
Abstract
Cocaine is habit-forming because of its ability to enhance dopaminergic neurotransmission in the forebrain. In addition to neuronal inputs, forebrain dopamine circuits are modulated by hormonal influences; one of these is leptin, an adipose-derived hormone that attenuates the rewarding effects of food- and hunger-associated brain stimulation reward. Here we report reciprocal inhibition between the reward-related effects of leptin and the reward-related effects of cocaine in rats. First, we report that cocaine and the expectancy of cocaine each depresses plasma leptin levels. Second, we report that exogenous leptin, given systemically or directly into the ventral tegmental area, attenuates the ability of cocaine to elevate dopamine levels in the nucleus accumbens, the ability of cocaine to establish a conditioned place preference, and the ability of cocaine-predictive stimuli to prolong responding in extinction of cocaine-seeking. Thus, whereas leptin represents an endogenous antagonist of the habit-forming and habit-sustaining effects of cocaine, this antagonism is attenuated by cocaine and comes to be attenuated by the expectancy of cocaine.
Similar content being viewed by others

Introduction
Recent attempts to understand the mechanisms of addiction have focused largely on the development of compensatory changes within the brain—neuroadaptations—that result from chronic exposure to addictive drugs (Kalivas, 2009; Lüscher and Malenka, 2011; Nestler, 1992) and that are thought to modify the habit-forming impact of those drugs (Koob and Le Moal, 2001). Among these are changes in excitatory transmission to the ventral tegmental area (VTA; Saal et al, 2003), origin of the mesocorticolimbic dopamine system, and a critical link between reward-predictive cues and brain reward circuitry (You et al, 2007). This system is responsive not only to a variety of glutamatergic inputs from local (Dobi et al, 2010) and distal (Geisler et al, 2007) neuronal sources, but also to a number of state variables mediated by blood-borne factors including leptin (Figlewicz et al, 2003; Krugel et al, 2003; Hommel et al, 2006; Liu et al, 2011; Thompson and Borgland, 2013). Leptin is an endogenous inhibitor of food reward (Figlewicz et al, 2007; Domingos et al, 2011) that has both direct (Fulton et al, 2006a; Krugel et al, 2003; Leinninger et al, 2009; Davis et al, 2011; Domingos et al, 2011) and indirect (Leinninger et al, 2009) effects on brain reward function and that is depressed in heroin addicts (Housová et al, 2005) and fluctuates abnormally during craving for alcohol (Kiefer et al, 2005) or nicotine (al'Absi et al, 2011).
The aim of the present study was to determine whether leptin levels fluctuate in relation to cocaine seeking and to determine whether such fluctuations, if present, affect the rewarding effects of cocaine or cocaine-associated predictive cues. We report (i) that plasma leptin levels are acutely depressed by self-administered or yoked cocaine injections, (ii) that plasma leptin levels become depressed in advance of daily cocaine availability under regular self-administration training conditions, (iii) that these depressions of endogenous leptin levels are accompanied by depressions of leptin receptor-mediated signaling in the VTA, and (iv) that exogenous leptin, given systemically or microinjected directly into the VTA, attenuates rewarding effects of cocaine and cocaine-predictive stimuli in laboratory rats.
Materials and methods
Subjects
Male Long-Evans male rats (Charles-River, Raleigh, NC), weighing 350–400 g at the time of the surgery, were used throughout. They were housed individually under a reversed light–dark cycle (light on at 2000 hours), with free access to food and water and randomly assigned to testing or treatment groups of 8–10 animals per group. All procedures were approved by the Animal Care and Use Committee of the NIDA IRP and were consistent with the ‘Principles of Laboratory Animal Care’ published by the National Institutes of Health (NIH publication 86-23, 1996).
Surgery
Surgeries were conducted using pentobarbital (30 mg/kg i.p.) and chloral hydrate (140 mg/kg, i.p.) anesthesia. Rats to be used in microinjection experiments were implanted first with angled bilateral guide cannulae dorsal to the VTA. Flat-skull (Paxinos and Watson, 2013) stereotaxic coordinates for cannula entry were 5.6 mm posterior to the bregma and 2.2 mm lateral to the midline; the cannulae were lowered at a 12° angle toward the midline to a depth of 6.8 mm. Rats for microdialysis experiments were implanted with one guide cannula 1 mm dorsal to the nucleus accumbens, 1.2 mm anterior to the bregma, 1.5 lateral to the midline, and 6.0 ventral to the skull surface, and one cannula 1 mm dorsal to the VTA (as above). The cannulae were secured to the skull with four stainless steel screws and dental acrylic.
Each rat was implanted with chronic indwelling jugular catheters. A small incision was made to the right of the midline of the neck and the external jugular vein was externalized. An intravenous (i.v.) silastic catheter (Dow Corning, Midland, MI) was inserted to just reach the entry to the right atrium. The catheter was secured to the vein with silk suture. The catheter was fed subcutaneously around the back of the neck to exit near the back of the skull. The end was then connected to a bent 22-gauge stainless steel cannula (Plastic One, Roanoke, VA) with a threaded head used to secure a dummy cannula and, during testing, an infusion line. Catheters and cannula assemblies were secured to the skull with stainless steel screws and dental cement.
Each rat was given 0.25 ml of 2.27% enrolfloxacine once daily for 3 days as precaution against infection. For five days before testing, the catheters were flushed daily with 0.2 ml of gentamicin in sterile saline and 0.05 ml of heparin (30 U/ml in sterile saline).
Self-Administration Training
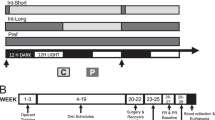
All rats were initially trained to self-administer i.v. cocaine or saline. Unless otherwise specified, ‘cocaine’ designates a cocaine salt that readily crosses the blood–brain barrier and is traditionally used for i.v. self-administration: cocaine HCl. Fourteen consecutive 4-h-daily training sessions were given, starting (with one exception) at the same time each day (1100 hours, 3 h into the 12-h dark cycle). A lever press on one of two levers—a retractable ‘active’ lever that was inserted into the cage to start each session—caused illumination of a white cue light above the lever and concurrent delivery of i.v. cocaine (1 mg/kg per injection in a volume of 0.13 ml over 4.5 s) or saline (equal volume) on a fixed ratio-1 schedule of reinforcement. A 20-s timeout period, during which the cue light remained on and additional lever pressing was ineffective, accompanied each injection. In order to avoid many of the pre-session cues involved in animal handling and transport from the colony room to the testing room, one group was housed in the testing apparatus where it was given 14 days of normal testing followed by 14 days of testing at irregular times from 1 day to the next (sometimes 0900 hours, sometimes 1100 hours, sometimes 1300 hours in irregular sequence).
Blood Sample Collection
For assays of plasma leptin, blood samples (0.3 ml) were taken at various stages of testing via the i.v. catheter that was used for drug self-administration. Before withdrawal, the catheters were flushed with 0.2 ml of heparinized saline. Samples were then gently withdrawn, immediately transferred into tubes containing EDTA, and centrifuged at 4000 g for 15 min. The supernatant from each sample was stored at −80 °C until they were assayed.
Blood samples were initially taken from three groups of cocaine-trained and three groups of saline-trained rats under various conditions at the usual training time on the day following the 14-day training period. One cocaine-trained group and one saline-trained group were simply given another day in their training conditions. The remaining groups received unearned injections ‘yoked’ to the earned injections of an executive animal in the cocaine self-administration group. One cocaine-trained group and one saline-trained group received yoked injections of normal cocaine; one cocaine-trained group and one saline-trained group received yoked injections of cocaine methiodide (MI). Cocaine MI is a synthetic cocaine analog that does not cross the blood–brain barrier but that can serve as a conditioned reinforcer in cocaine-trained animals (Wang et al, 2013). Blood samples were drawn at the start of the session and at 30, 120, and 240 min into the sessions.
Two groups were tested under extinction conditions after cocaine self-administration training. They were placed in the chambers on Day 15, and their levers were inserted as usual. However, now each lever press resulted in injection of saline rather than cocaine, given along with normal illumination of the cue light. Blood samples were drawn at the start of the session and at 30 and 60 min into the session from one group during the first extinction trial and from the other group during the fourteenth extinction trial.
Post-Training Behavioral Tests
Four groups of cocaine-trained rats were pretreated, two with i.v. injection of leptin (0.6 mg/kg) and another two with i.v. saline (0.6 ml/kg). Forty minutes later, one group from each treatment was given another 4-h drug self-administration (drug-taking) session. The remaining two groups were placed in their regular training chambers; however, for these animals responses on the active levers were rewarded by saline instead of by cocaine (seeking condition). Four additional groups (prepared with bilateral VTA guide cannulae) were pretreated with VTA microinjections of leptin (0.5 μg/side) or aCSF (0.5 μl/side) and similarly tested 40 min later. Injections were made over 30 s through microdialysis probes that had the membrane removed from the tip of the probe. Responding on active and inactive levers and the numbers of infusions were recorded.
Seven additional groups of cocaine-trained rats were tested for the ability of various intra-VTA microinjection pretreatments to block the development of a cocaine-conditioned place preference. The pretreatments included leptin (0.125, 0.25, or 0.5 μg/side); the leptin agonist DesI2-E1/aca (0.01 μg/side); the leptin antagonist Allo-aca (0.25 μg/side); the combination of leptin and Allo-aca (0.25 μg each in 0.5 μl/side); or aCSF (0.5 μl/side). Forty minutes after pretreatment, the animals were given i.p. cocaine injections (10 mg/kg) and placed for 15 min in one of the closed end chambers of a three-chamber-conditioned place preference apparatus (Med Associates, St Albans, VT). On the following day the animals were returned to the apparatus drug-free, placed in the center chamber, and allowed free access to the three open chambers for 15 min. Time in each chamber was recorded.
Plasma Leptin and Brain Protein Assays
Leptin levels were assayed according to the manufacturer’s instructions using ELISA kits (Millipore, Billerica, MA). The intra- and inter-assay variations for leptin were less than 4%. Hormone levels were interpolated using the four-parameter logic equation from standard curve running for each plate
Leptin binds to the long form of the leptin receptor (LepRb; Figlewicz et al, 2003), leading to activation of three intracellular signaling pathways: JAK-STAT, PI3K, and MAPK pathways (Niswender et al, 2001; Trinko et al, 2011). We assayed VTA tissue for STAT3 and Akt (protein kinase B), markers for the two primary pathways, in groups killed just before or 6 h after normal self-administration testing. We also monitored STAT3 in additional groups following VTA microinjections of the agents used in the conditioned place preference experiment: aCSF (0.5 μl/side), leptin (0.25 μg/side), Allo-aca (0.25 μg/side), or leptin+Allo-aca (0.25 μg each in 0.5 μl/side). For the brains that did not have VTA cannulae, bilateral VTA tissues were obtained. VTA tissue samples included the paranigral, parabrachial pigmented, and interfascicular nuclei, and the ventral half of the rostral linear nucleus. For the remaining brains, tissue collections were restricted to a radius of 0.5 mm from those injection tips that correctly terminated in the VTA. Tissue samples were then homogenized and processed in duplicate.
STAT3 proteins were separated after loading the samples on 4–12% NuPAGE neutral polyacrylamide gel using 1XMOPS buffer under constant voltage (150 V) electrophoresis. The proteins on each gel were then transferred to PVDF membranes, blocked for 60 min in TBS containing 0.1% Tween 20 and 5% nonfat dry milk. Blocked membranes were first incubated with mouse monoclonal anti-phospho-STAT3 antibody (Cell Signaling, Cat #4113) at 4 °C overnight, followed on the next day by another 2 h of incubation with rabbit STAT3 and β-tubulin (an internal control) antibodies. The blots were then washed and incubated for 1 h with anti-rabbit and anti-mouse secondary antibodies labeled with IRDyes 680 and 800, respectively. Fluorescence intensity from each band was assessed using an Odyssey IR fluorescence scanner and quantified using the Odyssey 2.0 software. Protein levels were calculated using a ratio of fluorescence intensity of STAT3 or p-STAT3 normalized to that of β-tubulin obtained from the same samples.
The procedures for Akt separation and analysis were similar to those described above for STAT3 proteins, except that Akt antibody pair was used (Akt Rabbit mAb #4691, Phospho-Akt (Ser473) Mouse mAb #4051, Cell Signaling).
Microdialysis
Four groups of cocaine-trained rats with unilateral VTA and ipsilateral NAS cannulae were connected to microdialysis systems (dialysis probes inserted through the NAS guide cannulae) on the evening following completion of cocaine self-administration training and maintained in the testing chamber overnight. The next morning, the perfusion syringes were refilled and the flow rate was increased to 1.2 μl/min for 2 h before initiation of testing. Three 20-min samples were collected and then the rats were given VTA microinjections of the agents used in the conditioned place preference experiment: aCSF (0.5 μl/side), leptin (0.25 μg/side), Allo-aca (0.25 μg/side), or leptin+Allo-aca (0.25 μg each in 0.5 μl/side). Forty minutes after the pretreatments an i.p. cocaine injection (10 mg/kg) was given and 20-min dialysis samples were collected for two additional hours. Dopamine levels in the dialysate were assayed using an HPLC system with electrochemical detection as described previously (You et al, 2008).
Body Weight, Food Intake, and Fat Pad Measurements
Body weights of cocaine- and saline-trained rats were taken daily. Food intake was measured at times 7, 10, 13, 16, and 19 h following the last training session in one group of cocaine-trained and one group of saline-trained rats. Following an additional self-administration session on the following day, the animals were decapitated, blood samples were collected, and fat pads from three major body deposits (inguinal, gonadal, and retroperitoneal) were taken and weighed.
Histology
After the completion of the microinjection and microdialysis experiments, the rats were decapitated under deep anesthesia and the brains were removed and fixed in 10% formalin. Fifty-micrometer coronal sections were sliced with a cryostat. The probe location was examined under microscope at low magnification in wet tissue that allowed differentiation of fiber tracts and cell groups.
Data Analysis
Protein levels measured with western blot were calculated as a ratio of the protein fluorescence intensities to that obtained from β-tubulin. The levels of the assayed dopamine were expressed as the concentrations found in the perfusates. Basal values refer to those obtained before VTA injection. The data were analyzed with either one-way or two-way analysis of variance (ANOVA) with or without repeated measures, as appropriate. Post hoc multiple comparisons were performed using the Newman–Keuls test. A level of P<0.05 for a two-tailed test was considered critical for statistical significance.
Drugs
Cocaine hydrochloride, cocaine MI, pentobarbital, and chloral hydrate were obtained from the pharmacy within the Institute. Rat recombinant leptin and AG490 were purchased from Sigma-Aldrich (St Louis, MO). Allo-aca and DesI2-E1/aca were synthesized and their characteristics were reported previously (Otvos et al, 2008, 2011).
Results
Depression of Plasma Leptin Levels by Actual and Anticipated Cocaine
Following 14 days of cocaine self-administration, plasma leptin levels just before the normal time of daily testing were depressed by ~40% (Figure 1). Leptin levels fell another 40–50% within the self-administration sessions (red solid line) or within sessions where self-administration-trained animals were given yoked injections of cocaine (orange solid line) or cocaine MI (blue solid line) instead of the normal opportunity to self-administer cocaine. Leptin levels were normal at the start of sessions in saline-trained animals, but they fell by a similar 40% when such animals were given yoked cocaine injections (orange dashed line). In saline-trained groups that were given yoked injections of cocaine MI (dashed blue line) or were allowed to self-administer saline (black dashed line), leptin levels fell ~2% per hour during the 4-h period without food.

Leptin levels during cocaine or saline self-administration or during receipt of yoked injections of cocaine or cocaine methiodide (MI) following 14 days of cocaine or saline self-administration training. Solid lines indicate data from animals previously trained to self-administer cocaine HCl; dashed lines indicate data from control animals, given similar training sessions with saline rather than cocaine. Data expressed as actual concentration; the insert expresses the same data as a percent change from pre-test baseline. Analysis of variance (ANOVA) revealed significant effects of Group (F5,36=8.56, P<0.0001), time (F3,108=35.75, P<0.0001), and Group × Time interaction (F15,108=2.10, P<0.02). Leptin levels were significantly lower in cocaine-trained than in saline-trained groups across all corresponding time points (P<0.01) and were significantly decreased 2 h into sessions (P<0.05) when animals were trained with cocaine HCl (solid lines) or received yoked cocaine HCl injections during the test sessions (dashed orange line). N=6–8 per group.
The difference in prior-to-session leptin levels seen between cocaine-trained and cocaine-naive (saline-trained) animals on Day 15 developed progressively over the first 14 days of training (Figure 2a). Prior-to-session leptin levels were similarly depressed in animals switched to extinction testing on Day 15 (where non-rewarding saline was substituted for rewarding cocaine); the prior-to-session baseline returned to normal baseline over the 14 days of subsequent extinction testing (Figure 2b). We subsequently took blood samples before testing in two groups of animals that were housed in the testing chambers with their IV lines connected for remote blood withdrawals under two additional conditions. One group was trained at the same time (1100 hours) every day. Their plasma leptin levels were normal 2 h before testing time, but fell by 40% as the regular testing time approached (Figure 2c). The comparison group was tested at irregular times from day to day, and their plasma leptin levels remained normal up until the start of daily testing (Figure 2c).

Plasma leptin levels at various times relative to cocaine self-administration training. (a) Pre-test leptin levels decreased progressively across the 14 days of cocaine self-administration training (F5,32=7.23, P<0.01). (b) Plasma leptin levels were depressed before and further depressed during the first day of extinction testing but recovered by the fourteenth day of such testing (main effect of Group F1,12=13.41, P<0.01). (c) Plasma leptin levels decreased progressively over the 2 h before regular but not irregular daily self-administration sessions (main effect of Group: F1,14=4.57, P<0.05; Group × Time interaction: F3,42=3.07, P<0.05). (d) Cumulative intake was normal by 7 h after the sessions (main effect of group: F1,35=0.88, P=0.36, n.s.). (e) Cocaine-trained and saline-trained animals gained weight equally over the training period.
Central Leptin Signaling
The depressed plasma leptin levels just before the time of regular cocaine self-administration sessions were accompanied by decreased central leptin signaling. Phosphorylated STAT3 protein, a marker for the primary signaling cascade associated with leptin receptor activation, was depressed in ventral tegmental tissue punches taken just before the normal time of testing but was normal several hours later, after the self-administered cocaine was metabolized and feeding had resumed; Akt, a marker for one of the other signaling cascades did not differ between conditions (Figure 3).

Effects of cocaine self-administration on ventral tegmental area (VTA) STAT3 and Akt activities. Samples were taken just before or 7 h after daily training sessions. Analysis of variance for the phosphorylated STAT3 protein revealed significant main effects of training history (F1,24=5.29, P<0.05) and time (F1,24=6.94, P<0.02); post hoc comparisons revealed a significant effect only for phosphorylated STAT3 (P<0.01) before self-administration testing. N=7–8 per group.
Effects of a Leptin Agonist and an Antagonist on Cocaine-Seeking
Neither i.v. nor intraventral tegmental leptin infusion affected cocaine-taking per se (Figure 4a). Cocaine-seeking was, however, depressed by leptin on the first day when non-rewarding saline was unexpectedly substituted for rewarding cocaine (Figure 4b).

Effects of intravenous (0.6 mg/kg: left panels) or intra-ventral tegmental area (VTA; 0.5 μg/side: right panels) leptin on lever pressing for intravenous cocaine (‘taking,’ top panels) or for saline- and cocaine-associated cues in unexpected extinction testing (‘seeking,’ bottom panels). Note that cocaine-taking scores are for 4-h self-administration sessions, whereas cocaine-seeking scores are for 2-h extinction sessions. (a) There was no significant effect of either leptin treatment on lever pressing for cocaine (F1,11=9.48, P<0.02 and F1,11=19.53, P<0.001, respectively). (b) Leptin by either route of injection significantly decreased lever pressing for cocaine-predictive cues in the extinction tests (F1,11=9.48, P<0.02 and F1,11=19.53, P<0.001, respectively).
Leptin infusions into the VTA attenuated cocaine-conditioned place preference in a dose orderly manner (Figure 5a). The effects of leptin were blocked by the leptin antagonist Allo-aca, which did not alter the effectiveness of cocaine when given on its own (Figure 5a). The synthetic leptin agonist DesI2-E1/aca also blocked cocaine-conditioned place preference (Figure 5a). The leptin infusions elevated phosphorylated STAT3 protein levels in the VTA (Figure 5b) and attenuated the cocaine-induced elevations of nucleus accumbens dopamine (Figure 5c). The ability of leptin to elevate phosphorylated STAT3 levels and to counter the dopamine-elevating effects of cocaine were attenuated by Allo-aca, which had no effect on its own (Figure 5b and c).

Effects of intra-ventral tegmental area (VTA) leptin agonists and antagonists on conditioned place preference and neurochemical effects of intraperitoneal cocaine (10 mg/kg). (a) Leptin and the leptin agonist DesI2-E1/aca (0.01 μg/side) blocked cocaine-conditioned place preferences (F6,40=9.45, P<0.0001). (b) Leptin increased P-STAT3 but not STAT3 (F6,40=9.45, P<0.0001) in rats given similar cocaine injections. (c) Cocaine (C) elevated nucleus accumbens dopamine levels (F10,190=46.56, P<0.0001); the effect was significantly attenuated by leptin (L) pretreatment (F30,190=2.30, P<0.001). Antag=the leptin antagonist Allo-aca (0.25 μG/side); Agonist=the synthetic leptin agonist DesI2-E1/aca (0.01 μg/side). Color coding is consistent; protein and dopamine levels were assessed only in the group receiving the leptin dose of 0.25 μg/side. Starred groups were significantly different from the aCSF comparison condition (Newman–Keuls, P<0.05).
Food Intake, Body Weight, and Fat Pad Measures
Neither food intake measured across various time points following the last training session (Figure 2d) nor body weights measured across the 14 days of training (Figure 2e) differed significantly between cocaine-trained and saline-trained animals. Fat pad weights, measured from three major body deposits, also failed to differ—despite the differences in plasma leptin levels at the time of being killed—between cocaine-trained and saline-trained rats (Table 1). Injection sites and probe placements are shown in Figure 6.

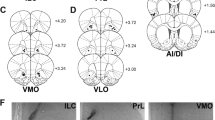
Locations of injector tips and microdialysis probes. (a) Ventral tegmental area (VTA) leptin injections during cocaine self-administration, (b) during saline substitution, (c) during cocaine-conditioned place preference, or (d) for STAT3 protein level determination. Injector tips terminated mainly in the parabrachial pigmented and paranigral nuclei, and occasionally the rostral region of interpeduncular nucleus. Two rats in c were excluded for bad placements, one in red nucleus and one in substantia nigra. Dialysis membranes (e) usually penetrated the core and ventral shell regions of nucleus accumbens. The number of dots for injector tips and of lines for dialysis membranes is less than the number of animals because of overlap in the placements. Drawings were adapted from the atlas of Paxinos and Watson (2013). Numbers on sides of the drawings indicate the distance (in millimeters) relative to the bregma.
Discussion
These studies reveal reciprocal antagonistic effects between cocaine and leptin. Leptin inhibited the mesolimbic dopamine system—the target of cocaine’s rewarding effects (Wise, 2004)—reducing the elevation of nucleus accumbens dopamine by cocaine, the rewarding effects of cocaine, and the effectiveness of cocaine-predictive cues in maintaining cocaine seeking. Conversely, cocaine and cocaine expectancy depressed plasma leptin levels. The effects of leptin on cocaine reward were expected on the basis of earlier evidence that leptin attenuates food reward (Figlewicz et al, 2001, 2004, 2006) as well as the rewarding effects of hunger-related (but not hunger-insensitive) brain stimulation (Fulton et al, 2000, 2006b).
Leptin acts at the long form of the leptin receptor (LepRb), which is expressed by diverse cell types in many brain regions (Scott et al, 2009), including the VTA, where they are expressed by dopamine neurons (Figlewicz et al, 2003; Hommel et al, 2006; Leshan et al, 2010). Leptin’s direct actions on reward function are thought to involve the mesolimbic dopamine system: leptin inhibits the firing of ventral tegmental dopamine neurons by mechanisms involving each of three intracellular signaling pathways linked to LepRb (Hommel et al, 2006; Trinko et al, 2011; Thompson and Borgland, 2013). Leptin actions in the hypothalamus can also affect reward function and the dopamine system (Leinninger et al, 2009). Leptin effects on the dopamine system appear to be modulated by the physiological state; intracerebroventricular leptin decreases both basal and feeding-induced nucleus accumbens dopamine levels in food-deprived rats (Krugel et al, 2003), whereas systemic leptin fails to alter nucleus accumbens dopamine levels in sated rats (Perry et al, 2010).
In contrast to our finding that leptin antagonized the elevation of dopamine levels by cocaine, Perry et al (2010) found that systemic leptin enhanced the elevation of dopamine levels by amphetamine (Perry et al, 2010). One possibility is that the differences between the cocaine and amphetamine effects resulted from differences in the states of the animals; whereas Perry et al, 2010 tested animals in the normal state, we tested our animals at a time when plasma leptin levels were depressed by 40% as a result of the animals’ cocaine self-administration history. Another possibility is that the differences in leptin effect are due to the different mechanisms of action of amphetamine and cocaine; whereas amphetamine causes impulse-independent release of dopamine (Sulzer et al, 2005), cocaine elevates dopamine levels by impulse-dependent blockade of dopamine reuptake (Heikkila et al, 1975). A third possibility is that the differences are due to route of administration; Perry et al (2010) gave leptin systemically, where it would act at several central sites, whereas our effects were caused by leptin injections targeted to the VTA.
Our finding that PSTAT3 was depressed with depressed plasma leptin levels confirms that ventral tegmental JAK-STAT signaling was sensitive to the plasma leptin depressions seen in our animals; it does not, however, rule out the local involvement of other signaling pathways. The actions of leptin are multiple and complex and the inhibition of dopamine neurons in normal animals is reported to be influenced by leptin-induced signaling in each of three pathways: the JAK-STAT pathway (Hommel et al, 2006; Morton et al, 2009), the MAPK pathway (Trinko et al, 2011), and the PI3K pathway (Thompson and Borgland, 2013). The relative importance of these pathways for leptin signaling appears to depend on the animal’s state of energy balance (Krugel et al, 2003; Perry et al, 2010) and may be affected by cocaine experience. The involvement of the PI3K pathway in the inhibition of dopaminergic activity involves the presynaptic inhibition of glutamatergic inputs to the dopamine system (Thompson and Borgland, 2013) and glutamate input to the dopamine system is known to be altered by cocaine self-administration training (You et al, 2007; Wang et al, 2005, 2013). Finally, there may have been undetected changes in Akt—particularly if they were localized to presynaptic terminal segments—in our homogenized tissue samples.
In our conditioned place preference experiment, the leptin antagonist did not affect dopamine levels or acquisition of preference when given by itself. This finding may be unique to our testing conditions; the place preference study was conducted in cocaine-trained animals, at the time of day previously associated with cocaine availability, and thus the antagonist was given to animals that already had severely depressed leptin levels. This might account for the animals’ insensitivity to further leptin antagonism.
The decrease in leptin levels was not secondary to major differences in food intake or body weight. As has been our experience in studies of rats given limited (4 h per day) access to i.v. cocaine, the animals in the present study ate and gained weight normally. Food was not available during the sessions and was depressed during the period of cocaine intoxication following the sessions; however, cumulative intake had returned to normal within 7 h of each session and remained normal overnight (until 2 h before the next day’s testing). The decrease in leptin levels that can be attributed to within-session food deprivation was ~2% per hour; this was the degree of decrease in the saline-trained animals receiving saline as well as in those receiving cocaine MI as shown in Figure 1.
Although leptin did not affect cocaine intake itself in the present study, this was not surprising as cocaine intake is a relatively insensitive measure of cocaine reward in well-trained animals (Arnold and Roberts, 1997), one that is strongly influenced by slowly modified habit variables (Yokel and Pickens, 1976), including its own peripheral conditioned stimulus properties (Wang et al, 2013). Leptin did attenuate the establishment of cocaine-conditioned place preferences, a well-established consequence of drug reward (Tzschentke, 2007), as well as cocaine-seeking under unexpected extinction conditions, a behavior that is significantly prolonged by conditioned reinforcers (Mote and Finger, 1942). Conditioned reinforcers—reward-predictive cues such as the cocaine-associated cue light in the present study—drive the dopamine system almost immediately (Schultz, 1998; Phillips et al, 2003; Stuber et al, 2005) and have a critical role in behaviors that involve a significant delay in primary reinforcement (Spence, 1947), as is the case with i.v. cocaine (Wise and Kiyatkin, 2011).
The present experiments reveal both unconditioned and conditioned depressions of plasma leptin levels in animals regularly self-administering cocaine. One possibility is that unconditioned effect may be a simple consequence of cocaine-induced autonomic arousal; cocaine is a stressor (Moldow and Fischman, 1987; Rivier and Vale, 1987) that elevates plasma NE levels (Sofuoglu et al, 2001), and NE is known to inhibit leptin gene expression and leptin release from adipose tissue (Rayner and Trayhurn, 2001). The conditioned effects do not yield to any such simple hypothesis. First, there are two experience-dependent suppressions to be understood. One is the suppression that develops before the normal testing time in animals that are tested at the same time each day but is not seen in animals that are tested at irregular times. Regular testing began 3 h after the onset of the dark cycle; thus, light change was not an immediate trigger of leptin changes. It is possible, however, that dark onset triggered a meal or activity sequence that, in turn, led to the decrease in leptin release beginning 1–2 h before the time of normal testing. It seems most likely that the leptin fluctuations become entrained, through conditioning, to a circadian pacemaker (Amir and Stewart, 1996; Luo and Aston-Jones, 2009). The second conditioned effect is reflected in the within-session depression of leptin levels seen in the first extinction session when the animals received only the light cue and i.v. saline (Figure 2b) and also seen in the effect of yoked cocaine MI in cocaine-trained animals (Figure 1); cocaine MI is known to serve (temporarily) as a reward-predictive cue and a conditioned reward in its own right in cocaine-trained animals (Wise et al, 2008; Wang et al, 2013).
The present data add to the growing evidence that physiological state variables, including stress (Heilig and Koob, 2007) and feeding-related hormonal signals (Carroll et al, 1979; Aston-Jones et al, 2010; Dickson et al, 2011; Graham et al, 2013), can influence the motivation for drug-seeking in drug-experienced individuals. They are consistent with the emerging view that the compulsive overeating of sweet and high-fat foods is under the influence of some of the same mechanisms as are involved in addiction (Wise, 1988, 2001; Volkow and Wise, 2005; Kenny, 2011). To the degree that release of feeding- and stress-related hormones is altered by repeated drug use (Housová et al, 2005; Heilig and Koob, 2007), therapies that have been developed in relation to excessive food intake may prove useful in treating the critical early stage of drug detoxification, a stage when the probability of relapse is high and when the effects of drug-associated cues is considerable (Gawin and Kleber, 1986).
Funding and disclosure
This study was funded by the Intramural Research Program of the National Institute on Drug Abuse.
References
al'Absi M, Hooker S, Fujiwara K, Kiefer F, von der Goltz C, Cragin T et al (2011). Circulating leptin levels are associated with increased craving to smoke in abstinent smokers. Pharmacol Biochem Behav 97: 509–513.
Amir S, Stewart J (1996). Resetting of the circadian clock by a conditioned stimulus. Nature 379: 542–545.
Arnold JM, Roberts DC (1997). A critique of fixed and progressive ratio schedules used to examine the neural substrates of drug reinforcement. Pharmacol Biochem Behav 57: 441–447.
Aston-Jones G, Smith RJ, Sartor GC, Moorman DE, Massi L, Tahsili-Fahadan P et al (2010). Lateral hypothalamic orexin/hypocretin neurons: a role in reward-seeking and addiction. Brain Res 1314: 74–90.
Carroll ME, France CP, Meisch RA (1979). Food deprivation increases oral and intravenous drug intake in rats. Science 205: 319–321.
Davis JF, Choi DL, Schurdak JD, Fitzgerald MF, Clegg DJ, Lipton JW et al (2011). Leptin regulates energy balance and motivation through action at distinct neural circuits. Biol Psychiatry 69: 668–674.
Dickson SL, Egecioglu E, Landgren S, Skibicka KP, Engel JA, Jerlhag E (2011). The role of the central ghrelin system in reward from food and chemical drugs. Mol Cell Endocrinol 340: 80–87.
Dobi A, Margolis EB, Wang HL, Harvey BK, Morales M (2010). Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J Neurosci 30: 218–229.
Domingos AI, Vaynshteyn J, Voss HU, Ren X, Gradinaru V, Zang F et al (2011). Leptin regulates the reward value of nutrient. Nat Neurosci 14: 1562–1568.
Figlewicz DP, Bennett J, Evans SB, Kaiyala K, Sipols AJ, Benoit SC (2004). Intraventricular insulin and leptin reverse place preference conditioned with high-fat diet in rats. Behav Neurosci 118: 479–487.
Figlewicz DP, Bennett JL, Naleid AM, Davis C, Grimm JW (2006). Intraventricular insulin and leptin decrease sucrose self-administration in rats. Physiol Behav 89: 611–616.
Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG (2003). Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res 964: 107–115.
Figlewicz DP, Higgins MS, Ng-Evans SB, Havel PJ (2001). Leptin reverses sucrose-conditioned place preference in food-restricted rats. Physiol Behav 73: 229–234.
Figlewicz DP, MacDonald Naleid A, Sipols AJ (2007). Modulation of food reward by adiposity signals. Physiol Behav 91: 473–478.
Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN et al (2006a). Leptin regulation of the mesoaccumbens dopamine pathway. Neuron 51: 811–822.
Fulton S, Woodside B, Shizgal P (2000). Modulation of brain reward circuitry by leptin. Science 287: 125–128.
Fulton S, Woodside B, Shizgal P (2006b). Potentiation of brain stimulation reward by weight loss: evidence for functional heterogeneity in brain reward circuitry. Behav Brain Res 174: 56–63.
Gawin FH, Kleber HD (1986). Abstinence symptomatology and psychiatric diagnosis in cocaine abusers. Clinical observations. Arch Gen Psychiatry 43: 107–113.
Geisler S, Derst C, Veh RW, Zahm DS (2007). Glutamatergic afferents of the ventral tegmental area in the rat. J Neurosci 27: 5730–5743.
Graham DL, Erreger K, Galli A, Stanwood GD (2013). GLP-1 analog attenuates cocaine reward. Mol Psychiatry 18: 961–962.
Heikkila RE, Orlansky H, Cohen G (1975). Studies on the distinction between uptake inhibition and release of (3H)dopamine in rat brain tissue slices. Biochem Pharmacol 24: 847–852.
Heilig M, Koob GF (2007). A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci 30: 399–406.
Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB et al (2006). Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51: 801–810.
Housová J, Wilczek H, Haluzík MM, Kremen J, Krízová J, Haluzík M (2005). Adipocyte-derived hormones in heroin addicts: the influence of methadone maintenance treatment. Physiol Res 54: 73–78.
Kalivas PW (2009). The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10: 561–572.
Kenny PJ (2011). Common cellular and molecular mechanisms in obesity and drug addiction. Nat Rev Neurosci 12: 638–651.
Kiefer F, Jahn H, Otte C, Demiralay C, Wolf K, Wiedemann K (2005). Increasing leptin precedes craving and relapse during pharmacological abstinence maintenance treatment of alcoholism. J Psychiatry Res 39: 545–551.
Koob GF, Le Moal M (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24: :97–129.
Krugel U, Schraft T, Kittner H, Kiess W, Illes P (2003). Basal and feeding-evoked dopamine release in the rat nucleus accumbens is depressed by leptin. Eur J Pharmacol 482: 185–187.
Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG et al (2009). Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab 10: 89–98.
Leshan RL, Opland DM, Louis GW, Leinninger GM, Patterson CM, Rhodes CJ et al (2010). Ventral tegmental area leptin receptor neurons specifically project to and regulate cocaine- and amphetamine-regulated transcript neurons of the extended central amygdala. J Neurosci 30: 5713–5723.
Liu J, Perez SM, Zhang W, Lodge DJ, Lu XY (2011). Selective deletion of the leptin receptor in dopamine neurons produces anxiogenic-like behavior and increases dopaminergic activity in amygdala. Mol Psychiatry 16: 1024–1038.
Luo AH, Aston-Jones G (2009). Circuit projection from suprachiasmatic nucleus to ventral tegmental area: a novel circadian output pathway. Eur J Neurosci 29: 748–760.
Lüscher C, Malenka RC (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69: 650–663.
Moldow RL, Fischman AJ (1987). Cocaine induced secretion of ACTH, beta-endorphin, and corticosterone. Peptides 8: 819–822.
Morton GJ, Blevins JE, Kim F, Matsen M, Figlewicz DP (2009). The action of leptin in the ventral tegmental area to decrease food intake is dependent on Jak-2 signaling. AJ Physiol Endocrinol Metab 297: E202–E210.
Mote FA, Finger FW (1942). Exploratory drive and secondary reinforcement in the acquisition and extinction of a simple running response. J Exp Psychol 31: 57–68.
Nestler EJ (1992). Molecular mechanisms of drug addiction. J Neurosci 12: 2439–2450.
Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG Jr, Schwartz MW (2001). Key enzyme in leptin-induced anorexia. Nature 413: 794–795.
Otvos L Jr, Kovalszky I, Riolfi M, Ferla R, Olah J, Sztodola A et al (2011). Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. European journal of cancer 47: 1578–1584.
Otvos L Jr, Terrasi M, Cascio S, Cassone M, Abbadessa G, De Pascali F et al (2008). Development of a pharmacologically improved peptide agonist of the leptin receptor. Biochimica et biophysica acta 1783: 1745–1754.
Paxinos G, Watson C (2013). The rat brain in stereotaxic coordinates, 7 Edition. New York: Academic Press.
Perry ML, Leinninger GM, Chen R, Luderman KD, Yang H, Gnegy ME et al (2010). Leptin promotes dopamine transporter and tyrosine hydroxylase activity in the nucleus accumbens of Sprague-Dawley rats. J Neurochem 114: 666–674.
Phillips PE, Stuber GD, Heien ML, Wightman RM, Carelli RM (2003). Subsecond dopamine release promotes cocaine seeking. Nature 422: 614–618.
Rayner DV, Trayhurn P (2001). Regulation of leptin production: sympathetic nervous system interactions. J Mol Med 79: 8–20.
Rivier C, Vale W (1987). Cocaine stimulates adrenocorticotropin (ACTH) secretion through a corticotropin-releasing factor (CRF)-mediated mechanism. Brain Res 422: 403–406.
Saal D, Dong Y, Bonci A, Malenka RC (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582.
Schultz W (1998). Predictive reward signal of dopamine neurons. J Neurophysiol 80: 1–27.
Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM et al (2009). Leptin targets in the mouse brain. J Comp Neurol 514: 518–532.
Sofuoglu M, Nelson D, Babb DA, Hatsukami DK (2001). Intravenous cocaine increases plasma epinephrine and norepinephrine in humans. Pharmacol Biochem Behav 68: 455–459.
Spence KW (1947). The role of secondary reinofrcement in delayed reward learning. Psychol Rev 54: 1–8.
Stuber GD, Wightman RM, Carelli RM (2005). Extinction of cocaine self-administration reveals functionally and temporally distinct dopaminergic signals in the nucleus accumbens. Neuron 46: 661–669.
Sulzer D, Sonders MS, Poulsen NW, Galli A (2005). Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol 75: 406–433.
Thompson JL, Borgland SL (2013). Presynaptic leptin action suppresses excitatory synaptic transmission onto ventral tegmental area dopamine neurons. Biol Psychiatry 73: 860–868.
Trinko R, Gan G, Gao XB, Sears RM, Guarnieri DJ, DiLeone RJ (2011). Erk1/2 mediates leptin receptor signaling in the ventral tegmental area. PLoS One 6: e27180.
Tzschentke TM (2007). Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol 12: 227–462.
Volkow ND, Wise RA (2005). How can drug addiction help us understand obesity? Nat Neuroscience 8: 555–560.
Wang B, Shaham Y, Zitzman D, Azari S, Wise RA, You ZB (2005). Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci 25: 5389–5396.
Wang B, You ZB, Oleson EB, Cheer JF, Myal S, Wise RA (2013). Conditioned contribution of peripheral cocaine actions to cocaine reward and cocaine-seeking. Neuropsychopharmacology 38: 1763–1769.
Wise RA (1988). The neurobiology of craving: implications for understanding and treatment of addiction. J Abnorm Psychol 97: 118–132.
Wise RA (2001) Neural circuitry of desire. In: Hetherington M (ed). Food Cravings and Addiction. Leatherhead Press: London, pp 33–58.
Wise RA (2004). Dopamine, learning and motivation. Nat Rev Neurosci 5: 483–494.
Wise RA, Kiyatkin EA (2011). On the speed of cocaine. Nat Rev Neurosci 12: 700.
Wise RA, Wang B, You ZB (2008). Cocaine serves as a peripheral interoceptive conditioned stimulus for central glutamate and dopamine release. PLoS One 3: e2846.
Yokel RA, Pickens R (1976). Extinction responding following amphetamine self-administration: determination of reinforcement magnitude. Physioll Psychol 4: 39–42.
You ZB, Wang B, Zitzman D, Azari S, Wise RA (2007). A role for conditioned ventral tegmental glutamate release in cocaine seeking. J Neurosci 27: 10546–10555.
You ZB, Wang B, Zitzman D, Wise RA (2008). Acetylcholine release in the mesocorticolimbic dopamine system during cocaine seeking: conditioned and unconditioned contributions to reward and motivation. J Neurosci 28: 9021–9029.
Acknowledgements
We thank Stephanie Myal, Stephan Steidl, and Maggie Bruck for comments on earlier versions of the manuscript and Stephanie Myal for help with the surgeries and Jeanne Pieper for training and testing of animals.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
You, ZB., Wang, B., Liu, QR. et al. Reciprocal Inhibitory Interactions Between the Reward-Related Effects of Leptin and Cocaine. Neuropsychopharmacol 41, 1024–1033 (2016). https://doi.org/10.1038/npp.2015.230
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.230
This article is cited by
-
Involvement of the ghrelin system in the maintenance and reinstatement of cocaine-motivated behaviors: a role of adrenergic action at peripheral β1 receptors
Neuropsychopharmacology (2022)
-
Involvement of the ghrelin system in the maintenance of oxycodone self-administration: converging evidence from endocrine, pharmacologic and transgenic approaches
Molecular Psychiatry (2022)
-
A role for leptin and ghrelin in the augmentation of heroin seeking induced by chronic food restriction
Psychopharmacology (2020)
-
The dopamine motive system: implications for drug and food addiction
Nature Reviews Neuroscience (2017)
-
Changes in gene expression and sensitivity of cocaine reward produced by a continuous fat diet
Psychopharmacology (2017)
