition to the role of BBB endothelial cells to ‘catch’ and recruit peripheral immune cells into the brain, an interesting concept is that the BBB relays inflammatory signaling from circulation into the CNS. For instance, BBB endothelial cells express key cytokine receptors (IL-1R1, IL-6, TNF receptor), and binding of these receptors leads to transport or secretion of additional proinflammatory cytokines and secondary mediators (prostaglandins) that propagate inflammatory signaling into the CNS (Bebo and Linthicum, 1995; Ericsson et al, 1995; Vallieres and Rivest, 1999). Following activation by cytokines, resident microglia further secretes proinflammatory cytokines to directly influence neuronal pathways and behavior (Quan and Banks, 2007). Evidence for this type of communication comes from a recent study that targets endothelial cell-dependent cytokine signaling. In this study, transgenic mice were created to genetically knock out the IL-1 receptor specifically on endothelial cells (eIL-1R1KD) (Li et al, 2011). These mice developed RSD-induced accumulation of primed monocytes in circulation that traffic to the brain. However, eIL-1R1KD reduced proinflammatory cytokine expression in microglia isolated from mice exposed to RSD and prevented RSD-induced anxiety-like behavior. That is to say, primed monocytes were still able to traffic to the brain, but eliminating endothelial IL-1 signaling reduced proinflammatory signaling from microglia and prevented anxiety-like behavior. These results indicate that dynamic interactions between peripheral monocytes, endothelial cells, and microglia converge to promote a proinflammatory environment in the CNS that affects behavior (Figure 3). Certainly, many of the effects observed in RSD should be replicated in other stress models.

Reactive endothelium facilitates stress-induced monocyte trafficking to the CNS. Exposure to stress upregulates adhesion molecules that assist in the capture and extravasation of peripherally derived monocytes into perivascular and parenchymal monocytes. Rolling/capture occurs via PSGL-1/P-selectin and L-selectin/E-selectin interactions. Activated microglia release cytokines (IL-1β) and chemokines (CCL2) that promote monocyte arrest via ICAM/VCAM-mediated events. Accumulation of peripherally derived monocytes in the CNS converge with resident microglia to increase neuroinflammatory signaling (ie, IL-1β). In addition, endothelial cells secrete cytokines and prostaglandins that propagate neuroinflammation. Excess release of IL-1β is implicated in the prolonged behavioral response of stress.
Long-term consequences of RSD on neuroinflammation and behavior
An important clinical component of stress research is the length that stress-induced behavior persists. Within the RSD model, anxiety-like behavior in common behavioral tasks like the open field, light/dark preference, and elevated plus maze occurs immediately after the last cycle of social defeat and persists for at least 8 days, but is no longer apparent after 24 days (Wohleb et al, 2014a). This pattern temporally correlates with neuroinflammatory signaling and the presence of peripherally derived monocytes in the brain, as elevated cytokines and monocytes are detected at 8 days but not 24 days after RSD. However, the organism is now altered in such a way that subsequent exposure to a subthreshold stressor reengages the inflammatory and behavioral processes discussed above, a phenomenon referred to as ‘stress-sensitization’ (Wohleb et al, 2014a). For example, a single cycle of social defeat is considered to be subthreshold because it does not traffic monocytes to the brain, activates microglia, or induces prolonged anxiety-like behavior. However, reexposure to a single cycle of social defeat 24 days after RSD reactivated the neuroimmune axis, leading to increased monocyte trafficking to the brain and elevated proinflammatory cytokine signaling (Wohleb et al, 2014a; McKim et al, 2015). Coinciding with reactivation of neuroimmune processes, exposure to subthreshold stress reestablished anxiety-like behavior and reinforced social avoidance.
Interestingly, trafficking of peripheral monocytes to the brain was not primarily driven by monocyte production and egress from the BM, such as what occurs following initial exposure to RSD. Instead, reexposure to subthreshold stress caused the release of primed monocytes from a splenic monocyte reservoir (Wohleb et al, 2014a; McKim et al, 2015). Although not fully understood, a current area of exploration is that initial exposure to RSD causes egress of primed monocyte progenitor cells from the BM that is housed in the spleen. Upon exposure to subsequent stress, splenetic monocyte progenitor cells differentiate into primed monocytes that enter circulation and traffic to the brain to influence neuroinflammatory processes and behavior. In support of this view, the number of splenetic monocytes in RSD-sensitized mice (24 days after RSD) before stress reexposure are similar to those in naive mice. However, splenetic monocytes are significantly increased following reexposure to subthreshold stress, suggesting that monocytes are newly differentiated from progenitor cells. A more direct role for the spleen comes from a study in which control and RSD-sensitized mice were splenectomized before subthreshold stress exposure. Stress-sensitized mice with their spleen removed were protected against the effects of stress reexposure to traffic monocytes to the brain and reestablish anxiety-like behaviors (Wohleb et al, 2014a). Support for the role of spleen-derived monocytes to influence the inflammatory state of resident microglia comes from the finding that splenectomy attenuated microglial cytokine expression (Wohleb et al, 2014a; McKim et al, 2015). Thus, reestablished anxiety-like behavior, monocyte trafficking to the brain, and microglia cytokine expression were all dependent on an intact spleen.
Similar to the egress of monocytes from the BM after RSD, differentiation and release of splenic monocytes following stress reexposure is likely mediated by noradrenergic signaling via circulating or direct sympathetic nervous system innervation of the spleen (McKim et al, 2015). For example, guanethidine is a peripheral sympathetic inhibitor that prevents release of NE. Guanethidine intervention before stress reexposure prevented monocyte egress from the spleen and trafficking to the brain. This blockade corresponded with reduced microglial cytokine expression and prevented anxiety-like behavior. Because guanethidine does not cross the BBB, the protective effects of guanethidine derive from its ability to prevent monocyte trafficking to the brain. Furthermore, a similar protective effect was observed with pretreatment of propranolol, a β-adrenergic receptor antagonist, before stress reexposure. Therefore, sympathetic activation of NE is an early mediator in stress-sensitized spleen-to-brain monocyte trafficking.
Human research on chronic stress and inflammation, and translational implications of stress-induced primed and GC-resistant monocytes
An important clinical component of rodent research is how well a particular phenomenon translates to the human population. There is a long history of human research that acknowledges the impact of stress on immune function. Although many historical studies demonstrate that stress, and the resulting endocrine changes, suppresses multiple aspects of immune function (Khansari et al, 1990; Kiecolt-glaser et al, 1991, 1995), more recent advances increasingly show that stress is not globally immunosuppressive. A meta-analysis of human studies found that the severity and chronicity of stressors played an important role in the outcome of several immunological parameters (Segerstrom and Miller, 2004). Important to this review, a similar GC-resistant and primed monocyte phenotype is reported in human populations subject to chronic stress. A direct human analog was provided by Miller et al (2008). These investigators performed genome-wide expression microarrays on peripheral blood monocytes from familial caregivers of brain cancer patients vs matched control subjects. Importantly, familial caregivers are at increased risk of developing depressive or anxiety symptoms (Cannuscio et al, 2002). Similar to the RSD rodent model, circulating monocytes from caregivers showed diminished GR-mediated transcription and heightened NF-κB-mediated transcription that was associated with an approximately twofold increase of circulating C-reactive protein (CRP). This cannot be attributed to dysregulated GC rhythmicity, as caregivers showed higher cortisol than control subjects 4 h after waking, but did not differ any other time of day, indicating that the diurnal secretion of GCs was not affected (Miller et al, 2008). CD14+/CD16− monocytes in humans are the functional counterpart to immature/inflammatory Ly6Chi monocytes in mice (Yang et al, 2014). Further investigation revealed that the primary source of upregulated inflammatory genes among caretakers was derived from an increased CD14+/CD16− monocyte population (Powell et al, 2013; Miller et al, 2014). Similar to Ly6Chi monocytes in mice, CD14+/CD16− monocytes produced higher levels of IL-6 when stimulated with LPS ex vivo (Miller et al, 2014), suggesting that similar immune process occur in humans. This led to a direct comparison between peripheral mononuclear cells from humans of low socioeconomic status, a risk factor for depression (Lorant et al, 2003), and monocytes from RSD-exposed mice. Although humans from low socioeconomic status differ from mice exposed to RSD in many respects, there is substantial overlap between the inflammatory gene expression profiles resulting from stress exposure between the two species. Approximately one-third of total gene expression differences observed in low socioeconomic status humans were also observed in RSD mice, including CD163, MMP9, and TNF to highlight a few (Powell et al, 2013). Identifying CD14+/CD16− monocytes as the primary source of elevated inflammatory gene expression and IL-6 production implicates this cell type as particularly sensitive to chronic stress, and may provide a measurable biomarker to identify vulnerable human subpopulations. Children might be particularly vulnerable to the ability of chronic stress to reprogram innate immune function, as immune changes can persist throughout the majority of their lifetime. For example, circulating monocytes from healthy adult subjects (25–40 years old) raised in unfavorable socioeconomic circumstances had decreased transcriptional activity of GR and increased transcriptional activity of NF-κB. Furthermore, the GR-insensitive monocytes were primed to immune activation, as they produced elevated IL-6 following immune stimulation ex vivo (Miller et al, 2009). These data indicate that hematopoiesis of inflammatory and GC-resistant monocytes similarly occurs in chronically stressed humans.
Crosstalk between the peripheral immune system and the brain can have a detrimental influence on mental and physical health. Miller and Cole (2012) published a recent study in which they interviewed 147 healthy young women every 6 months for 2.5 years who were high risk for depression because of family history or cognitive vulnerability. At the same time, blood was collected to assess systemic inflammation. Childhood adversity promoted clustering of depressive episodes along with elevated circulating IL-6 and CRP. Furthermore, high circulating IL-6 predicted depressive episodes 6 months later (Miller and Cole, 2012). Similar predictive susceptibility was ascribed to IL-6 from mice before being exposed to a different model of RSD. Before stress, leukocytes were isolated from blood and stimulated with LPS ex vivo. The mice with leukocytes that had the largest IL-6 response to LPS produced the most IL-6 after stress and had the largest stress-induced behavioral deficits (social avoidance) (Hodes et al, 2014). Interestingly, transplanting the BM from a ‘susceptible’ mouse to a ‘control’ mouse promoted susceptibility. That is to say, the control mouse had an immune and behavioral phenotype similar to the ‘susceptible’ donor mouse. In opposition, transplantation from an IL-6-deficient mouse, or administration of IL-6 neutralizing antibodies, promoted stress resilience in host mice. As such, IL-6 may be a useful biomarker and potential therapeutic target, particularly in treatment-resistant depressive patients (Hodes et al, 2014).
Despite increasing evidence linking neuroinflammation and mood disorders, little is known about CNS macrophages in mood disorder pathology. The first study examining this issue was by Torres-Platas et al (2014). The dorsal anterior cingulate cortex (dACC) is repeatedly implicated in mood disorders and the behavioral response to inflammation (Miller et al, 2013). These investigators observed high densities of macrophages and increased gene expression of CCL2 along blood vessels in the dACC of depressed suicide victims compared with controls who died suddenly in the absence of psychiatric, neurological, or inflammatory illness (Torres-Platas et al, 2014). This is the first evidence supporting the existence of peripheral macrophages in the central inflammatory response of human depressed patients. Certainly, more studies are need before conclusions can be made regarding the role of leukocyte recruitment to the CNS in chronically stressed and/or depressed individuals.
Future research directions: stress-induced danger signals propagate neuroinflammation
The research highlighted above illustrates the dual process of GC-resistant/primed monocytes and microglia/endothelial cells in the CNS to converge and promote a heightened neuroinflammatory environment that directly influences the behavioral response to stress. Under this context, stress-reactive proinflammatory signaling molecules that activate microglia, promote trafficking of peripheral monocytes, and propagate neuroinflammation are of significant interest to understand the biological mechanisms underlying stress-induced neuroinflammation. As already noted, inhibiting microglia blunts the stress-induced increase in proinflammatory signaling molecules and subsequent changes to behavior, an effect that is secondary to neuronal activation. However, the mechanisms that drive neuron-to-microglia communication are not completely understood and are a topic of active investigation by many research groups.
Recently, ‘danger signals’ have emerged as an intriguing mediator in a variety of neuroinflammatory conditions, including stress. Danger signals are endogenous signaling molecules that are released in response to ‘danger’ and activate immune processes to help the organism respond to the adverse event, even in the absence of foreign pathogens (Matzinger, 2002). Immunologically speaking, danger is a broad term that refers to the capacity of an event to induce tissue stress or damage (Pradeu and Cooper, 2012). This concept originally references peripheral immune processes, such as sterile injury, but similar processes have been observed in the CNS, including ischemia (Liesz et al, 2015), and recently, psychological stress. The current hypothesis is that the brain responds to a stressor as ‘danger,’ thereby releasing danger signals to prepare innate immune processes to respond to the adverse event. Here we will explore two danger signals that have recently been implicated in neuroinflammatory effects of stress. It should be noted that each signal is not mutually exclusive and, in fact, likely works in conjunction to promote a heightened neuroinflammatory environment (Figure 4).

Stress-induced danger signals propagate neuroinflammation. Danger signals are released in stress-responsive brain regions to activate resident microglia. HMGB1 signals through Toll-like receptors to translocate NF-κB to the nucleus that drives transcription of pro-IL-1β and NLRP3. ATP binds to P2X7 and initiates K+ efflux from the cell. In turn, NLRP3 forms a multiprotein complex with ASC and pro-caspase-1 termed the inflammasome. Pro-caspase-1 is cleaved to the bioactive caspase-1 that cleaves pro-IL-1β to mature IL-1β. Released IL-1β interacts with IL-1 receptors located on (1) endothelial cells to induce a reactive endothelium that facilities recruitment of monocytes and (2) neurons to promote the behavioral response of stress.
ATP as a Danger Signal that Stimulates IL-1β
ATP is an activity-dependent signaling molecule that can be released by neurons, astrocytes, and microglia. Accordingly, ATP is ascribed multiple roles, including immune modulator via neuron-to-glia and glia-to-glia communication. The link between ATP and stress-induced neuroinflammation comes from the ability of ATP to regulate IL-1β synthesis and release. As mentioned above, IL-1β is critical for the neuroinflammatory and behavioral response to RSD (Wohleb et al, 2011, 2014b). In a seminal study published in 2002, Martinon et al (2002) found that IL-1β is tightly regulated and processed differently than other cytokines. IL-1β is transcribed into the precursor protein pro- IL-1β and requires enzymatic cleavage by caspase-1 into the bioactive and mature IL-1β. Caspase-1 is also regulated in a similar manner and requires cleavage from pro-caspase-1. This occurs via the formation of a multimeric protein complex termed the inflammasome. The primary component of the inflammasome is the nucleotide-binding oligomerization receptor (Nod-like receptor (NLR)). Critically, two separate signals are needed to release IL-1β; the first to drive NLR and pro-IL-1β expression, and the second to form the inflammasome complex and cleave pro-IL-1β to IL-1β. Assembly and activation of the inflammasome, and the resulting synthesis and release of IL-1β, is a current focus of stress-related neuroinflammation and depressive-like behaviors (for more information see Pan et al, 2014; Frank et al, 2015a). Importantly, inflammasome formation is driven by ligand-sensing leucine-rich repeat (LRR) domain located on the NLR. The LRR domain is sensitive to a diverse array of danger signals, including ATP (Leemans et al, 2011). Under this context, ATP is an activating signal to the NLRP3 inflammasome. In support of this, direct treatment of ATP on primed microglia is sufficient for maturation and release of IL-1β in vitro (Halle et al, 2008) and does so via the P2X7 receptor (Colomar et al, 2003) expressed on microglia (Monif et al, 2009).
Recent studies have begun to explore ATP as a stress-induced danger signal that modulates microglia activation and IL-1β maturation/secretion. For example, ATP is rapidly released in the hippocampus within the first 30 min of immobilization stress, preceding the release of IL-1β and subsequently TNF-α (Iwata et al, 2015). The release of IL-1β was prevented by pharmacological blockage of P2X7 with the receptor specific antagonist A-804598, supporting an earlier finding that A-804598 reduces footshock-induced IL-1β gene expression (Catanzaro et al, 2014). Furthermore, daily treatment with A-804598 during 28 days of chronic unpredictable stress prevented behavioral deficits in sucrose preference, forced swim, and elevated plus maze, suggesting that ATP is necessary for the associated behavioral consequences of stress. The cellular source of stress-induced ATP likely involves glutamate-mediated neuron-to-astrocyte communication as glutamate was rapidly released before ATP. Astrocytes reside in close proximity to neuronal synapses and express glutamate receptors that regulate CA2+ influx (Cornell-Bell et al, 1990) to influence ATP signaling (Cotrina et al, 2000). In support of this, in vitro treatment of glutamate stimulates the release of ATP on cultured astrocytes (Iwata et al, 2015). Thus, stress-induced ATP provides a direct neuron-to-astrocyte-to-microglia communication pathway that enhances IL-1β signaling and affects anxiety-like and depressive-like behaviors.
HMGB-1 as a Danger Signal that Stimulates IL-1β
HMGB-1 is a constitutively expressed protein in the nucleus of all cell types, including neurons, astrocytes, and microglia. Past research conducted on peripheral immune cells demonstrates that HMGB-1 is a potent proinflammatory signaling molecule that is released under inflammatory conditions, including sepsis and ischemia (Yang and Tracey, 2009). Recently, HMGB-1 was found to be highly expressed in the CNS and was implicated in a variety of neuroinflammatory conditions (Kang et al, 2014), including stress (Weber et al, 2015). Indeed, direct administration of HMGB-1 into the CNS activates NF-κB-dependent transcription, primes the NLRP3 inflammasome, and increases proinflammatory cytokines, including IL-1β (O'Connor et al, 2003a; Yang et al, 2005; Xiang et al, 2011; Frank et al, 2015a). Furthermore, this potentiated neuroinflammatory response had behavioral consequences, as HMGB-1 was sufficient to decrease social exploration toward a conspecific juvenile following an LPS injection (Frank et al, 2015a). Therefore, if HMGB-1 is released in the CNS in response to stress, it would result in an enhanced proinflammatory environment.
This idea is supported by recent studies using tail shock as a stressor. For example, HMGB-1 protein is increased in the hippocampus following a series of repeated tail shock and persists for at least 24 h later (Weber et al, 2015). Microglia are likely a primary source of stress-induced HMGB-1, as microglia isolated from rats exposed to tail shock actively secreted HMGB-1 in the absence of cell death. Receptor targets for HMGB-1 include TLR2 and TLR4 to elicit a proinflammatory response, and the receptor for advanced glycation end products (RAGE) to initiate cell migration and proliferation. Pharmacological antagonism of the receptor targets for HMGB-1 blocked the effect of stress to prime microglia (Weber et al, 2013, 2015). These studies suggest that stress-induced HMGB-1 signals in an autocrine/paracrine manner to activate microglia and propagate proinflammatory signaling in the CNS (for more information see Frank et al, 2015b). Importantly, signaling at TLR2 and TLR4 initiates an NF-κB-dependent signaling cascade that transcribes pro-IL-1β and NLRP3 (Hanamsagar et al, 2012). Indeed, HMGB-1 increased NLRP3 mRNA and protein (Xiang et al, 2011; Frank et al, 2015a) and acts as a priming stimulus for the NLRP3 inflammasome (Frank et al, 2015a). Conceptually, HMGB-1 primes the NLRP3 inflammasome and exposure to an activating signal, such as ATP, would form the inflammasome complex and cleave pro-IL-β into mature IL-1β, thereby releasing IL-1β. It should be noted that HMGB-1 is highly expressed in neurons and astrocytes. Typically, neuronal release of HMGB-1 occurs during necrotic conditions, such as ischemia (Yang and Tracey, 2009) and sepsis (Yang et al, 2004). Repeated stress has been linked with neuronal atrophy in stress-reactive areas including the hippocampus (McEwen, 1999), and hence neurons may also be a significant source of HMGB-1. Taken together, stress-induced HMGB-1 represents a molecular mechanism that modulates microglia activation to promote stress-induced neuroinflammation.
Summary
Exposure to repeated or chronic stressors activates bidirectional communication pathways between the CNS and peripheral innate immune system. This includes sympathetic-mediated accumulation of GC-resistant and inflammatory Ly6Chi monocytes in circulation. In conjunction, CNS-mediated neuroinflammatory events promote trafficking of peripherally derived monocytes to the CNS via interactions with BBB endothelial cells that converge with resident microglia to promote a heightened neuroinflammatory environment that affects behavior. We identified potential biomarkers that can be measured in plasma (IL-6 and CD14+/CD16− monocytes) to identify stress-susceptible human populations following trauma. Furthermore, we discussed current research investigating chemotactic signaling pathways (CCL2–CCR2 and CX3CL1–CX3CR1) that recruit monocytes and specific neuron–glia communication pathways that mediate stress-induced immune processes. This includes investigating endogenous danger signals that respond to stress as ‘danger’ to activate resident microglia and recruit monocytes to the CNS. These findings help elucidate the complex communication that occurs in response to stress and highlights novel therapeutic targets to protect against the neuroinflammatory responses that contribute to long-lasting behavioral complications.
Funding and disclosure
The authors declare no conflict of interest.
References
Aloisi F (2001). Immune function of microglia. Glia 36: 165–179.
Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH (2000). Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab 20: 1681–1689.
Ataka K, Asakawa A, Nagaishi K, Kaimoto K, Sawada A, Hayakawa Y et al (2013). Bone marrow-derived microglia infiltrate into the paraventricular nucleus of chronic psychological stress-loaded mice. Plos One 8: e81744.
Audoy-Remus J, Richard JF, Soulet D, Zhou H, Kubes P, Vallieres L (2008). Rod-shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J Neurosci 28: 10187–10199.
Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S et al (2007). Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317: 666–670.
Avitsur R, Kinsey SG, Bidor K, Bailey MT, Padgett DA, Sheridan JF (2007). Subordinate social status modulates the vulnerability to the immunological effects of social stress. Psychoneuroendocrinology 32: 1097–1105.
Avitsur R, Padgett DA, Dhabhar FS, Stark JL, Kramer KA, Engler H et al (2003). Expression of glucocorticoid resistance following social stress requires a second signal. J Leukoc Biol 74: 507–513.
Avitsur R, Stark JL, Dhabhar FS, Padgett DA, Sheridan JF (2002). Social disruption-induced glucocorticoid resistance: kinetics and site specificity. J Neuroimmunol 124: 54–61.
Avolio C, Ruggieri M, Giuliani F, Liuzzi GM, Leante R, Riccio P et al (2003). Serum MMP-2 and MMP-9 are elevated in different multiple sclerosis subtypes. J Neuroimmunol 136: 46–53.
Bailey MT, Engler H, Powell ND, Padgett DA, Sheridan JF (2007). Repeated social defeat increases the bactericidal activity of splenic macrophages through a Toll-like receptor-dependent pathway. Am J Physiol Regul Integrd Comp Physiol 293: R1180–R1190.
Bauer ME, Perks P, Lightman SL, Shanks N (2001). Restraint stress is associated with changes in glucocorticoid immunoregulation. Physiol Behav 73: 525–532.
Bebo BF Jr, Linthicum DS (1995). Expression of mRNA for 55-kDa and 75-kDa tumor necrosis factor (TNF) receptors in mouse cerebrovascular endothelium: effects of interleukin-1 beta, interferon-gamma and TNF-alpha on cultured cells. J Neuroimmunol 62: 161–167.
Bechmann I, Galea I, Perry VH (2007). What is the blood-brain barrier (not)? Trends Immunol 28: 5–11.
Bian YQ, Pan Z, Hou ZY, Huang C, Li W, Zhao BH (2012). Learning, memory, and glial cell changes following recovery from chronic unpredictable stress. Brain Res Bull 88: 471–476.
Bianchi ME (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81: 1–5.
Blandino P Jr, Barnum CJ, Deak T (2006). The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress. J Neuroimmunol 173: 87–95.
Brevet M, Kojima H, Asakawa A, Atsuchi K, Ushikai M, Ataka K et al (2010). Chronic foot-shock stress potentiates the influx of bone marrow-derived microglia into hippocampus. J Neurosci Res 88: 1890–1897. First evidence that stress exposure causes trafficking of peripehral monocytes into the brain parenchyma.
Calhoon GG, Tye KM (2015). Resolving the neural circuits of anxiety. Nat Neurosci 18: 1394–1404.
Cannuscio CC, Jones C, Kawachi I, Colditz GA, Berkman L, Rimm E (2002). Reverberations of family illness: a longitudinal assessment of informal caregiving and mental health status in the Nurses’ Health Study. Am J Public Health 92: 1305–1311.
Catanzaro JM, Hueston CM, Deak MM, Deak T (2014). The impact of the P2X7 receptor antagonist A-804598 on neuroimmune and behavioral consequences of stress. Behav Pharmacol 25: 582–598.
Chijiwa T, Oka T, Lkhagvasuren B, Yoshihara K, Sudo N (2015). Prior chronic stress induces persistent polyI:C-induced allodynia and depressive-like behavior in rats: possible involvement of glucocorticoids and microglia. Physiol Behav 147: 264–273.
Colomar A, Marty V, Medina C, Combe C, Parnet P, Amedee T (2003). Maturation and release of interleukin-1beta by lipopolysaccharide-primed mouse Schwann cells require the stimulation of P2X7 receptors. J Biol Chem 278: 30732–30740.
Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ (1990). Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247: 470–473.
Cotrina ML, Lin JH, Lopez-Garcia JC, Naus CC, Nedergaard M (2000). ATP-mediated glia signaling. J Neurosci 20: 2835–2844.
Coutinho AE, Chapman KE (2011). The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 335: 2–13.
D’Mello C, Le T, Swain MG (2009). Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factor alpha signaling during peripheral organ inflammation. J Neurosci 29: 2089–2102.
Dantzer R, Bluthe RM, Gheusi G, Cremona S, Laye S, Parnet P et al (1998). Molecular basis of sickness behavior. Ann NY Acad Sci 856: 132–138.
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9: 46–57.
Davies TH, Ning YM, Sanchez ER (2002). A new first step in activation of steroid receptors - Hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem 277: 4597–4600.
Dhabhar FS, Malarkey WB, Neri E, McEwen BS (2012). Stress-induced redistribution of immune cells—from barracks to boulevards to battlefields: a tale of three hormones—Curt Richter Award winner. Psychoneuroendocrinology 37: 1345–1368.
Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV (2006). Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab 26: 797–810.
Diserbo M, Agin A, Lamproglou I, Mauris J, Staali F, Multon E et al (2002). Blood-brain barrier permeability after gamma whole-body irradiation: an in vivo microdialysis study. Can J Physiol Pharmacol 80: 670–678.
Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA et al (2011). Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci 31: 9910–9922.
Duits FH, Hernandez-Guillamon M, Montaner J, Goos JD, Montanola A, Wattjes MP et al (2015). Matrix metalloproteinases in Alzheimer’s disease and concurrent cerebral microbleeds. J Alzheimers Dis 48: 711–720.
Eash KJ, Means JM, White DW, Link DC (2009). CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood 113: 4711–4719.
Engler H, Bailey MT, Engler A, Sheridan JF (2004). Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. J Neuroimmunol 148: 106–115.
Ericsson A, Liu C, Hart RP, Sawchenko PE (1995). Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol 361: 681–698.
Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW (2006). CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: A potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci 26: 1098–1106.
Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S (1985). Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol 135 (2 Suppl): 755s–765s.
Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF (2007). Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun 21: 47–59.
Frank MG, Thompson BM, Watkins LR, Maier SF (2012). Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav Immun 26: 337–345.
Frank MG, Weber MD, Fonken LK, Hershman SA, Watkins LR, Maier SF (2015a). The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: a role for the NLRP3 inflammasome. Brain Behav Immun 55: 215–224.
Frank MG, Weber MD, Watkins LR, Maier SF (2015b). Stress sounds the alarmin: the role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav Immun 48: 1–7.
Galigniana NM, Ballmer LT, Toneatto J, Erlejman AG, Lagadari M, Galigniana MD (2012). Regulation of the glucocorticoid response to stress-related disorders by the Hsp90-binding immunophilin FKBP51. J Neurochem 122: 4–18.
Gehrmann J, Matsumoto Y, Kreutzberg GW (1995). Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20: 269–287.
Gilman SE, Trinh NH, Smoller JW, Fava M, Murphy JM, Breslau J (2013). Psychosocial stressors and the prognosis of major depression: a test of Axis IV. Psychol Med 43: 303–316.
Gomez-Nicola D, Schetters ST, Perry VH (2014). Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia 62: 1041–1052.
Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, Engelhardt B (2011). Leucocyte-endothelial cell crosstalk at the blood-brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathol Appl Neurobiol 37: 24–39.
Grozdanov V, Bliederhaeuser C, Ruf WP, Roth V, Fundel-Clemens K, Zondler L et al (2014). Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol 128: 651–663.
Haegeman G, De Bosscher K, Vanden Berghe W (2003). The interplay between the glucocorticoid receptor and nuclear factor-kappa B or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24: 488–522.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T et al (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9: 857–865.
Hamilton JA, Achuthan A (2013). Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol 34: 81–89.
Hammond MD, Taylor RA, Mullen MT, Ai YX, Aguila HL, Mack M et al (2014). CCR2(+)Ly6C(hi) inflammatory monocyte recruitment exacerbates acute disability following intracerebral hemorrhage. J Neurosci 34: 3901–3909.
Hanamsagar R, Hanke ML, Kielian T (2012). Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol 33: 333–342.
Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF (2012). Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun 26: 1150–1159.
Haroon E, Raison CL, Miller AH (2012). Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 37: 137–162.
He X, Zhang L, Yao X, Hu J, Yu L, Jia H et al (2013). Association studies of MMP-9 in Parkinson’s disease and amyotrophic lateral sclerosis. PLoS One 8: e73777.
Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A et al (2014). Chronic variable stress activates hematopoietic stem cells. Nat Med 20: 754–758.
Hinwood M, Morandini J, Day TA, Walker FR (2012). Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex 22: 1442–1454.
Hinwood M, Tynan RJ, Charnley JL, Beynon SB, Day TA, Walker FR (2013). Chronic stress induced remodeling of the prefrontal cortex: structural re-organization of microglia and the inhibitory effect of minocycline. Cereb Cortex 23: 1784–1797.
Hodes GE, Kana V, Menard C, Merad M, Russo SJ (2015). Neuroimmune mechanisms of depression. Nat Neurosci 18: 1386–1393. Elegant article implicating circulating IL-6 as a biomarker and therapeutic target against treatment-resistant depressed patient.
Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D et al (2014). Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci USA 111: 16136–16141.
Hunzeker J, Padgett DA, Sheridan PA, Dhabhar FS, Sheridan JF (2004). Modulation of natural killer cell activity by restraint stress during an influenza A/PR8 infection in mice. Brain Behav Immun 18: 526–535.
Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S et al (2015). Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol Psychiatry 80: 12–22. Clear evidence that stress-induced ATP can stimulate IL-1β release to influence behavior and does so via the NLRP3 inflammasome.
Jin J, Wang X, Wang Q, Guo X, Cao J, Zhang X et al (2013). Chronic psychological stress induces the accumulation of myeloid-derived suppressor cells in mice. PLoS One 8: e74497.
Johnson JD, O’Connor KA, Hansen MK, Watkins LR, Maier SF (2003). Effects of prior stress on LPS-induced cytokine and sickness responses. Am J Physiol Regul Integr Comp Physiol 284: R422–R432.
Johnson JD, Zimomra ZR, Stewart LT (2013). Beta-adrenergic receptor activation primes microglia cytokine production. J Neuroimmunol 254: 161–164.
Jung SH, Wang YF, Kim T, Tarr A, Reader B, Powell N et al (2015). Molecular mechanisms of repeated social defeat-induced glucocorticoid resistance: role of microRNA. Brain Behav Immun 44: 195–206.
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L et al (2014). HMGB1 in health and disease. Mol Aspects Med 40: 1–116.
Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA et al (2006). Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124: 407–421.
Kessler RC (1997). The effects of stressful life events on depression. Annu Rev Psychol 48: 191–214.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011). Physiology of microglia. Physiol Rev 91: 461–553.
Khansari DN, Murgo AJ, Faith RE (1990). Effects of stress on the immune-system. Immunol Today 11: 170–175.
Kiecolt-glaser JK, Marucha PT, Malarkey WB, Mercado AM, Glaser R (1995). Slowing of wound-healing by psychological stress. Lancet 346: 1194–1196.
Kiecolt-glaser JK, Speicher CE, Glaser R, Dura JR, Trask OJ (1991). Spousal caregivers of dementia victims - longitudinal changes in immunity and health. Psychosom Med 53: 345–362.
Kierdorf K, Katzmarski N, Haas CA, Prinz M (2013). Bone marrow cell recruitment to the brain in the absence of irradiation or parabiosis bias. PLoS One 8: e58544.
Kim M, Carman CV, Springer TA (2003). Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301: 1720–1725.
Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R (2007). Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun 21: 458–466.
Kopp BL, Wick D, Herman JP (2013). Differential effects of homotypic vs. heterotypic chronic stress regimens on microglial activation in the prefrontal cortex. Physiol Behav 122: 246–252.
Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV et al (2014). Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry 19: 699–709.
LeDoux J (2003). The emotional brain, fear, and the amygdala. Cell Mol Neurobiol 23: 727–738.
Leemans JC, Cassel SL, Sutterwala FS (2011). Sensing damage by the NLRP3 inflammasome. Immunol Rev 243: 152–162.
Levine J, Barak Y, Chengappa KN, Rapoport A, Rebey M, Barak V (1999). Cerebrospinal cytokine levels in patients with acute depression. Neuropsychobiology 40: 171–176.
Li QM, Powell N, Zhang H, Belevych N, Ching S, Chen Q et al (2011). Endothelial IL-1R1 is a critical mediator of EAE pathogenesis. Brain Behav Immun 25: 160–167.
Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W et al (2015). DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci 35: 583–598.
Liu YN, Peng YL, Liu L, Wu TY, Zhang Y, Lian YJ et al (2015). TNFalpha mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur Cytokine Netw 26: 15–25.
Lorant V, Deliege D, Eaton W, Robert A, Philippot P, Ansseau M (2003). Socioeconomic inequalities in depression: a meta-analysis. Am J Epidemiol 157: 98–112.
Maier SF, Watkins LR (1998). Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol Rev 105: 83–107. Clear overview explaining how cytokines affect behavior, mood, and cognition.
Maier SF, Watkins LR (2003). Immune-to-central nervous system communication and its role in modulating pain and cognition: Implications for cancer and cancer treatment. Brain Behav Immun 17: S125–S131.
Martinon F, Burns K, Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10: 417–426. A seminal article that identified the inflammasome complex as the mediator of caspase-1 activation.
Matzinger P (2002). The danger model: a renewed sense of self. Science 296: 301–305.
McEwen BS (1999). Stress and hippocampal plasticity. Annu Rev Neurosci 22: 105–122.
McKim DB, Niraula A, Tarr A, Wohleb ES, Sheridan J, Godbout JP (2016). Neuroinflammatory dynamics underlie memory impairments following repeated social defeat. J Neurosci 36: 2590–2604.
McKim DB, Patterson JM, Wohleb ES, Jarrett BL, Reader BF, Godbout JP et al (2015). Sympathetic release of splenic monocytes promotes recurring anxiety following repeated social defeat. Biol Psychiatry 79: 803–813.
Miller AH, Haroon E, Raison CL, Felger JC (2013). Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety 30: 297–306.
Miller AH, Raison CL (2015). The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16: 22–34.
Miller GE, Chen E, Fok AK, Walker H, Lim A, Nicholls EF et al (2009). Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci USA 106: 14716–14721.
Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R et al (2008). A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry 64: 266–272.
Miller GE, Cole SW (2012). Clustering of depression and inflammation in adolescents previously exposed to childhood adversity. Biol Psychiatry 72: 34–40. Childhood adversity promotes clustering of depressive episodes along with elevated circulating IL-6 and CRP and high circulating IL-6 predicted depressive episodes 6 months later.
Miller GE, Murphy ML, Cashman R, Ma R, Ma J, Arevalo JM et al (2014). Greater inflammatory activity and blunted glucocorticoid signaling in monocytes of chronically stressed caregivers. Brain Behav Immun 41: 191–199.
Monif M, Reid CA, Powell KL, Smart ML, Williams DA (2009). The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci 29: 3781–3791.
Monteiro S, Roque S, de Sa-Calcada D, Sousa N, Correia-Neves M, Cerqueira JJ (2015). An efficient chronic unpredictable stress protocol to induce stress-related responses in C57BL/6 mice. Front Psychiatry 6: 6.
Nair A, Bonneau RH (2006). Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol 171: 72–85.
Nie YC, Han YC, Zou YR (2008). CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med 205: 777–783.
O’Carroll SJ, Kho DT, Wiltshire R, Nelson V, Rotimi O, Johnson R et al (2015). Pro-inflammatory TNFalpha and IL-1beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J Neuroinflammation 12: 131.
O’Connor KA, Hansen MK, Pugh CR, Deak MM, Biedenkapp JC, Milligan ED et al (2003a). Further characterization of high mobility group box 1 (HMGB1) as a proinflammatory cytokine: central nervous system effects. Cytokine 24: 254–265.
O’Connor KA, Johnson JD, Hansen MK, Wieseler Frank JL, Maksimova E, Watkins LR et al (2003b). Peripheral and central proinflammatory cytokine response to a severe acute stressor. Brain Res 991: 123–132.
Pan Y, Chen XY, Zhang QY, Kong LD (2014). Microglial NLRP3 inflammasome activation mediates IL-1beta-related inflammation in prefrontal cortex of depressive rats. Brain Behav Immun 41: 90–100.
Powell ND, Bailey MT, Mays JW, Stiner-Jones LM, Hanke ML, Padgett DA et al (2009). Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun 23: 225–231.
Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E et al (2013). Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA 110: 16574–16579. Direct comparison of inflammatory phenotype between monocytes from chronically stressed humans and mice after RSD.
Pradeu T, Cooper EL (2012). The danger theory: 20 years later. Front Immunol 3: 287.
Quan N, Avitsur R, Stark JL, He LL, Lai WM, Dhabhar F et al (2003). Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. J Neuroimmunol 137: 51–58.
Quan N, Banks WA (2007). Brain-immune communication pathways. Brain Behav Immun 21: 727–735.
Raison CL, Capuron L, Miller AH (2006). Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol 27: 24–31.
Ramirez K, Niraula A, Sheridan JF (2015a). GABAergic modulation with classical benzodiazepines prevent stress-induced neuro-immune dysregulation and behavioral alterations. Brain Behav Immun 51: 154–168.
Ramirez K, Shea DT, McKim DB, Reader BF, Sheridan JF (2015b). Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain Behav Immun 46: 212–220.
Ransohoff RM, Cardona AE (2010). The myeloid cells of the central nervous system parenchyma. Nature 468: 253–262.
Ransohoff RM, Kivisakk P, Kidd G (2003). Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol 3: 569–581.
Reader BF, Jarrett BL, McKim DB, Wohleb ES, Godbout JP, Sheridan JF (2015). Peripheral and central effects of repeated social defeat stress: monocyte trafficking, microglial activation, and anxiety. Neuroscience 289: 429–442.
Rose S, Misharin A, Perlman H (2012). A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytometry A 81A: 343–350.
Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL et al (2010). Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5: e13693.
Sawicki CM, McKim DB, Wohleb ES, Jarrett BL, Reader BF, Norden DM et al (2015). Social defeat promotes a reactive endothelium in a brain region-dependent manner with increased expression of key adhesion molecules, selectins and chemokines associated with the recruitment of myeloid cells to the brain. Neuroscience 302: 151–164. Adhesion molecules that recruit leukocytes are increased in brain vasculature after social defeat in an exposure- and brain region-dependent manner.
Segerstrom SC, Miller GE (2004). Psychological stress and the human immune system: a meta-analytic study of 30 years of inquiry. Psychol Bull 130: 601–630.
Serbina NV, Jia T, Hohl TM, Pamere EG (2008). Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol 26: 421–452.
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G et al (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72: 268–275.
Smith RS (1991). The macrophage theory of depression. Med Hypotheses 35: 298–306.
Sood AK, Bhatty R, Kamat AA, Landen CN, Han L, Thaker PH et al (2006). Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res 12: 369–375.
Stamatovic SM, Keep RF, Andjelkovic AV (2008). Brain endothelial cell-cell junctions: how to ‘Open’ the blood brain barrier. Curr Neuropharmacol 6: 179–192.
Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF (2001). Social stress induces glucocorticoid resistance in macrophages. Am J Physiol Regul Integr Comp Physiol 280: R1799–R1805.
Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N (2014). Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun 42: 50–59. The first clinical evidence that peripheral monocytes traffic to the brain in depressed patients.
Tseng RJ, Padgett DA, Dhabhar FS, Engler H, Sheridan JF (2005). Stress-induced modulation of NK activity during influenza viral infection: role of glucocorticoids and opioids. Brain Behav Immun 19: 153–164.
Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP et al (2007). Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117: 902–909.
Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV et al (2010). Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun 24: 1058–1068.
Ulrich-Lai YM, Herman JP (2009). Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 10: 397–409.
Vallieres L, Rivest S (1999). Interleukin-6 is a needed proinflammatory cytokine in the prolonged neural activity and transcriptional activation of corticotropin-releasing factor during endotoxemia. Endocrinology 140: 3890–3903.
Weber MD, Frank MG, Sobesky JL, Watkins LR, Maier SF (2013). Blocking toll-like receptor 2 and 4 signaling during a stressor prevents stress-induced priming of neuroinflammatory responses to a subsequent immune challenge. Brain Behav Immun. 32: 112–121.
Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF (2015). Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley Rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci 35: 316–324. First evidence that HMGB-1 primes the NLRP3 inflammasome in the CNS after stress exposure.
Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP (2012). Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 37: 1491–1505.
Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT et al (2011). beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci 31: 6277–6288.
Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF et al (2014a). Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry 75: 970–981. Stress-induced redistribution of myeloid cells to the spleen leads to long-term immune and behavioral sensitization to subthreshold stressors.
Wohleb ES, Patterson JM, Sharma V, Quan N, Godbout JP, Sheridan JF (2014b). Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J Neurosci 34: 2583–2591.
Wohleb ES, Powell ND, Godbout JP, Sheridan JF (2013). Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 33: 13820–13833. Clear evidence that RSD-induced monocytes are recruited to the CNS to promote anxiety-like behavior.
Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G et al (2011). Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol 187: 4809–4817.
Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB et al (2006). Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res 66: 10357–10364.
Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE et al (2004). Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA 101: 296–301.
Yang H, Tracey KJ (2009). Targeting HMGB1 in inflammation. Biochim Biophys Acta 1799: 149–156.
Yang H, Wang H, Czura CJ, Tracey KJ (2005). The cytokine activity of HMGB1. J Leukoc Biol 78: 1–8.
Yang J, Zhang L, Yu C, Yang XF, Wang H (2014). Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2: 1.
Zhou H, Andonegui G, Wong CHY, Kubes P (2009). Role of endothelial TLR4 for neutrophil recruitment into central nervous system microvessels in systemic inflammation. J Immunol 183: 5244–5250.
Acknowledgements
This work was supported by NIMH grant (R01-MH093473 and R01-MH097243) to John F Sheridan. Jonathan P Godbout was supported by NIA (R0-1AG033028). Michael D Weber was supported by NIDCR Training Grant T32-DE14320.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Weber, M., Godbout, J. & Sheridan, J. Repeated Social Defeat, Neuroinflammation, and Behavior: Monocytes Carry the Signal. Neuropsychopharmacol 42, 46–61 (2017). https://doi.org/10.1038/npp.2016.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.102
This article is cited by
-
Distinguishing features of depression in dementia from primary psychiatric disease
Discover Mental Health (2024)
-
SKA2 regulated hyperactive secretory autophagy drives neuroinflammation-induced neurodegeneration
Nature Communications (2024)
-
CCL2-mediated inflammatory pathogenesis underlies high myopia-related anxiety
Cell Discovery (2023)
-
Microglia depletion facilitates the display of maternal behavior and alters activation of the maternal brain network in nulliparous female rats
Neuropsychopharmacology (2023)
-
Oleoylethanolamide restores stress-induced prepulse inhibition deficits and modulates inflammatory signaling in a sex-dependent manner
Psychopharmacology (2023)
