
Abstract
Ketamine, a pan-NMDA receptor channel blocker, and CP-101,606, an NR2B-selective negative allosteric modulator, have antidepressant effects in humans that develop rapidly after the drugs are cleared from the body. It has been proposed that the antidepressant effect of ketamine results from delayed synaptic potentiation. To further investigate this hypothesis and potential mechanistic underpinnings we compared the effects of ketamine and CP-101,606 on neurophysiological biomarkers in rats immediately after drug administration and after the drugs had been eliminated. Local field and auditory-evoked potentials (AEPs) were recorded from primary auditory cortex and hippocampus in freely moving rats. Effects of different doses of ketamine or CP-101,606 were evaluated on amplitude of AEPs, auditory gating, and absolute power of delta and gamma oscillations 5–30 min (drug-on) and 5–6 h (drug-off) after systemic administration. Both ketamine and CP-101,606 significantly enhanced AEPs in cortex and hippocampus in the drug-off phase. In contrast, ketamine but not CP-101,606 disrupted auditory gating and increased gamma-band power during the drug-on period. Although both drugs affected delta power, these changes did not correlate with increase in AEPs in the drug-off phase. Our findings show that both ketamine and CP-101,606 augment AEPs after drug elimination, consistent with synaptic potentiation as a mechanism for antidepressant efficacy. However, these drugs had different acute effects on neurophysiological parameters. These results have implications for understanding the underlying mechanisms for the rapid-onset antidepressant effects of NMDA receptor inhibition and for the use of electrophysiological measures as translatable biomarkers.
Similar content being viewed by others

Introduction
It has been a long-standing interest to analyze the underlying neurophysiological mechanisms for the behavioral and cognitive effects of N-methyl-D-aspartate receptor antagonists. Ketamine, a pan-NMDA receptor channel blocker, elicits several symptoms of schizophrenia in healthy subjects, and disrupts normal behavior and cognitive function in humans and experimental animals (Javitt et al, 2012; Kocsis et al, 2013; Moghaddam and Krystal, 2012). In contrast to its disruptive effects, ketamine is also a unique antidepressant drug. Since the original observation (Berman et al, 2000), accumulating evidence has confirmed the significant therapeutic effect of brief exposure to ketamine in depressed patients (Krystal et al, 2013; McGirr et al, 2015). Remarkably, the antidepressant effect of ketamine has a very short onset, 2–3 h, whereas all currently used antidepressant drugs show clinical efficacy only after weeks of treatment. Furthermore, these antidepressant effects of ketamine develop and are sustained after the drug is cleared from the body. Determining the synaptic, cellular, and neuronal network mechanisms involved in ketamine’s psychotomimetic and antidepressant actions, and the interrelationship between these actions, would have a great impact on our understanding of these neuropsychiatric disorders and in developing novel therapies.
The neurophysiological changes caused by acute administration of ketamine and believed to be associated with the psychotomimetic effects are broadly described (Hong et al, 2010; Javitt et al, 2008, 2012; Kocsis, 2012a; Kocsis et al, 2013). In contrast, neurophysiological mechanisms that may underlie the antidepressive effects of ketamine are much less explored. Recently, a critical role of synaptic potentiation has been proposed for the antidepressant effects of ketamine in treatment-resistant depressed patients (Cornwell et al, 2012). In patients responding to a short, 40-min ketamine infusion, an increase in stimulus-evoked somatosensory cortical response was noted 6.5 h after the infusion. This time point is well after psychotomimetic effects associated with the drug infusion had waned and ketamine was presumed cleared from the body. Thus, a first aim of the present study was to explore whether this putative synaptic potentiation observed in humans responding to ketamine could be modeled as altered sensory-evoked potentials in awake, freely moving rats. Auditory-evoked potentials (AEPs) were recorded from the primary auditory cortex and hippocampus CA3 region and changes in AEP amplitudes were analyzed acutely (5–30 min after bolus drug administration) and after ketamine was eliminated (5–6 h).
A second aim of the present study was to begin to explore the underlying mechanisms using a comparative pharmacological approach. A negative allosteric modulator (NAM) of the NR2B subtype of NMDA receptor, CP-101,606 (Menniti et al, 1997; Mott et al, 1998), also evidenced an antidepressant response in patients (Preskorn et al, 2008). Compounds of the NR2B NAM class bind at the interface of the NR2B/NR1 amino terminal domains to allosterically reduce channel-opening probability to inhibit ion flux and functionally inhibit receptor activity (Karakas et al, 2011; Traynelis et al, 2010). Like ketamine, the response to CP-101,606 developed rapidly after a short drug infusion and was sustained well after the drug was cleared from the body. Thus, we also investigated the effects of CP-101,606 on AEPs in freely moving rats both acutely and after the drug was eliminated. An observation of increased AEPs after elimination for both ketamine and CP-101,606 would strengthen the argument that synaptic potentiation underlies the antidepressant response to these drugs.
A third aim of the present study was to investigate similarities and differences in the acute effects of ketamine and CP-101,606 on neurophysiological parameters to identify events that may trigger a delayed synaptic potentiation. Acute administration of ketamine impairs auditory gating, increases delta power and elicits aberrant, high-power gamma oscillation in rats (Kocsis, 2012a; Pinault, 2008; Zhang et al, 2012) and humans (Kocsis et al, 2013). We investigated whether CP-101,606 had acute neurophysiological effects similar to ketamine in an identical recording paradigm. Again, similarities, and differences, in the acute effects of these two drugs would provide insight into the mechanisms that may trigger the development of synaptic potentiation and antidepressant activity.
Materials and methods
Compounds
Ketamine HCl (Hospira; Lake Forest, IL) and CP-101,606, ((1S,2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol) mesylate salt, synthesized as described (Chenard et al, 1995) and dissolved in saline. Doses for both compounds are expressed as free base. Both CP-101,606 and ketamine were administered subcutaneously (SC); volumes were 2 ml/kg for ketamine and CP-101,606 at 20 mg/kg, and was 1 ml/kg for the 2 and 6.7 mg/kg doses of CP-101,606.
Electrophysiology
Experiments were performed on male Sprague–Dawley rats (Charles River, Wilmington, MA) weighing ~350–400 g, housed in a climate-controlled facility on a 12 : 12 light/dark cycle. Animals received food and water ad libitum. All experimental procedures were in line with an approved animal use protocol in compliance with the Animal Welfare Act Regulations and with the Guide for the Care and Use of Laboratory Animals, National Institutes of Health guidelines (NIH Publications No. 80–23, revised 1996). A total of 30 animals were implanted with recording electrodes and used in these studies.
Neurophysiological recordings were carried out as we described previously (Hajós et al, 2008; Harvey et al, 2013; Nagy et al, 2015; Siok et al, 2012). In brief, chronic recording electrodes were implanted into the primary auditory cortex (5 mm caudal, 4 mm lateral from Bregma) and hippocampus CA3 region (3.5 mm caudal, 3 mm lateral and 3.8 mm ventral from Bregma) under ketamine/xylazine anesthesia (Paxinos and Watson, 1998); control electrophysiological recording started no sooner than 1 week following surgery. During pharmacological experimental days, local field potentials (LFP) and AEPs were monitored continually over the light phase; signals were collected and analyzed using CED Spike2 system and software (Cambridge Electronic Design, Cambridge, UK). Auditory gating was determined using the same pattern of auditory stimulation that is used in clinical practice: delivery of two consecutive tone bursts (frequency of 5 kHz) of 10-msec duration with an intertone interval of 0.5 s (Hajós, 2006). The AEPs were determined by measuring the potential difference between the positive and the negative deflection 5–30 msec and 40–60 msec after stimulation (P20 and N40), respectively, as described previously (Hajós et al, 2008). Field potentials were analyzed by fast Fourier transform (Hajós et al, 2008; Kiss et al, 2011; Siok et al, 2012), with a particular attention to changes in cortical and hippocampal delta (0–4 Hz) and gamma (30–90 Hz) oscillations.
Drug Treatment
Doses of ketamine were chosen based on pharmacokinetic considerations to achieve clinically comparable brain exposures in rats (Shaffer et al, 2014) and also on previous findings reporting both effective and ineffective antidepressant doses of ketamine in experimental animals (Li et al, 2010). Doses of CP-101,606 were selected based on previous studies (Steece-Collier et al, 2000; Taniguchi et al, 1997). In these previous studies, CP-101,606 was effective at reducing haloperidol-induced catalepsy and neuropathic pain in rats over the dose range (2–20 mg/kg) used in the present study. Furthermore, comparative effects of ketamine and CP-101,606 over the dose ranges used in this study in a number of different behavioral paradigms have also been reported (Dix et al, 2010; Gilmour et al, 2012; Smith et al, 2011).
The study design followed a Latin square configuration, with the exception that the highest dose of ketamine was given at the end of the study. Over the course of the study, animals received saline vehicle, three doses of ketamine (5, 10, and 80 mg/kg) or three doses of CP-101,606 (2, 6.7, and 20 mg/kg). The 30 implanted animals were randomly assigned to treatment condition until data was collected for a total of 8 animals per treatment condition. For individual animals, there was at least 1 week between treatments. Collection of neurophysiological signals was initiated 2 h before drug administration, with data averaged over the 1-h period before drug administration serving as baseline. After drug administration, data was averaged over 5–30 min (drug-on) and 5–6 h (drug-off).
Statistical Analyses
Normal distributions of neurophysiological values, including amplitudes of AEPs, auditory gating (S2/S1 ratios), absolute delta and gamma-band powers were verified by Kolmogorov–Smirnoff probe. For all recording sessions for each animal, measures were converted to a percent difference from baseline except auditory gating, where absolute values were given. For each measure, an overall statistically significant difference in treatment effect was determined by one-way ANOVA (P<0.05, n=8 animals/treatment). Subsequently, statistically significant group treatment differences from vehicle were determined using the Dunnett’s test.
Pharmacokinetics
Plasma levels of ketamine and CP-101,606 were determined in satellite groups of animals matched to those used for neurophysiological recordings (n=4 per dose group). Blood samples (200 μl) were taken from the tail vein at 0.25, 0.5, 0.75, 1, 2, and 5 h after dosing into tubes containing disodium EDTA and centrifuged to obtain plasma. Subsequently, plasma proteins were precipitated by addition of 200 μl 2% tetrahydrofuran in acetonitrile, centrifuged, and supernatant removed for analysis. Ketamine and CP-101,606 levels were determined by LC-MS/MS. Analytes were separated on an Atlantis dC18 (50 × 4.6 mm, 3 μm) column using an acetonitrile/0.2% formic acid in water gradient. Detection limits for ketamine were 20.4–3000 ng/ml plasma and for CP-101,606 were 10.1 and 3064 ng/ml plasma. All values are expressed as ng/ml total plasma.
Results
Drug Plasma Exposure
In order to determine appropriate neurophysiological data collection epochs, plasma concentrations of ketamine and CP-101,606 were determined by tail vein sampling of animals receiving 5, 10, or 80 mg/kg SC ketamine or 2, 6.7, or 20 mg/kg SC CP-101,606. For the two lower doses of ketamine and all doses of CP-101,606, highest levels were observed at the first time point sampled (0.25 h; Figure 1). For both drugs at all doses, plasma levels were at or near the lower limit of detection at the 5 h sampling point. On the basis of these data, neurophysiological data were averaged over 5–30 min (drug-on) and 5–6 h (drug-off) after drug administration.

Time profile of plasma concentration of ketamine (5, 10 and 80 mg/kg, subcutaneously) and CP-101,606 (2, 6.7 and 20 mg/kg, subcutaneously) following their administration in rats. Data are presented as means±SEM.
Neurophysiological Data
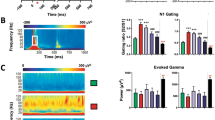
Typical recordings of cortical and hippocampal LFPs and AEPs following vehicle, ketamine (10 mg/kg, SC), and CP-101,606 (6.7 mg/kg, SC) administration are shown in Figure 2 at baseline, drug-on and drug-off periods in freely moving rats. Changes in LFPs and AEPs following ketamine and CP-101,606 administration were compared with changes in vehicle-treated rats. Corresponding power spectrograms (0–20 Hz) of primary auditory cortex LFPs, performed by means of fast Fourier transformation at baselines and after ketamine (10 mg/kg) and CP-101,606 (6.7 mg/kg) treatments are shown in Supplementary Figure 1.

Typical neurophysiological recordings from the primary auditory cortex in vehicle, ketamine (10 mg/kg, subcutaneously) and CP-101,606 (6.7 mg/kg, subcutaneously) treated rats at control period (baseline), and during drug-on (middle panels) and drug-off (right panel) periods. Traces show gamma activity following band-pass filtering (Gamma), unfiltered local field potentials (LFP) and auditory-evoked potentials within an auditory gating paradigm, triangles indicating auditory stimulation (Gating).
AEPs
Effects of ketamine
Ketamine significantly altered amplitude of AEPs both in the cortex and hippocampus compared with vehicle. During drug-on period, the highest dose of ketamine (80 mg/kg) significantly reduced AEP amplitudes in both the cortex and hippocampus, whereas there were no effects at lower doses of ketamine (Figures 2 and 3). In contrast, during the drug-off period AEPs were significantly augmented by ketamine at a dose of 10 mg/kg. The amplitudes of cortical AEPs were increased by ~50%, whereas hippocampal AEPs showed a threefold increase in this time period. The lower or higher doses of ketamine were without effect in either region in the drug-off period.

Dose–response effects of ketamine (a) and CP-101,606 (b) on auditory-evoked potentials in the primary auditory cortex and hippocampus compared with vehicle-treated rats during drug-on and drug-off phase. Data are presented as means±SEM. *P<0.05.
Effects of CP-101,606
There was no effect on AEP amplitudes in the drug-on period after administration of 2 or 6.7 mg/kg CP-101,606. However, there was a small, though significant increase in hippocampal AEPs after the highest dose of CP-101,606 in this period (Figures 2 and 3). In contrast, CP-101,606 robustly enhanced AEPs in the drug-off period. This effect was statistically significant for the 6.7 and 20 mg/kg doses in auditory cortex, and for all three doses in hippocampus. In both regions, the 6.7 mg/kg dose of CP-101,606 was most effective, indicating a potential bell-shape dose–response curve.
Auditory Gating
In line with previous observations, higher doses of ketamine significantly (P<0.05) disrupted auditory gating in the hippocampus during the drug-on period; S2/S1 ratios (±SEM) were at baselines and after ketamine treatment in the hippocampus: 0.49±0.03 and 0.61±0.04 (10 mg/kg), 0.47±0.03 and 0.70±0.07 (80 mg/kg), and in the cortex: 0.47±0.03 and 0.66±0.04 (10 mg/kg), 0.49±0.03 and 0.72±0.04 (80 mg/kg). During the drug-off period, degree of auditory gating in both the hippocampus and primary auditory cortex returned to baseline level following each tested dose of ketamine, except in the hippocampus following the 10 mg/kg dose (baseline 0.49±0.03 and 0.64±0.05 after 10 mg/kg). In contrast, neither vehicle nor CP-101,606 impacted auditory gating in the cortex or hippocampus either during the drug-on or drug-off periods (data not shown).
Gamma Power
Effects of ketamine
Ketamine dose-dependently increased gamma power in the cortex and hippocampus during the drug-on period. The greatest effect was observed with the 80 mg/kg dose, which caused a near threefold increase in cortex and an approximately fivefold increase in the hippocampus (Figure 4). Ketamine at 10 mg/kg also caused a smaller increase in gamma power during this period, reaching statistical significance in the cortex. Gamma power returned near baseline values during drug-off period, and only an ~25% increase was observed in the cortex and hippocampus after the highest dose, compared with the vehicle-treated group.

Changes in delta (a, b) and Gamma (c, d) power of local field potential in the primary auditory cortex and hippocampus following ketamine (a, c) and CP-101,606 (b, d) treatment during drug-on and drug-off phase. Data are presented as means±SEM. *P<0.05.
Effects of CP-101,606
Gamma power was not impacted at all by CP-101,606 in the cortex or hippocampus during either the drug-on or drug-off periods (Figure 4).
Delta Power
Effects of ketamine
Ketamine dose-dependently increased delta power during the drug-on period; the 10 and 80 mg/kg doses elicited a significant two to threefold increase in absolute delta power in both the cortex and hippocampus. Furthermore, a significantly enhanced delta power was also present in both regions after each tested dose of ketamine in the drug-off period, ~50–150% higher than in vehicle-treated rats (Figure 4).
Effects of CP-101,606
During the drug-on phase, CP-101,606 did not change delta power in the cortex or hippocampus compared with vehicle. In contrast, in the drug-off phase delta power was significantly enhanced in the cortex at each dose, whereas in the hippocampus only the highest dose resulted in a marginally elevated delta power (Figure 4).
The effects of ketamine and CP-101,606 on each neurophysiological end point are summarized in Figure 5.

Summary figure comparing the effects of various doses of ketamine and CP-101,606 on neurophysiological markers during drug-on and drug-off periods. Only significant changes, as compared with the appropriate vehicle-treated controls, are shown; AEP amplitudes, delta and gamma powers are normalized to baseline (100%). A full color version of this figure is available at the Neuropsychopharmacology journal online.
Discussion
The present findings confirm our prediction that ketamine leads to augmented sensory-evoked potentials in rats, consistent with the previously reported enhanced–somatosensory-evoked potentials in patients responding to ketamine (Cornwell et al, 2012). Increase in cortical and hippocampal AEP amplitudes occurred 5–6 h after ketamine administration, a time period when ketamine was shown to be fully eliminated in rats (this study and Shaffer et al, 2014) also paralleling the clinical observations. Interestingly, a greater effect size was observed in hippocampus compared with auditory cortex. The effective dose of ketamine to increase AEP amplitude was 10 mg/kg, SC; lower (5 mg/kg) and higher (80 mg/kg) doses were not active. Selection of ketamine doses was based on previous studies of effects in rat behavioral models of depression and neurobiological correlates of these effects (Li et al, 2010, 2011). In these studies, ketamine at 10 mg/kg was also the most effective, whereas lower or higher doses were not active. Importantly, this optimal dose in the rat experimental studies yields exposures that correlate closely to human exposures at the clinically used antidepressive dose of ketamine, based on pharmacokinetic modeling (Shaffer et al, 2014).
The NR2B NAM CP-101,606 has been shown to have an antidepressant response in patients similar to that of ketamine, namely, the response developed rapidly after drug infusion and was sustained in responders for days after the drug was presumed to be eliminated (Preskorn et al, 2008). Significantly, in the present study CP-101,606 also induced a robust increase in AEPs in both cortex and hippocampus in rats after the drug was eliminated. Similar to ketamine, the effect size of CP-101,606 was greater in hippocampus than in cortex. There was also evidence of a similar bell-shaped dose–response relationship, although the effects of all three doses of CP-101,606 reached statistical significance. Thus, the commonality of effects of CP-101,606 and ketamine on the drug-off increase in AEPs significantly strengthens the hypothesis that the rapid-onset antidepressant response to these NMDA receptor antagonists results from synaptic potentiation that is sustained after the drugs are eliminated.
In contrast to the similar effects of ketamine and CP-101,606 in drug-off augmentation of sensory-evoked potentials, these two drugs had significantly different effects on other neurophysiological signals, particularly during the acute phase immediately after administration. The acute effects of ketamine have been broadly studied, in part to investigate mechanisms by which this drug mimics numerous symptoms of schizophrenia (Hong et al, 2010; Javitt et al, 2011; Kocsis et al, 2013). Therefore, in our studies we measured additional neurophysiological markers; some, such as auditory gating, are clearly linked to information processing and known to be impacted in psychiatric patients (Hajós, 2006). In the present study ketamine impaired auditory gating, in line with published findings (Kocsis et al, 2013). It has also been demonstrated that ketamine enhances gamma-band power both in experimental animals and humans, a response which is frequently referred to as aberrant gamma activity, and which is considered a possible factor in the psychotomimetic effects of ketamine (Kittelberger et al, 2012; Kocsis et al, 2013; Pinault, 2008). Consistent with these previous findings, in the present study ketamine increased gamma power multiple times over baseline values compared with vehicle-treated rats in both cortical and hippocampal regions. However, in a sharp contrast to ketamine, CP-101,606 did not disrupt auditory gating, nor did the compound impact gamma-band power, in agreement with recent publications on CP-101,606 and other NR2B NAMs (Kocsis, 2012b; Sivarao et al, 2014).
The lack of effect of CP-101,606 on auditory gating and, in particular, gamma-band power, has implications for an understanding of the mechanisms underlying both the antidepressant and psychotomimetic effects of NMDA receptor inhibition. It has been hypothesized that the antidepressant response to ketamine results from induction of an acute hyperglutamatergic state accompanied by an increase in neuronal activity at gamma frequency that triggers an LTP-like synaptic potentiation (Gass et al, 2014). That CP-101,606 induces synaptic potentiation in the absence of change in gamma power suggests that the induction of gamma power may not be an obligatory step in the cascade from NMDA receptor inhibition to synaptic potentiation. The present results also raise further questions regarding the relationship of changes in gamma power to psychotomimetic effects. Recent studies of the low-trapping NMDA channel blocker lanicemine showed an enhanced gamma-band power both in rats and humans, but it did not induce psychotomimetic effects in patients (Sanacora et al, 2014). On the other hand, in humans, CP-101,606 was observed to evoke dissociative effects in some patients at higher doses (Nutt et al, 2008; Preskorn et al, 2008). It is not known at present whether the lack effects of CP-101,606 on gamma power in rats translates to humans. In fact, there may be subtle but important differences in the distribution of NR2B subunit containing receptors between rat and non-human primate, as suggested by Arnsten and colleagues (Wang et al, 2013) that could result in a lack of translation on some functional measures. Thus, it will be important to determine the neurophysiological signature of NR2B NAMs in humans and non-human primates to further clarify the role of gamma disruption in the psychotomimetic effects of NMDA receptor inhibition.
NMDA receptor channel blockers have additional neurophysiological effects, including induction of an increase in delta power (Dworak et al, 2011; Kiss et al, 2013; Zhang et al, 2012). In the present study, the higher doses of ketamine increased delta power both in the cortex and hippocampus during both drug-on and drug-off periods. Mechanisms underlying changes in delta power are not fully understood; previous studies analyzed acute, short-time effects of ketamine on EEG, demonstrating a transient increase in delta power (Fu et al, 2008; Zhang et al, 2012). Therefore, late-onset increase in delta power could be a rebound effect in response to sleep deprivation induced acutely by administration of NMDA receptor antagonists (Kocsis, 2012b). Interestingly, CP-101,606 also enhanced delta power but only during drug-off phase and most prominently in the cortex. The mechanism for this effect also bears further exploration. Nonetheless, it appears unlikely that the effects of CP-101,606 and ketamine on delta power can account for the delayed increase in AEPs. A limitation of the present study is the degree to which physiological responses in naive rats may be extrapolated to the physiology of the human brain of depressed patients. Back-translational studies, such as presented here using two drugs with demonstrated clinical antidepressant efficacy, may help to bridge this translational gap. We await further forward-translational clinical data with ketamine and NR2B NAMs to place the current findings in more definitive context.
In summary, the results of the present study support the hypothesis that the antidepressant response to brief NMDA receptor inhibition results in a rapidly developing synaptic potentiation that is sustained after drug–target engagement has ended. These findings recommend the use of long-lasting changes in sensory-evoked potentials as a translatable biomarker for this antidepressant effect. The relationship between antidepressant and psychotomimetic effects is one of the most important questions regarding the mode of action of NMDA antagonists and their further development as antidepressants. In so far as the increase in AEPs after drug elimination reflects an underlying mechanism for the antidepressant effect, the present study is informative in two ways. First, it is apparent from the clinical literature that the psychotomimetic effects of both types of NMDA antagonists are directly correlated with drug-exposure levels—higher exposures result in greater psychotomimetic effects. In contrast, in the present study dose responses for the synaptic potentiation for both ketamine and CP-101,606 exhibited bell-shaped relationships. This raises the possibility that optimal exposures for ketamine or an NR2B NAM may be identified that yield antidepressant efficacy with minimal or no psychotomimetic side effects. Indeed, the preliminary data reported by Preskorn et al (2008) with CP-101,606 supports this possibility. Second, although both ketamine and CP-101,606 caused robust increases in AEPs in the drug-off period, these drugs had distinct effects acutely during the period of target engagement. In so far as some of these acute, drug-on effects are related to the induction of psychotomimetic symptoms, these data suggest that it may be possible to identify mechanistically unique compounds that yield the desired rapid-onset antidepressant efficacy while effectively avoiding the psychotomimetic, and other, side effects associated with the NMDA receptor channel blockers.
Funding and disclosure
This work was partly supported by Mnemosyne Pharmaceuticals, Providence, RI, USA. MH has received research funding from Mnemosyne Pharmaceuticals, FORUM Pharmaceuticals, and CHDI Foundation; consulting fees from FORUM Pharmaceuticals and Lundbeck Pharmaceuticals. FSM is full-time employee and Chief Scientific Officer of Mnemosyne Pharmaceuticals. The remaining authors declare no conflict of interest.
References
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS et al (2000). Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47: 351–354.
Chenard BL, Bordner J, Butler TW, Chambers LK, Collins MA, De Costa DL et al (1995). (1S,2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol: a potent new neuroprotectant which blocks N-methyl-D-aspartate responses. J Med Chem 38: 3138–3145.
Cornwell BR, Salvadore G, Furey M, Marquardt CA, Brutsche NE, Grillon C et al (2012). Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry 72: 555–561.
Dix S, Gilmour G, Potts S, Smith JW, Tricklebank M (2010). A within-subject cognitive battery in the rat: differential effects of NMDA receptor antagonists. Psychopharmacology 212: 227–242.
Dworak M, McCarley RW, Kim T, Basheer R (2011). Delta oscillations induced by ketamine increase energy levels in sleep-wake related brain regions. Neuroscience 197: 72–79.
Fu Y, Guo L, Zhang J, Chen Y, Wang X, Zeng T et al (2008). Differential effects of ageing on the EEG during pentobarbital and ketamine anaesthesia. Eur J Anaesthesiol 25: 826–833.
Gass N, Schwarz AJ, Sartorius A, Schenker E, Risterucci C, Spedding M et al (2014). Sub-anesthetic ketamine modulates intrinsic BOLD connectivity within the hippocampal-prefrontal circuit in the rat. Neuropsychopharmacology 39: 895–906.
Gilmour G, Dix S, Fellini L, Gastambide F, Plath N, Steckler T et al (2012). NMDA receptors, cognition and schizophrenia—testing the validity of the NMDA receptor hypofunction hypothesis. Neuropharmacology 62: 1401–1412.
Hajós M (2006). Targeting information-processing deficit in schizophrenia: a novel approach to psychotherapeutic drug discovery. Trends Pharmacol Sci 27: 391–398.
Hajós M, Hoffmann WE, Kocsis B (2008). Activation of cannabinoid-1 receptors disrupts sensory gating and neuronal oscillation: relevance to schizophrenia. Biol Psychiatry 63: 1075–1083.
Harvey BD, Siok CJ, Kiss T, Volfson D, Grimwood S, Shaffer CL et al (2013). Neurophysiological signals as potential translatable biomarkers for modulation of metabotropic glutamate 5 receptors. Neuropharmacology 75: 19–30.
Hong LE, Summerfelt A, Buchanan RW, O'Donnell P, Thaker GK, Weiler MA et al (2010). Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology 35: 632–640.
Javitt DC, Schoepp D, Kalivas PW, Volkow ND, Zarate C, Merchant K et al (2011). Translating glutamate: from pathophysiology to treatment. Sci Transl Med 3: 102mr102.
Javitt DC, Spencer KM, Thaker GK, Winterer G, Hajós M (2008). Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev Drug Discov 7: 68–83.
Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D (2012). Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull 38: 958–966.
Karakas E, Simorowski N, Furukawa H (2011). Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 475: 249–253.
Kiss T, Feng J, Hoffmann WE, Shaffer CL, Hajós M (2013). Rhythmic theta and delta activity of cortical and hippocampal neuronal networks in genetically or pharmacologically induced N-methyl-D-aspartate receptor hypofunction under urethane anesthesia. Neuroscience 237: 255–267.
Kiss T, Hoffmann WE, Hajós M (2011). Delta oscillation and short-term plasticity in the rat medial prefrontal cortex: modelling NMDA hypofunction of schizophrenia. Int J Neuropsychopharmacol 14: 29–42.
Kittelberger K, Hur EE, Sazegar S, Keshavan V, Kocsis B (2012). Comparison of the effects of acute and chronic administration of ketamine on hippocampal oscillations: relevance for the NMDA receptor hypofunction model of schizophrenia. Brain Struct Funct 217: 395–409.
Kocsis B (2012a). Differential role of NR2A and NR2B subunits in N-methyl-D-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biol Psychiatry 71: 987–995.
Kocsis B (2012b). State-dependent increase of cortical gamma activity during REM sleep after selective blockade of NR2B subunit containing NMDA receptors. Sleep 35: 1011–1016.
Kocsis B, Brown RE, McCarley RW, Hajós M (2013). Impact of ketamine on neuronal network dynamics: translational modeling of schizophrenia-relevant deficits. CNS Neurosci Ther 19: 437–447.
Krystal JH, Sanacora G, Duman RS (2013). Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry 73: 1133–1141.
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M et al (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329: 959–964.
Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H et al (2011). Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry 69: 754–761.
McGirr A, Berlim MT, Bond DJ, Fleck MP, Yatham LN, Lam RW (2015). A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol Med 45: 693–704.
Menniti F, Chenard B, Collins M, Ducat M, Shalaby I, White F (1997). CP-101,606, a potent neuroprotectant selective for forebrain neurons. Eur J Pharmacol 331: 117–126.
Moghaddam B, Krystal JH (2012). Capturing the angel in "angel dust": twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr Bull 38: 942–949.
Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P et al (1998). Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat Neurosci 1: 659–667.
Nagy D, Tingley FD, Stoiljkovic M, Hajós M (2015). Application of neurophysiological biomarkers for Huntington's disease: evaluating a phosphodiesterase 9A inhibitor. Exp Neurol 263: 122–131.
Nutt JG, Gunzler SA, Kirchhoff T, Hogarth P, Weaver JL, Krams M et al (2008). Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and Parkinsonism. Mov Disord 23: 1860–1866.
Paxinos G, Watson C (1998) The Rat Brain in Stereotaxic Coordinates, 4th edn. Academic Press: San Diego, CA, USA.
Pinault D (2008). N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatry 63: 730–735.
Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW (2008). An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol 28: 631–637.
Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ et al (2014). Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry 19: 978–985.
Shaffer CL, Osgood SM, Smith DL, Liu J, Trapa PE (2014). Enhancing ketamine translational pharmacology via receptor occupancy normalization. Neuropharmacology 86: 174–180.
Siok CJ, Cogan SM, Shifflett LB, Doran AC, Kocsis B, Hajós M (2012). Comparative analysis of the neurophysiological profile of group II metabotropic glutamate receptor activators and diazepam: effects on hippocampal and cortical EEG patterns in rats. Neuropharmacology 62: 226–236.
Sivarao DV, Chen P, Yang Y, Li YW, Pieschl R, Ahlijanian MK (2014). NR2B antagonist CP-101,606 abolishes pitch-mediated deviance detection in awake rats. Front Psychiatry 5: 96.
Smith JW, Gastambide F, Gilmour G, Dix S, Foss J, Lloyd K et al (2011). A comparison of the effects of ketamine and phencyclidine with other antagonists of the NMDA receptor in rodent assays of attention and working memory. Psychopharmacology 217: 255–269.
Steece-Collier K, Chambers LK, Jaw-Tsai SS, Menniti FS, Greenamyre JT (2000). Antiparkinsonian actions of CP-101,606, an antagonist of NR2B subunit-containing N-methyl-d-aspartate receptors. Exp Neurol 163: 239–243.
Taniguchi K, Shinjo K, Mizutani M, Shimada K, Ishikawa T, Menniti FS et al (1997). Antinociceptive activity of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br J Pharmacol 122: 809–812.
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK et al (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496.
Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA et al (2013). NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron 77: 736–749.
Zhang Y, Yoshida T, Katz DB, Lisman JE (2012). NMDAR antagonist action in thalamus imposes delta oscillations on the hippocampus. J Neurophysiol 107: 3181–3189.
Acknowledgements
We acknowledge Patricia Seymour, Mnemosyne Pharmaceuticals, for useful discussions and comments and Bill Billing, BioPharma Works, Groton, CT for help with statistics.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article

Cite this article
Nagy, D., Stoiljkovic, M., Menniti, F. et al. Differential Effects of an NR2B NAM and Ketamine on Synaptic Potentiation and Gamma Synchrony: Relevance to Rapid-Onset Antidepressant Efficacy. Neuropsychopharmacol 41, 1486–1494 (2016). https://doi.org/10.1038/npp.2015.298
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.298
This article is cited by
-
Therapeutic potential of N-methyl-D-aspartate receptor modulators in psychiatry
Neuropsychopharmacology (2024)
-
Plasticity of synapses and reward circuit function in the genesis and treatment of depression
Neuropsychopharmacology (2023)
-
In vivo electrophysiological recordings of the effects of antidepressant drugs
Experimental Brain Research (2019)
-
Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects
Molecular Psychiatry (2017)
-
Prefrontal-hippocampal coupling by theta rhythm and by 2–5 Hz oscillation in the delta band: The role of the nucleus reuniens of the thalamus
Brain Structure and Function (2017)

{kind=link}