
Abstract
Fear expression is mediated by an activation of the centromedial amygdala (CEm), the major output nucleus of the amygdaloid complex. Consistently, fear extinction is associated with an increased synaptic inhibition as well as a suppression of the excitability of the CEm neurons. However, little is known about the role of CEm glutamatergic synapses in fear regulation and anxiety-like behaviors. The BDNF Val66Met, a single-nucleotide polymorphism in the human BDNF gene, impairs fear extinction and leads to anxiety-like symptoms. To determine whether the BDNF Val66Met polymorphism affects the CEm excitatory synapses, we examined basal glutamatergic synaptic transmission and plasticity in the CEm neurons of BDNF Val66Met knock-in (BDNFMet/Met) mice. The BDNF Val66Met single-nucleotide polymorphism exerted an opposite effect on non-NMDA and NMDA receptor transmission with a potentiation of the former and a suppression of the latter. In addition, the decay time of NMDA currents was decreased in BDNFMet/Met mice, suggesting a modification of NMDA receptor subunit composition. Unlike the wild-type mice that exhibited a potentiation of non-NMDA receptor transmission following fear conditioning and a depotentiation upon fear extinction, BDNFMet/Met mice failed to show this experience-dependent synaptic plasticity in the CEm neurons. Our results suggest that the elevated non-NMDA receptor transmission, the suppression of NMDA receptor transmission, and an impairment of synaptic plasticity in the CEm neurons might contribute to the fear extinction deficit and increased anxiety-like symptoms in BDNF Val66Met carriers.
Similar content being viewed by others


Introduction
Several factors including developmental stage, environment, and genetics influence fear extinction, which is a form of inhibitory learning believed to regulate anxiety-like behaviors. This modulation of fear extinction might involve changes in synaptic function in the medial prefrontal cortex (mPFC), hippocampus, and amygdala, the three major components of the fear circuit (Galvin and Ninan, 2014; Izquierdo et al, 2006; Koppensteiner et al, 2014; Pattwell et al, 2012a; Pattwell et al, 2012b). Among the few known genetic variations that have a profound effect on anxiety-like symptoms, the BDNF Val66Met single-nucleotide polymorphism (SNP), which leads to a valine to methionine substitution in the BDNF protein, affects fear extinction in both humans and rodents (Chen et al, 2006; Hartley and Casey, 2013; Montag et al, 2010; Pattwell et al, 2012a; Soliman et al, 2010). Consistent with the effect of the Val66Met SNP on neuronal development and regulated release of BDNF (Chen et al, 2006; Egan et al, 2003), a knock-in mice carrying the BDNF Val66Met SNP (BDNFMet/Met) showed an impairment of synaptic plasticity in the mPFC and hippocampus, which might interfere with the top-down control of the amygdala, a mechanism necessary for fear regulation (Bath et al, 2012; Ninan, 2014; Ninan et al, 2010; Pattwell et al, 2012a). However, given the role of BDNF in amygdala plasticity (Rattiner et al, 2005; Rosas-Vidal et al, 2014; Roth et al, 2014), it is also plausible that the BDNF Val66Met SNP exerts a direct effect on amygdala synapses. Human BDNF Val66Met carriers continued to recruit the amygdala despite fear extinction training, suggesting an altered amygdala plasticity (Soliman et al, 2010). Furthermore, BDNF Val66Met carriers exhibit an elevated amygdala activity in response to emotional stimuli (Montag et al, 2008). Heightened amygdala activity in BDNF Val66Met carriers could contribute to attentional bias for threat stimuli (Carlson et al, 2014).
Recent studies suggest that the central amygdala plays a key part in fear regulation (Duvarci et al, 2011; Li et al, 2013; Wilensky et al, 2006). The central amygdala comprises two major divisions: lateral (CEl) and medial (CEm) nuclei (Ehrlich et al, 2009). CEm, the output nucleus, projects to periaqueductal gray and regulates the freezing behavior associated with fear response (LeDoux et al, 1988). Congruent with an increased CEm activity in fear expression (Duvarci et al, 2011), an enhanced GABAergic inhibition and a suppressed excitability of CEm neurons were observed after fear extinction (Amano et al, 2010). Apart from the GABAergic modulation, the CEm receives glutamatergic input from the basolateral amygdala (BLA) (Pape and Pare, 2010; Pare et al, 1995). However, the role of CEm glutamatergic synapses in fear regulation is largely unknown. Given the effect of the BDNF Val66Met on fear extinction (Pattwell et al, 2012a; Soliman et al, 2010), we asked whether the BDNF Val66Met affects glutamatergic synapses in the CEm. We found an alteration of both NMDA and non-NMDA receptor transmission and a suppression of synaptic plasticity in the CEm neurons of BDNFMet/Met mice, which might have a role in impaired fear extinction in BDNF Val66Met carriers.
Materials and Methods
Animals
Male 2- to 5-month-old BDNFMet/Met and BDNFVal/Val mice were derived by crossing BDNFMet/+ mice with the C57BL/6 background (Chen et al, 2006). Mice (C57BL/6) used for experiments described in Figure 4 were purchased from the Jackson Laboratory. Animals were maintained on a 12 : 12 light–dark cycle at 23 oC with ad libitum access to food and water. Mice were handled for 3 days before the behavioral experiments. All procedures were approved by the Institutional Animal Care and Use Committee of the New York University School of Medicine.
Electrophysiology
Mice were intracardially perfused with carbogenated ice-cold artificial cerebrospinal fluid containing (in mM) NaCl (118), glucose (10), KCl (2.5), NaH2PO4 (1), CaCl2 (1), and MgSO4 (2) (325 mOsm, pH 7.4) for 60 s under pentobarbital anesthesia. Brains were removed, and 300 μm coronal slices containing the amygdala were prepared on a vibratome. Slices were allowed to recover for at least 1 h before being transferred to a recording chamber maintained at 32 °C and perfused with the aforementioned artificial cerebrospinal fluid containing 2 mM CaCl2. CEm neurons were visualized using an Olympus BX50WI upright microscope. Miniature excitatory postsynaptic currents (mEPSCs) and spontaneous excitatory postsynaptic currents (sEPSCs) were recorded as previously described (Galvin and Ninan, 2014; Pattwell et al, 2012a). NMDA mEPSCs were recorded at −50 mV in the presence of tetrodotoxin (1 μM), bicuculline (10 μM), NBQX (10 μM), glycine (10 μM), and strychnine (1 μM) in the absence of magnesium and analyzed with a detection threshold of 5 pA. Excitatory postsynaptic currents (EPSCs) were recorded at −60 mV in the presence of bicuculline by stimulating the BLA using an extracellular electrode (Figure 1b). NMDA EPSCs were recorded at +40 mV in the presence of bicuculline and NBQX. The non-NMDA/NMDA ratio was calculated by dividing the peak EPSC at −60 mV by the NMDA receptor current measured at 50 ms after the peak at 40 mV. The composition of the electrode solution was (in mM): CsCl (130), HEPES (10), EGTA (0.5), QX-314 (5), GTP (0.2), and MgATP (5) (osmolarity 290–300 mOsm, pH 7.4). Input resistance was monitored throughout the experiment. Data were acquired using Axopatch 200 A amplifier (Molecular Devices). Clampfit (Molecular Devices) and Mini Analysis programs were used for data analysis.

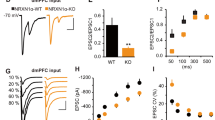
Non-NMDA receptor transmission is enhanced in the CEm neurons of BDNFMet/Met mice. (a) mEPSC frequency and amplitude in BDNFVal/Val (10 neurons/5 mice) and BDNFMet/Met (12 neurons/5 mice) groups. Upper panel shows examples of mEPSCs. Lower right panel shows event histogram for mEPSC amplitude. (b) EPSC amplitude in BDNFVal/Val (12 neurons/5 mice) and BDNFMet/Met (10 neurons/6 mice) groups. Left panel shows schematic presentation of the positions of stimulating and recording electrodes in the amygdala slice preparation. Middle panel shows examples of EPSCs evoked by 150 μA stimulation. (c) Paired pulse ratio in BDNFVal/Val (6 neurons/3 mice) and BDNFMet/Met (7 neurons/3 mice) groups. Left panel shows examples of EPSCs evoked at an interval of 60 ms. Asterisk denotes a statistically significant difference. BA, basal amygdala; CEl, latero-capsular subdivision of central amygdala; CEm, medial subdivision of central amygdala; LA, lateral amygdala.
Behavior
A fear conditioning apparatus consisting of a shock chamber placed in a sound-attenuated box (Coulbourn Instruments) was used. On day 1, after a 2-min acclimation period and two 30-s habituation tones at an interval of 30 s, mice were fear-conditioned with three tone-shock pairings, each one comprised 30-s tones (5 kHz, 50 dB) co-terminating with a 0.5-s foot shock (0.7 mA) with an inter-trial interval of 30 s. Mice were removed from the conditioning chamber 1 min after the last tone-shock pairing. Extinction training was carried out 24 h later (day 2) by exposing the mice to 30 presentations of 30-s tones at an interval of 30 s on a nonshock floor with a distinct design and olfactory cue from the shock floor. Freezing was tested 24 h later (day 3) on the nonshock floor by three presentations of 30-s tones at an interval of 30 s. The fear-conditioned group underwent fear conditioning on day 1, remained in the home cage on day 2, and was tested for fear memory on day 3. The control tone-alone group was presented with 5 tones without shock on day 1, remained in the home cage on day 2, and was tested on day 3. Brain slices for electrophysiology experiments were prepared 2–4 h after the fear memory test on day 3. Data were acquired and analyzed using the Freeze frame system.
Statistics
Data are presented as Mean±SEM. mEPSCs were compared using t-test. sEPSCs and freezing were compared using one-way ANOVA followed by least significant difference. EPSCs were compared using two-way repeated measures ANOVA followed by Bonferroni’s test. Greenhouse–Geisser correction was applied in case of violation of sphericity. p<0.05 was considered statistically significant.
Results
Increased non-NMDA Receptor Transmission in the CEm Neurons of BDNFMet/Met Mice
To assess whether the BDNF Val66Met polymorphism affects basal glutamatergic transmission in the CEm neurons, we compared the frequency and amplitude of mEPSCs in 3- to 5-month-old BDNFVal/Val and BDNFMet/Met mice. Although we did not observe any difference in the frequency of mEPSCs (t(20)=−0.18, p=0.85), the BDNFMet/Met group exhibited a statistically significant increase in mEPSC amplitude compared with the BDNFVal/Val group (t(20)=3.32, p=0.003) (Figure 1a). To confirm this increase in non-NMDA receptor transmission in the BDNFMet/Met group, we examined the amplitude of evoked EPSCs in the CEm neurons by an extracellular stimulation of the BLA. The BDNFMet/Met group showed a significantly higher EPSC amplitude compared with the BDNFVal/Val group, as revealed by a main effect of the group (F(1,20)=6.95, p=0.016), stimulus intensity (F(1.2,24.4)=22.9, p<0.001), and a nonsignificant interaction between factors (F(1.2,24.4)=3.89, p=0.053) (Figure 1b). However, the paired pulse ratio was not affected, as shown by no effect of the group (F(1,11)=0.039, p=0.847) and interaction between factors (F(1,11)=0.34, p=0.573) (Figure 1c), suggesting that the increased non-NMDA receptor transmission is unlikely to be mediated by enhanced glutamate release.
Decreased NMDA Receptor Transmission in the CEm Neurons of BDNFMet/Met Mice
To determine whether the BDNF Val66Met polymorphism affects NMDA receptor transmission in the CEm neurons, first we examined NMDA mEPSC frequency, amplitude, and decay time. We observed a significant decrease in frequency (t(17)=3.17, p=0.005) and decay time (t(17)=3.8, p=0.001) of NMDA mEPSCs in the BDNFMet/Met group compared with the BDNFVal/Val group without affecting the amplitude (t(17)=0.37, p=0.71) (Figure 2a). These results suggested that the BDNF Val66Met polymorphism causes a decrease in the number of glutamatergic synapses with functional NMDA receptors and an alteration of NMDA receptor subunit composition indicated by a decrease in the decay time (Cull-Candy et al, 2001).

NMDA receptor transmission is decreased in the CEm neurons of BDNFMet/Met mice. (a) Frequency, amplitude, and decay time of NMDA mEPSCs in BDNFVal/Val (9 neurons/3 mice) and BDNFMet/Met (10 neurons/4 mice) groups. Upper panel shows examples of NMDA mEPSCs. Lower right panel shows event histogram for mEPSC amplitude. NMDA EPSC amplitude (b) and decay time (c) in BDNFVal/Val (7 neurons/3 mice) and BDNFMet/Met (8 neurons/3 mice) groups. Left panel shows examples of EPSCs evoked by 150 μA stimulation. (d) Paired pulse ratio of NMDA EPSCs in BDNFVal/Val (10 neurons/4 mice) and BDNFMet/Met (9 neurons/3 mice) groups. Asterisk denotes a statistically significant difference.
To confirm the effect of the BDNF Val66Met SNP on NMDA receptor transmission, we studied the amplitude and decay time of NMDA EPSCs in the CEm neurons of BDNFMet/Met and BDNFVal/Val mice. The NMDA EPSC amplitude was decreased in the BDNFMet/Met group compared with the BDNFVal/Val group, as revealed by a significant effect of the group (F(1,13)=12.17, p=0.004), stimulus intensity (F(1.19,15.57)=16.55, p<0.001), and an interaction between factors (F(1.19,15.57)=6.73, p=0.016) (Figure 2b). Consistent with the decrease in NMDA mEPSC decay time, NMDA EPSC decay time was also decreased in the BDNFMet/Met group compared with the BDNFVal/Val group (t(13)=2.53, p=0.025) (Figure 2c). Similar to the paired pulse ratio of non-NMDA EPSCs, we did not observe any difference in paired pulse ratio of NMDA EPSCs (Figure 2d). These results further confirm that the BDNF Val66Met polymorphism impairs NMDA receptor transmission and affects the subunit composition of NMDA receptors.
The modulation of NMDA receptor subunit composition caused by changes in the GluN2B subunit could be tested using a highly selective GluN2B antagonist ifenprodil (Delaney et al, 2013; Galvin and Ninan, 2014; Williams, 1993). Therefore, to test whether the lack of GluN2B-mediated NMDA receptor transmission accounts for the reduced NMDA current decay time in the BDNFMet/Met group, we compared NMDA EPSC decay time and amplitude in BDNFMet/Met and BDNFVal/Val groups before and after perfusion with ifenprodil (5 and 10 μM, 15 min). Despite the notable decrease in decay time following ifenprodil (5 μM) (F(1,12)=51, p<0.001), we did not observe a statistically significant effect of group (F(1,12)=3.73, p=0.08) or interaction between factors (F(1,12)=1.25, p=0.28) (Figure 3b). Similarly, ifenprodil (10 μM) did not show a statistically significant effect of group (F(1,15)=2.91, p=0.109) or interaction between factors (F(1,15)=2.82, p=0.114) (Figure 3d). A comparison of the effect of ifenprodil (5 μM) on NMDA EPSC amplitude revealed a nonsignificant effect of group (F(1,12)=0.56, p=0.47) and interaction between factors (F(1,12)=0.15, p=0.9) (Figure 3c). Ifenprodil (10 μM) showed a nonsignificant effect of group (F(1,15)=0.002, p=0.966) but a significant effect of an interaction between factors (F(1,15)=5.3, p=0.036) (Figure 3e). In spite of the robust decrease in NMDA current decay time in the BDNFMet/Met group suggesting a reduction in GluN2B-containing NMDA receptors, this lack of a statistically significant group-dependent effect of ifenprodil on NMDA currents in BDNFMet/Met and BDNFVal/Val mice suggests the possibility that CEm neurons might express triheteromeric NMDA receptors, as ifenprodil shows less sensitivity for triheteromeric NMDA receptors compared with diheteromeric NMDA receptors (Hatton and Paoletti, 2005). Nevertheless, our results suggest that the BDNF Val66Met SNP alters NMDA receptor transmission, which includes a modification of the receptor subunit composition.

Effect of ifenprodil on NMDA receptor transmission in the CEm neurons of BDNFVal/Val and BDNFMet/Met mice. (a) Examples of NMDA EPSCs before and after perfusion with ifenprodil (5 μM). NMDA EPSC decay time (b) and amplitude (c) before and after ifenprodil (5 μM) perfusion in BDNFVal/Val (7 neurons/3 mice) and BDNFMet/Met (7 neurons/3 mice) groups. NMDA EPSC decay time (d) and amplitude (e) before and after ifenprodil (10 μM) perfusion in BDNFVal/Val (8 neurons/4 mice) and BDNFMet/Met (9 neurons/4 mice) groups. Empty circles represent raw data and filled circles represent mean±SEM.
CEm Synaptic Plasticity Involved in Fear Extinction is Impaired in BDNFMet/Met Mice
Given that the BDNF Val66Met polymorphism alters NMDA receptor transmission, which is critical for synaptic plasticity and fear extinction (Dalton et al, 2012), we tested whether the fear behavior-dependent plasticity in the CEm is affected by the BDNF Val66Met polymorphism. An earlier study showed that both fear conditioning and extinction modulate synaptic transmission and excitability in the CEm neurons (Amano et al, 2010). To establish the nature of plasticity at the glutamatergic synapses in the CEm neurons in response to fear conditioning and extinction, we compared the frequency and amplitude of sEPSCs and the amplitude of EPSCs in control tone-alone, fear-conditioned, and fear-extinguished wild-type mice. One-way ANOVA followed by post-hoc comparison revealed overall significant difference in fear memory on day 3 (F(2,83)=18.28, p<0.001), with the fear-conditioned group showing a significantly higher freezing compared with the tone-alone group (p<0.001) and the fear extinction group showing a significant decrease in freezing compared with the fear-conditioned group (p<0.001) (Figure 4a). In agreement with a recent report (Young and Williams, 2013), we consistently observed a modest freezing in the control tone-alone group. Despite the decrease in both the frequency and amplitude of sEPSCs in the fear-extinguished group compared with the fear-conditioned and tone-alone mice, one-way ANOVA did not reveal an overall significant difference in either frequency (F(2,58)=2.6, p=0.076) or amplitude (F(2,58)=2.9, p=0.058) (Figure 4b). However, comparison of the EPSC amplitude showed an enhanced non-NMDA receptor transmission in the fear-conditioned group compared with the tone-alone group. Furthermore, the fear-extinguished group showed a significant decrease in EPSC amplitude compared with the fear-conditioned group, as shown by a main effect of group (F(2,54)=6.2, p=0.004), significant effect of stimulus intensity (F(1.24,66.8)=18.3, p<0.001), and a nonsignificant interaction between factors (F(2.47,66.8)=1.67, p=0.19) (Figure 4c). Consistent with the potentiation and depotentiation of CEm non-NMDA receptor transmission following fear conditioning and fear extinction, respectively, the non-NMDA/NMDA ratio was significantly higher in the fear-conditioned group compared with the tone-alone and fear extinction groups (F(2,24)=6.4, p=0.006) without an effect on the paired pulse ratio (Figure 4d and e). These results demonstrate a synaptic plasticity mechanism in fear regulation that involves an increase in AMPA receptor transmission in the CEm neurons after fear conditioning and its depotentiation upon fear extinction.

Fear-dependent synaptic plasticity in the CEm neurons. (a) Percentage freezing in tone-alone (TA, 28 mice), fear-conditioned (FC, 31 mice), and fear extinction (FE, 27 mice) groups on days 1 (fear conditioning or tone alone), 2 (fear extinction learning, data points are average of 5 successive recordings), and 3 (fear recall test) of the experiment. (b) Frequency and amplitude of sEPSCs in the CEm neurons of TA (18 neurons/7 mice), FC (22 neurons/10 mice), and FE (21 neurons/9 mice) groups. Upper panel shows examples of sEPSCs. (c) EPSC amplitude in the CEm neurons of TA (15 neurons/7 mice), FC (20 neurons/10 mice), and FE (22 neurons/9 mice) groups. Upper panel shows examples of EPSCs. (d) Paired pulse ratio of non-NMDA EPSCs in TA (17 neurons/6 mice), FC (15 neurons/5 mice), and FE (15 neurons/5 mice) groups. Upper panel shows examples of EPSCs evoked at an interval of 60 ms. (e) Non-NMDA/NMDA ratio in the CEm neurons from TA (9 neurons/3 mice), FC (9 neurons/3 mice), and FE (9 neurons/3 mice) groups. Upper panel shows examples of EPSCs evoked at −60 mV and +40 mV.Asterisk denotes a statistically significant difference.
To test whether the BDNF Val66Met SNP affects the aforementioned CEm synaptic plasticity involved in fear extinction, we studied EPSC amplitude in 2- to 3-month-old BDNFVal/Val and BDNFMet/Met mice after fear conditioning and extinction training. As reported previously (Pattwell et al, 2012a; Soliman et al, 2010), fear extinction memory was reduced in BDNFMet/Met mice (F(2,23)=2.78, p=0.08, p=0.58 for the fear-conditioned group vs fear extinction group) despite significant fear conditioning (p=0.02), whereas BDNFVal/Val mice exhibited significant fear conditioning and extinction (F(2,20)=11.05, p=0.001) (Figure 5a and b). BDNFVal/Val mice exhibited an enhanced EPSC amplitude in the fear-conditioned group compared with the tone-alone group, and the fear-extinguished group showed a depotentiation of EPSC amplitude, as shown by a main effect of group (F(2,39)=29.3, p<0.001), significant effect of stimulus intensity (F(2.05,80.2)=57.3, p<0.001), and a significant interaction between factors (F(4.1,80.2)=18.6, p<0.001) (Figure 5c). However, the BDNFMet/Met group did not show a statistically significant potentiation of EPSC amplitude upon fear conditioning or depotentiation after fear extinction, as shown by a lack of effect of group (F(2,46)=0.99, p=0.38) and a nonsignificant interaction between factors (F(2.34,53.8)=1.6, p=0.21) (Figure 5d). These results suggest that the CEm synaptic plasticity involved in fear extinction is diminished in BDNFMet/Met mice.

CEm synaptic plasticity involved in fear regulation is impaired in BDNFMet/Met mice. (a) Percentage freezing on test day (day 3) in BDNFVal/Val mice (TA=6 mice, FC=10 mice, and FE=7 mice). (b) Percentage freezing on test day (day 3) in BDNFMet/Met mice (TA=6 mice, FC=11 mice, and FE=9 mice). (c) EPSC amplitude in the CEm neurons of TA (15 neurons/6 mice), FC (13 neurons/7 mice), and FE (14 neurons/7 mice) groups of BDNFVal/Val mice. (d) EPSC amplitude in the CEm neurons of TA (13 neurons/6 mice), FC (17 neurons/8 mice), and FE (19 neurons/9 mice) groups of BDNFMet/Met mice. Asterisk denotes a statistically significant difference.
Discussion
Our results suggest that the BDNF Val66Met polymorphism alters glutamatergic synapses in the CEm neurons, resulting in an enhancement and suppression of non-NMDA and NMDA receptor transmission, respectively. Given the lack of effect on the paired pulse ratio, the increase in non-NMDA receptor transmission in BDNFMet/Met mice is unlikely to be mediated by increased glutamate release. Although the lack of change in mEPSC frequency suggests an unaltered number of functional synapses, a postsynaptic structural plasticity that also involves an increase in synaptic AMPA receptors could have a role in the enhanced glutamatergic transmission in BDNFMet/Met mice. Our earlier studies have shown that AMPA receptor-mediated transmission in both the mPFC and hippocampus remained unaffected in BDNFMet/Met mice (Ninan et al, 2010; Pattwell et al, 2012a). The enhanced AMPA receptor transmission and the resulting increase in excitatory drive onto the CEm neurons might contribute to an elevated amygdala output, a potential mechanism for dysregulation of fear behavior. Consistently, BDNF Val66Met carriers exhibit an elevated amygdala activity in response to emotional stimuli, as well as an attentional bias for threat stimuli (Carlson et al, 2014; Montag et al, 2008).
Unlike the enhanced non-NMDA receptor transmission, a diminished NMDA receptor transmission was observed in the CEm neurons of BDNFMet/Met mice. In our earlier studies, we observed a decreased NMDA receptor transmission in the mPFC and hippocampus of BDNFMet/Met mice, suggesting that an altered BDNF signaling owing to the BDNF Val66Met polymorphism impairs NMDA receptor transmission in multiple brain regions (Ninan et al, 2010; Pattwell et al, 2012a). The selective attenuation of NMDA mEPSC frequency in BDNFMet/Met mice suggests a decrease in the number of glutamatergic synapses with functional NMDA receptors. BDNF affects phosphorylation, activity, expression, and trafficking of NMDA receptors (Caldeira et al, 2007; Crozier et al, 2008; Levine et al, 1998; Lin et al, 1998; Suen et al, 1997). Our current findings also show that the BDNF Val66Met polymorphism modifies the subunit composition of NMDA receptors, as indicated by the decrease in decay time of NMDA currents (Cull-Candy et al, 2001). These results are consistent with the recent observation that BDNF-TrkB signaling favors GluN2B-predominant NMDA receptor transmission (Otis et al, 2014). BDNF enhances the phosphorylation of GluN2B tyrosine 1472 by the activation of Fyn, resulting in changes in NMDA receptor activity (Xu et al, 2006). The diminished effect of ifenprodil on NMDA current decay suggests the possibility that the CEm neurons might express triheteromeric NMDA receptors, as ifenprodil shows less sensitivity for triheteromeric NMDA receptors compared with diheteromeric NMDA receptors (Hatton and Paoletti, 2005). Given the role of GluN2B-mediated NMDA receptor transmission in fear extinction, the reduction in GluN2B transmission might be a critical factor in the impairment of fear extinction in BDNFMet/Met mice (Dalton et al, 2012).
The enhanced inhibition of CEm neurons in fear extinction and its dependence on mPFC-mediated activation of GABAergic intercalated cell masses supported the notion that the suppression of CEm output is critical for mediating fear extinction (Amano et al, 2010). Our EPSC data suggest that the suppression of CEm output in fear extinction also involves a depotentiation of non-NMDA receptor transmission in the CEm neurons. The robust modulation of evoked non-NMDA currents compared with spontaneous glutamatergic transmission in response to fear learning and extinction suggests the predominant role of BLA-CEm glutamatergic synapses in fear regulation.
The present data demonstrate not only a hyperactive glutamatergic transmission mediated by AMPA receptors but also an impaired fear-dependent synaptic plasticity in the CEm neurons of BDNFMet/Met mice. The impairment of synaptic plasticity in the CEm might make it less amenable to top-down regulation by the mPFC and hippocampus. In addition, the potentiated glutamatergic drive onto the CEm neurons owing to increased AMPA receptor transmission might alter the amygdala output to the downstream structures such as the brain stem and hence cause an impairment of fear regulation. Furthermore, it is possible that the hyperactive CEm triggers a bottom-up modulation of the mPFC and hippocampus via indirect mechanisms, which might result in a generalization of fear memory in BDNF Val66Met carriers (Hopkins and Holstege, 1978; Muhlberger et al, 2014; Price and Amaral, 1981). Therefore, the BDNF Val66Met-induced dysfunction of BLA-CEm glutamatergic synapses could have a significant role in fear extinction deficit and anxiety-like symptoms. The elevated spontaneous glutamatergic transmission in the CEm neurons of BDNFMet/Met mice could also involve an altered thalamic input, which might have a role in deficits in fear regulation and anxiety-like behaviors in BDNFMet/Met mice (Ciocchi et al, 2010; Samson and Pare, 2005). In addition, an indirect modulation of CEm neurons through an effect on the BLA inputs in the CEl could have a role in anxiety-like behaviors in BDNF Val66Met carriers (Ciocchi et al, 2010). On the basis of our current findings and the previous studies showing the importance of GluN2B-mediated NMDA receptor transmission in fear extinction (Dalton et al, 2012), future studies will be necessary to test whether enhancing GluN2B-mediated NMDA receptor transmission is an effective approach to relieve fear extinction deficit and anxiety symptoms in BDNF Val66Met carriers.
Funding and Disclosure
The authors declare no conflict of interest.
References
Amano T, Unal CT, Pare D (2010). Synaptic correlates of fear extinction in the amygdala. Nat Neurosci 13: 489–494.
Bath KG, Jing DQ, Dincheva I, Neeb CC, Pattwell SS, Chao MV et al (2012). BDNF Val66Met impairs fluoxetine-induced enhancement of adult hippocampus plasticity. Neuropsychopharmacology 37: 1297–1304.
Caldeira MV, Melo CV, Pereira DB, Carvalho RF, Carvalho AL, Duarte CB (2007). BDNF regulates the expression and traffic of NMDA receptors in cultured hippocampal neurons. Mol Cell Neurosci 35: 208–219.
Carlson JM, Cha J, Harmon-Jones E, Mujica-Parodi LR, Hajcak G (2014). Influence of the BDNF genotype on amygdalo-prefrontal white matter microstructure is linked to nonconscious attention bias to threat. Cereb Cortex 24: 2249–2257.
Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ et al (2006). Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314: 140–143.
Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I et al (2010). Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468: 277–282.
Crozier RA, Bi C, Han YR, Plummer MR (2008). BDNF modulation of NMDA receptors is activity dependent. J Neurophysiol 100: 3264–3274.
Cull-Candy S, Brickley S, Farrant M (2001). NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11: 327–335.
Dalton GL, Wu DC, Wang YT, Floresco SB, Phillips AG (2012). NMDA GluN2A and GluN2B receptors play separate roles in the induction of LTP and LTD in the amygdala and in the acquisition and extinction of conditioned fear. Neuropharmacology 62: 797–806.
Delaney AJ, Sedlak PL, Autuori E, Power JM, Sah P (2013). Synaptic NMDA receptors in basolateral amygdala principal neurons are triheteromeric proteins: physiological role of GluN2B subunits. J Neurophysiol 109: 1391–1402.
Duvarci S, Popa D, Pare D (2011). Central amygdala activity during fear conditioning. J Neurosci 31: 289–294.
Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A et al (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112: 257–269.
Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A (2009). Amygdala inhibitory circuits and the control of fear memory. Neuron 62: 757–771.
Galvin C, Ninan I (2014). Regulation of the mouse medial prefrontal cortical synapses by endogenous estradiol. Neuropsychopharmacology 39: 2086–2094.
Hartley CA, Casey BJ (2013). Risk for anxiety and implications for treatment: developmental, environmental, and genetic factors governing fear regulation. Ann NY Acad Sci 1304: 1–13.
Hatton CJ, Paoletti P (2005). Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron 46: 261–274.
Hopkins DA, Holstege G (1978). Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res Exp Hirnforsch 32: 529–547.
Izquierdo A, Wellman CL, Holmes A (2006). Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci 26: 5733–5738.
Koppensteiner P, Aizawa S, Yamada D, Kabuta T, Boehm S, Wada K et al (2014). Age-dependent sensitivity to glucocorticoids in the developing mouse basolateral nucleus of the amygdala. Psychoneuroendocrinology 46: 64–77.
LeDoux JE, Iwata J, Cicchetti P, Reis DJ (1988). Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci 8: 2517–2529.
Levine ES, Crozier RA, Black IB, Plummer MR (1998). Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci USA 95: 10235–10239.
Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, Li B (2013). Experience-dependent modification of a central amygdala fear circuit. Nature Neurosci 16: 332–339.
Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB (1998). BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res 55: 20–27.
Montag C, Basten U, Stelzel C, Fiebach CJ, Reuter M (2010). The BDNF Val66Met polymorphism and anxiety: support for animal knock-in studies from a genetic association study in humans. Psychiatry Res 179: 86–90.
Montag C, Reuter M, Newport B, Elger C, Weber B (2008). The BDNF Val66Met polymorphism affects amygdala activity in response to emotional stimuli: evidence from a genetic imaging study. Neuroimage 42: 1554–1559.
Muhlberger A, Andreatta M, Ewald H, Glotzbach-Schoon E, Troger C, Baumann C et al (2014). The BDNF Val66Met polymorphism modulates the generalization of cued fear responses to a novel context. Neuropsychopharmacology 39: 1187–1195.
Ninan I (2014). Synaptic regulation of affective behaviors; role of BDNF. Neuropharmacology 76: 684–695.
Ninan I, Bath KG, Dagar K, Perez-Castro R, Plummer MR, Lee FS et al (2010). The BDNF Val66Met polymorphism impairs NMDA receptor-dependent synaptic plasticity in the hippocampus. J Neurosci 30: 8866–8870.
Otis JM, Fitzgerald MK, Mueller D (2014). Infralimbic BDNF/TrkB enhancement of GluN2B currents facilitates extinction of a cocaine-conditioned place preference. J Neurosci 34: 6057–6064.
Pape HC, Pare D (2010). Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev 90: 419–463.
Pare D, Smith Y, Pare JF (1995). Intra-amygdaloid projections of the basolateral and basomedial nuclei in the cat: Phaseolus vulgaris-leucoagglutinin anterograde tracing at the light and electron microscopic level. Neuroscience 69: 567–583.
Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I (2012a). The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci 32: 2410–2421.
Pattwell SS, Duhoux S, Hartley CA, Johnson DC, Jing D, Elliott MD et al (2012b). Altered fear learning across development in both mouse and human. Proc Natl Acad Sci USA 109: 16318–16323.
Price JL, Amaral DG (1981). An autoradiographic study of the projections of the central nucleus of the monkey amygdala. J Neurosci 1: 1242–1259.
Rattiner LM, Davis M, Ressler KJ (2005). Brain-derived neurotrophic factor in amygdala-dependent learning. Neuroscientist 11: 323–333.
Rosas-Vidal LE, Do-Monte FH, Sotres-Bayon F, Quirk GJ (2014). Hippocampal—prefrontal BDNF and memory for fear extinction. Neuropsychopharmacology 39: 2161–2169.
Roth TL, Matt S, Chen K, Blaze J (2014). Bdnf DNA methylation modifications in the hippocampus and amygdala of male and female rats exposed to different caregiving environments outside the homecage. Dev Psychobiol 56: 1755–1763.
Samson RD, Pare D (2005). Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci 25: 1847–1855.
Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS et al (2010). A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science 327: 863–866.
Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY et al (1997). Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-D-aspartate receptor subunit 1. Proc Natl Acad Sci USA 94: 8191–8195.
Wilensky AE, Schafe GE, Kristensen MP, LeDoux JE (2006). Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J Neurosci 26: 12387–12396.
Williams K (1993). Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol 44: 851–859.
Xu F, Plummer MR, Len GW, Nakazawa T, Yamamoto T, Black IB et al (2006). Brain-derived neurotrophic factor rapidly increases NMDA receptor channel activity through Fyn-mediated phosphorylation. Brain Res 1121: 22–34.
Young EJ, Williams CL (2013). Differential activation of amygdala arc expression by positive and negatively valenced emotional learning conditions. Front Behav Neurosci 7: 191.
Acknowledgements
This work was supported by NIH (MH96899 to IN and NS052819 to FSL).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Galvin, C., Lee, F. & Ninan, I. Alteration of the Centromedial Amygdala Glutamatergic Synapses by the BDNF Val66Met Polymorphism. Neuropsychopharmacol 40, 2269–2277 (2015). https://doi.org/10.1038/npp.2015.76
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.76
This article is cited by
-
N-Methyl D-aspartate receptor subunit signaling in fear extinction
Psychopharmacology (2019)
-
Basolateral amygdala and stress-induced hyperexcitability affect motivated behaviors and addiction
Translational Psychiatry (2017)
