
Abstract
Fear promotes adaptive responses to threats. However, when the level of fear is not proportional to the level of threat, maladaptive fear-related behaviors characteristic of anxiety disorders result. Post-traumatic stress disorder develops in response to a traumatic event, and patients often show sensitized reactions to mild stressors associated with the trauma. Stress-enhanced fear learning (SEFL) is a rodent model of this sensitized responding, in which exposure to a 15-shock stressor nonassociatively enhances subsequent fear conditioning training with only a single trial. We examined the role of corticosterone (CORT) in SEFL. Administration of the CORT synthesis blocker metyrapone prior to the stressor, but not at time points after, attenuated SEFL. Moreover, CORT co-administered with metyrapone rescued SEFL. However, CORT alone without the stressor was not sufficient to produce SEFL. In these same animals, we then looked for correlates of SEFL in terms of changes in excitatory receptor expression. Western blot analysis of the basolateral amygdala (BLA) revealed an increase in the GluA1 AMPA receptor subunit that correlated with SEFL. Thus, CORT is permissive to trauma-induced changes in BLA function.
Similar content being viewed by others


INTRODUCTION
A single emotionally traumatic event can have a lifelong impact on the psychological well-being of an organism, including triggering anxiety-related disorders like post-traumatic stress disorder (PTSD). PTSD leads to hyperactivity to mild stressors, especially when it bears similarity to the traumatic event, and to a propensity to form new fears. PTSD patients often exhibit co-morbid depression and high rates of alcohol and drug abuse (Brady et al, 2000). Exposure therapy, the major mode of treatment for anxiety disorders, is relatively ineffective in treating PTSD (Hembree et al, 2004). Given the high prevalence of PTSD and its toll on society, it is essential to understand the mechanisms by which stress induces changes in brain function, as well as the mechanisms that perpetuate long-term expression of maladaptive behavior.
We have developed an animal model of acute stress that parallels many symptoms of PTSD (Rau et al, 2005; Rau and Fanselow, 2009; Long and Fanselow, 2012). In this model, rats receive a series of 15 shocks, randomly distributed over 90 min. This experience produces behavioral changes lasting at least 90 days unabated (Rau and Fanselow, 2009). After this experience, rats acquire exaggerated contextual and cued fear in novel situations using a single shock. This reflects a permanent sensitization of fear-conditioning circuitry because it does not depend on an explicit memory of the stressful situation, does not reflect fear generalization from the stress situation, and is not affected by extinction of the stress context (Rau et al, 2005; Rau and Fanselow, 2009; Long and Fanselow, 2012). From a clinical standpoint, nonassociative sensitization of fear is particularly problematic, as it is not bound by the trauma context or stimuli specifically associated with it. Therefore, we have labeled this phenomenon stress-enhanced fear learning (SEFL).
The primary goals of this review are to first describe PTSD and how it is a form of aberrant fear learning, as well as to illustrate SEFL as a robust animal model of PTSD in terms of phenotypic, biochemical, and neuroanatomical similarities. Moreover, we report the necessary conditions for the induction and expression of SEFL because such knowledge should suggest potential treatment targets for PTSD. We first describe the necessity of corticosterone (CORT) to, in part, cause SEFL during a severe stressor. Using the CORT synthesis blocker metyrapone, we determined that CORT changes during stress are necessary for SEFL induction. Moreover, we show that SEFL expression relies on long-term CORT-dependent changes in receptors mediating excitatory neurotransmission in the basolateral amygdala (BLA), a region crucial for fear learning and memory. Lastly, we discuss future directions of PTSD research that will be necessary in order to develop appropriate treatments and potential cures.
POST-TRAUMATIC STRESS DISORDER
When a fear response is disproportionate to the severity of a threat, it can interfere with behaviors serving other adaptive functions, compromising function and fitness (Fanselow and Lester, 1988). Inappropriate fear responses in humans can result in the development of anxiety disorders (Rosen and Schulkin, 1998), including PTSD. PTSD develops in some individuals who experience a traumatic event and affects 4–7% of the United States population. Symptoms include avoiding stimuli associated with the traumatic event, constant re-experiencing of the event, and increased arousal, exhibited by exaggerated startle responses (American Psychiatric Association, 2013). In threatening situations, these symptoms are adaptive for coping with the causes of trauma (Eberly et al, 1991; Bonne et al, 2004; Charney, 2004; Christopher, 2004). For instance, avoiding stimuli associated with the traumatic event lessens the probability of encountering the threat or others like it. Lastly, hypervigilance may help increase awareness of surroundings and detect potential threats.
However, patients with PTSD lose normal daily functioning because these responses become dysfunctional and exaggerated by occurring in situations that do not demand such intense reactions. As a result, re-experiencing the event can lead to sleep disturbances, avoiding trauma-related cues can lead to a stifled life, and hypervigilance can lead to exhaustion (Eberly et al, 1991). One feature of PTSD is an exaggerated reaction to a mild stressor or reminder of the trauma, a response more suitable for the original traumatic event that is too intense for the current, normal conditions (Friedman, 1994; Bremner et al, 1995; Dykman et al, 1997). Additionally, PTSD is co-morbid with phobias and depression, and other reports have also shown that PTSD leads to a predisposition to drug and alcohol abuse (Goisman et al, 1998; van Dam et al, 2013; Dutton et al, 2013; Stander et al, 2014). Therefore, PTSD poses itself as a serious mental illness, and there is a need for developing novel and effective treatments for this disorder.
STRESS RESPONSE
Exposure to stress appears to sensitize the biological system involved in generating fear responses; therefore, it is likely that biological mediators of enhanced fear responses are stress hormones (Johnson et al, 1992; Carrasco and Van de Kar, 2003). Stress activates both the sympathetic nervous system and the neuroendocrine stress cascade, also called the hypothalamic-pituitary-adrenal (HPA) axis. The activation of these systems causes the body to undergo a set of responses that facilitate dealing with a challenge and restore homeostasis after the threat has passed. These responses include mobilizing energy and other resources to sustain the brain, heart, and muscles (ie, increased blood pressure and heart rate), preparing the immune system, enhancing cognitive functioning, and inhibiting behaviors that are currently unnecessary for survival (Johnson et al, 1992; Sapolsky, 2000; Christopher, 2004).
Activation of the HPA axis is coordinated by stress-induced activation of corticotropin-releasing hormone in the paraventricular nucleus of the hypothalamus, which causes the release of adrenocorticotropic hormone from the anterior pituitary gland. Adrenocorticotropic hormone then travels through the bloodstream and initiates glucocorticoid release from the adrenal cortex (Vale et al, 1981; Antoni, 1986; Owens and Nemeroff, 1991). Glucocorticoids (ie, cortisol in humans, corticosterone in rodents; CORT) aid in energy mobilization and help to restore homeostasis via negative feedback mechanisms after a threat has passed (Munck et al, 1984; Sapolsky et al, 2000). Glucocorticoids are lipophilic and are actively transported into the brain, where they bind to receptors or cross-neuronal cell membranes (McEwen and Weiss, 1970; De Kloet et al, 1998). They bind to either mineralocorticoid or glucocorticoid receptors (GRs), which can translocate into the nucleus and alter gene transcription; some of these mechanisms help exert negative feedback control over the stress response (McEwen and Weiss, 1970; Reichardt and Schutz, 1998).
Mimicking HPA axis activation with injections of CORT was shown to have consistent results in studies where animals were exposed to stress before fear conditioning. Both chronic and acute administration of CORT enhances fear conditioning in rats (Cordero et al, 2003; Thompson et al, 2004). Animals that were given CORT injections for 5 days before fear conditioning and animals given just a single injection of CORT after fear conditioning both showed increased conditional fear during a context test, as opposed to vehicle-treated animals. Together, these animal studies suggest a role for stress hormones in mediating stress-induced enhancement of behavioral responding.
Altered HPA axis responsiveness may contribute to the sensitized responses PTSD patients experience to innocuous events that are perceived as threatening; that is, PTSD symptoms may develop via a sensitization process involving the HPA axis that causes less intense stressors to be perceived as stronger than they are (Rasmusson and Charney, 1997; Yehuda, 1997). The initial traumatic event activates the stress response, but upon receiving reminders of the trauma or mild but similar stressors, the stress response reactivates. Repeated activation modifies the HPA axis negative feedback system and makes the stress response become more easily triggered. Sensitization results in a lowered activation threshold for subsequent stimuli, facilitating higher responding to neutral stimuli now perceived as threatening (Rosen and Schulkin, 1998; Hagemen et al, 2001). Moreover, re-experiencing aspects of the traumatic event sensitizes fear systems leading to exaggerated reactions, as observed in PTSD patients (Post et al, 1997; Hagemen et al, 2001). This may occur via sensitization of amygdalar nuclei (discussed in the next section), a process that is mediated by stress hormones (Rosen and Schulkin, 1998). Sensitized activity in fear circuitry due to repeated activation of the stress response may contribute to dysregulation of the HPA axis and perhaps to PTSD symptomatology.
NEURAL SYSTEMS INVOLVED IN FEAR LEARNING
The neural systems mediating associative fear learning are well-known, which further validates using such a procedure to model fear responses and related symptoms in PTSD (Fendt and Fanselow, 1999). During learning, sensory input relating to both the conditional stimulus (CS) and unconditional stimulus (US) converge on the BLA complex, a subregion of the amygdala, where a CS-US association is encoded via long-term potentiation at BLA synapses (Rogan et al, 1997; Kim and Jung, 2006). This plasticity is dependent upon excitatory N-methyl-D aspartate receptors (NMDAR) (Miserendino et al, 1990; Fanselow and Kim, 1994) and is modulated through inhibitory γ-aminobutyric acid (GABA)-ergic neurons (Ehrlich et al, 2009; Makkar et al, 2010). Under normal conditions, the BLA, consisting of lateral and basal nuclei, is critical for fear learning (Maren, 1998; Fanselow and LeDoux, 1999; Gale et al, 2004; McDannald and Galarce, 2011). Specifically, the BLA is necessary for encoding the memory of the US as either pleasant or aversive, as well as for storing it long-term (Fanselow and Gale, 2003; Gale et al, 2004). Indeed, functional magnetic resonance imaging studies show enhanced amygdala activity in PTSD patients during encoding and exposure to negative stimuli (Rauch et al, 2000; Shin et al, 2006; Brohawn et al, 2010).
The BLA projects to central amygdala both directly and indirectly, via a link through the intercalated cell masses that lie between these two regions (Pitkanen et al, 1997; Pare et al, 2004). Specifically, BLA neurons project to the lateral subdivision of the central amygdala, which sends GABAergic projections to the medial subdivision of the central amygdala (Haubensak et al, 2010). The fear response (ie, freezing) is controlled by projections from medial subdivision of the central amygdala to the periaqueductal grey (Fanselow, 1991). Besides the central amygdala, the BLA projects to the bed nucleus of the stria terminalis, which in turn projects to the periaqueductal grey for fear responding (Walker et al, 2003; Waddell et al, 2006).
Other important regions in the fear learning and memory network involve cortical regions such as the prefrontal cortex (Milad and Quirk, 2002; Santini et al, 2004). Descending projections of the medial prefrontal cortex modulate the behavioral outputs of this circuit—the prelimbic cortex projects to the BLA to enhance fear responding, while the infralimbic cortex indirectly projects to the medial subdivision of the central amygdala via intercalated cells to promote extinction of fear (Quirk et al, 2003). Moreover, prelimbic and infralimbic cortexes receive amygdala projections originating mainly from the basal amygdala. Infralimbic and prelimbic cortices have opposing roles in the expression of fear following extinction learning (Senn et al, 2014), which suggests that these reciprocal connections influence the outcome of fear and extinction learning. Moreover, inputs from the ventral hippocampus onto the BLA, either directly or indirectly through the prelimbic cortex, mediate contextual control of fear and fear renewal after extinction (Orsini et al, 2011).
BIOCHEMICAL SUBSTRATES OF FEAR LEARNING AND THEIR ASSOCIATED CHANGES
Within the amygdala, glutamatergic modifications in excitatory neurotransmission, including glutamate receptor-regulated synaptic plasticity, have been implicated in fear conditioning. Glutamate NMDAR and alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors (AMPAR) within the amygdala participate in different components of fear learning, including acquisition, expression, and extinction (Kim et al, 1993; Fanselow and Kim, 1994; Walker and Davis, 2002; Jasnow et al, 2004). In particular, it has been shown that blockade of NMDAR in the BLA prevented acquisition of fear learning (Miserendino et al, 1990; Fanselow and Kim, 1994). Moreover, intra-BLA infusions of an AMPAR antagonist blocked expression of fear (Kim et al, 1993). It has also been shown that altered activity patterns due to stress can change the distribution of AMPAR in the BLA, increasing the density of AMPAR on dendritic spines (Hubert et al, 2013).
The neural structures, pathways, and systems discussed in these sections are highly integrated. This is a requirement of the fear conditioning system that is advantageous to survival; the system is able to rapidly discern and encode relevant associative relationships over a multitude of environmental stimuli that could signal a major threat. Furthermore, an important survival system could not afford to rely upon a single locus of function in case of damage; there would need to be alternate and compensatory structures and pathways to continue operating. One implication of this complexity is that the temporal contiguity of the occurrence of the CS and US is not enough to establish a predictive relationship between a CS and US (Kamin, 1968; Fanselow, 1998). Instead, associative learning within a complex environment is dynamic so that CS-US associations are strengthened for selected stimuli, producing competition between flexible neural circuits for CS associations with the US (Fanselow, 2010).
STRESS-ENHANCED FEAR LEARNING MODEL
We have developed a model using fear conditioning procedures to examine how exposure to a traumatic stressor can affect future responding (Rau et al, 2005). In the SEFL procedure, animals are given a series of 15 randomized, unsignaled shocks in a distinct context. Animals are then given a single context-shock pairing in a novel context, with different grid floor, lighting, and scent from the stressor context. Animals exposed to the 15 shocks show an enhanced fear response to the single shock in the second context compared with animals that did not receive the 15 shocks. Prior to the single shock, previously stressed rats show no generalized freezing to the second context, arguing against an account of associative generalization. Rather, the effect of the 15-shock stressor appears to be nonassociative, as it occurs in a novel situation and only after the animal receives a milder version of the previous trauma (Poulos et al, 2015). Sensitization is a nonassociative process in which there is increased reactivity to a potent stimulus after repeated exposure to that stimulus, producing a lowered activation for subsequent stimulation (Groves and Thompson, 1970; Rosen and Schulkin, 1998). We believe this effect is similar to that experienced by PTSD patients in which exposure to a traumatic event causes sensitized reactions to less intense but similar stressors (Bremner et al, 1995; Dykman et al, 1997).
There are several important features of SEFL indicating that it is a very long-lasting nonassociative enhancement of fear learning. Firstly, SEFL is indeed an enduring phenomenon. We have separated the 15-shock treatment from the single shock treatment by as long as 90 days with no diminution in the enhancement of conditioning (Rau and Fanselow, 2009). The presence of symptoms at least 30 days after trauma is required for a diagnosis of PTSD; hence, the longevity of SEFL is an important factor for establishing its relevance to PTSD. The most obvious associative interpretation of SEFL is generalization between the stress and test contexts. However, there are multiple converging lines of evidence that SEFL is not mediated by generalization between the two contexts. As mentioned earlier, there is no evidence of fear in the conditioning context prior to the single shock. Additionally, eliminating fear of the stress context does not impact SEFL. For example, imposing extinction of the stressor context before conditioning, while effective in eliminating fear of the stressor context, has no impact on the enhancement of new conditioning. This may correspond to the reduced effectiveness of extinction in treating PTSD (eg, Peri et al, 2000) in that it suggests that extinguishing fear of trauma-related events will not reduce the nonassociative effects of trauma. Additionally, blocking contextual fear learning to the stressor context by delivering an amnestic agent to the hippocampus during the 15 shocks does not alter the enhancement. This may correspond to observation of PTSD in patients who have amnesia for the traumatic episode (eg, Krikorian and Layton, 1998). Furthermore, whereas we have most often looked at the enhancement in new context fear learning, the fear enhancement is also found in auditory conditioning (Rau et al, 2005). As there is no auditory cue during the 15 shocks, there is no basis for generalization to influence tone fear learning. Lastly, SEFL reflects a change in fear learning: the order of the 15 shocks and single shock matters. SEFL occurs only when the single shock conditioning is given after, not before, the stressor. If SEFL were due to summation of fear expression (or, for that matter, generalization between contexts), the order should not matter. If SEFL alters the fear learning circuit, the stressor would necessarily have to come before the single-shock conditioning, and that is what is observed.
SEFL is extremely robust. Collapsing over many experiments we have trained well over 500 rats with just a single shock in the conditioning context that have either received or did not receive the prior 15-shock treatment. Over 90% of the rats receiving the 15-shock treatment freeze more than 2 standard deviations above the mean of the rats without prior stress (eg, Rau et al, 2005; Rau and Fanselow, 2009). One could point to this robustness as a deviation from PTSD, where it is estimated that 10–25% of people who experience trauma go on to develop PTSD. However, based on previous research and in our own unpublished data, we find that if we reduce the number of pre-shocks, a smaller percentage of rats meet the 2 standard deviation criterion (Fanselow and Bolles, 1979). If we reduce the pre-shock treatment to three or four, we find only 20% of the rats develop SEFL by this criteria—which is more in line with what is observed in human trauma cases. However, we use the 15-shock stressor because it simply makes experiments more tractable and efficient when we use parameters that cause a more robust conversion to SEFL.
ANIMAL MODEL TRANSLATION TO PTSD
The SEFL model captures multiple aspects of PTSD, including exaggerated fear as seen through freezing, and blunted emotional reactivity, as measured through reactivity to the shock (the first of which will be the primary measure of the presented studies). These animal studies also show that HPA axis activation can enhance fear responding, and their results are consistent with the stress response of PTSD patients. For instance, Vietnam combat veterans diagnosed with PTSD show increased levels of corticotropin-releasing hormone in their cerebrospinal fluid (Baker et al, 1999; Bremner et al, 1997). However, there seems to be controversy about alterations in CORT levels. Both increased levels (Lemieux and Coe, 1995; Maes et al, 1998) and decreased levels (Mason et al, 1986; Pitman and Orr, 1990; Yehuda et al, 1990, 1995; Boscarino, 1996) of CORT have been found in PTSD patients compared with individuals without PTSD. The disagreement of these results suggests that a simple change in CORT levels in itself cannot explain the symptoms of the disorder. One theory is that PTSD patients experience enhanced negative feedback responding of the HPA axis during activation of the stress response, which would cause low CORT levels to be observed (Yehuda, 1997, 2001). Using the SEFL model, we discovered that one long-term consequence of exposure to the stressor is a dysregulation in the diurnal rhythm of basal CORT. Rather than having a single peak shortly after dark, rats showed a double peak once during the light cycle and once at dark. Therefore, the stressed rats were either elevated or suppressed relative to controls depending on the hour of the day at which CORT was determined. Additionally, as will be argued below, it is possible that CORT changes during stress are the critical changes for the induction of exaggerated fear and that CORT level changes at other times, such as after the trauma, are less critical to the sensitized behavior. Obviously, CORT levels are rarely taken during a traumatic episode in humans.
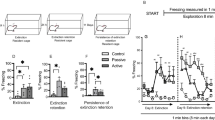
Rats exhibiting SEFL also show decreased exploratory behavior in open fields (Figure 1a and b), increased consumption of alcohol (Meyer et al, 2013), potentiated startle reactivity (Figure 1c and d), and a depression-like phenotype in the forced swim test (Figure 1e and f). Moreover, SEFL causes an anxiety profile on the elevated plus maze, a long-lasting dysregulation of the diurnal cycle for CORT, and an increase of GR in the BLA (Poulos et al, 2013). These findings show that SEFL behavior reflects several of the symptoms of PTSD (Table 1).

SEFL causes anxiety phenotype on the open field test, exaggerated startle to white noise, and a depressive-like phenotype in the forced swim test. (a) Experimental design. We developed a modified version of the open field test that has been validated for anxiety testing (Godsil and Fanselow, 2004; Godsil et al, 2005). The open field test consisted of three phases: (1) 4 min of dark, (2) 4 min of light and (3) 4 min of dark. Locomotion, defined as the number of crossovers, was quantified during the 12-min test. (b) Open field test. Pre-exposure to shock significantly decreased the number of crossovers during phase 1, the first four dark minutes of the open field, p<0.005. There was no effect of pre-exposure to shock on the number of crossovers during phase 2, minutes 5–8. Pre-exposure to shock decreased in the number of crossovers during phase 3, p<0.05. Therefore, previously shocked rats showed reduced exploratory activity than controls when placed in a dark open field. When bright lights turned on at one end, the rats retreated to the dark end and reduced activity similar to controls. However, when the lights went out, unlike controls, they remained in the dark corner and did not increase exploration. The open field test did not affect the context tests in either the stress or conditioning contexts (data not shown). (c) Experimental design for exaggerated startle test. Instead of 1-shock in conditioning context on Day 2, white noise (92 dB, 1 s) was given. (d) Freezing (+SEM) for baseline on Day 2, 5 min post-noise on Day 2, and context test on Day 3. Previously stressed rats showed a pronounced freezing response to a loud noise; this reaction was not seen in unstressed controls, **p<0.01 (mixed-factorial ANOVA). Upon return to the noise context the following day without noise, the stressed rats showed a small but statistically reliable increase in freezing relative to controls, *p<0.05. (e) Experimental design. Training (Day 4) and testing (Day 11) sessions for forced swim test were 5 min. Water temperature was 77 °F, and the apparatus dimensions are 74 cm × 36.5 cm. (f) Time spent immobilized (sec+SEM) on Day 4, and time spent immobilized (sec+SEM) on Day 11. *p<0.05 (one-way ANOVA).
STRESS-ENHANCED FEAR INDUCTION
In this section, we describe the neural and biological mechanisms necessary for stress sensitization to occur, including the role of stress hormones in the BLA. In the first experiment, we examined the role of CORT in SEFL induction. Administration of the CORT synthesis blocker metyrapone prior to the 15-shock stressor, but not at time points after, attenuated SEFL. Next, we determined the necessity of CORT in SEFL by co-administering metyrapone and CORT pre-stressor. CORT restored SEFL after metyrapone administration, but CORT alone without the 15-shock stressor was not sufficient to produce SEFL. We then examined the role of the BLA in SEFL with intra-BLA infusions of muscimol, a GABAA receptor (GABAAR) agonist, pre- or post-stressor. The BLA must be functional during the 15 shocks but not after in order for SEFL to occur. Lastly, to determine whether CORT’s action was on the BLA, we infused the GR antagonist mifepristone directly into the BLA immediately prior to the 15-shock stressor, which attenuated SEFL. The data from these experiments indicate that CORT activation of GRs in the BLA is necessary for SEFL induction.
Pre-Stressor Administration of Metyrapone Attenuates SEFL
A total of 102 experimentally naïve adult male Long Evans rats, approximately 300 g, were used in this experiment. Rats were randomly assigned to one of the two groups: those receiving 15 shocks randomly distributed over a 90-min period in the stress context (‘stressed’) and those who remained in the chambers of this context for the same duration without receiving any shocks (‘unstressed’). Both groups were composed of four subgroups: 1 h prior to the trauma, animals were given a 1.0 ml/kg intraperitoneal injection of either vehicle (of 60% saline and 40% propylene glycol) (Sigma Aldrich, St. Louis, MO), 50 mg/kg metyrapone, 100 mg/kg metyrapone, or 150 mg/kg metyrapone (Tocris Bioscience, Ellisville, MO). All groups had an n of 12, except for stressed/vehicle (n=14) and unstressed/vehicle (n=16). Animals receiving no stress during exposure were placed in chambers for an equivalent amount of time as the stressed animals—90 min. Immediately after the stressor, animals were placed in a restraining tube for a maximum of 5 min and approximately 0.5 ml of tail blood was collected into heparinized tubes. Animals were then brought back to their homecages and returned to their housing area. CORT plasma levels were measured by enzyme immunoassay (AssayPro, St Charles, MO; Correlate-EIA corticosterone enzyme immunoassay kit).
Animals then underwent the rest of the 7-day SEFL procedure (see Figure 2a). Forty-five minutes after the context test on Day 7, tail blood was collected again in the same manner as was for Day 1. Freezing was recorded using the VideoFreeze program (Med Associates, St Albans, VT).

Pre-stressor administration of metyrapone attenuates SEFL, and co-administration of metyrapone and CORT restores SEFL. (a) Experimental design for metyrapone administration. (b) Plasma CORT levels (ng/ml; mean+SEM) after the 15-shock trauma on Day 1. Main effect of stress, p<0.001; linear trend analysis for stressed animals, p<0.05 (two-way ANOVA, followed by planned contrasts). (c) The mean+SEM of freezing percentage in stress context test on Day 4. Drug × stress, p<0.0001 (two-way ANOVA), linear trend analysis for stressed animals, p<0.0001. (d) The mean+SEM of freezing percentage during the generalization test in the conditioning context on Day 5. No significant differences were found for any main effect or interaction (two-way ANOVA). (e) Freezing (mean+SEM) in conditioning context on Day 7. Drug × stress interaction, p<0.05, linear trend analysis for stressed animals, p<0.0005 (two-way ANOVA, followed by planned contrasts). (f) Experimental design for metyrapone and CORT co-administration. (g) Freezing (mean+SEM) in the stress context on Day 4. First digit in group designations=metyrapone dose (0 or 150 mg/kg); second=CORT dose (0 or 10 mg/kg). ***p<0.0001 (overall one-way ANOVA, followed by planned contrasts). (h) Freezing (mean+SEM) in conditioning context on Day 7. First digit in group designations=metyrapone dose (0 or 150 mg/kg); second=CORT dose (0 or 10 mg/kg). **p<0.01; ***p<0.0001 (overall one-way ANOVA, followed by planned contrasts).
Two-way analysis of variance (ANOVA) was used to analyze percent freezing as influenced by both drug dose and stress condition during Day 4 in the stress context and Days 5 and 7 in the conditioning context, as well as to compare plasma CORT levels. Moreover, trend analysis was performed to determine whether the data follow linear dose-dependent functions.
As expected, pre-stress administration of metyrapone dose-dependently reversed the stress-induced rise in plasma CORT levels after the 15-shock stressor on Day 1 (Figure 2b). Overall, stressed animals had higher CORT levels than did unstressed animals, F (1, 94)=11.509, p<0.001. Trend analysis showed that in stressed but not unstressed animals, metyrapone linearly dose-dependently decreased plasma CORT levels after the 15-shock stressor on Day 1 in a linear manner, F (1, 94)=5.19, p<0.05. However, CORT levels during the Day 7 context test showed no reliable group differences (data not shown; p’s>0.05).
The Day 4 stress context test showed that metyrapone given pre-stressor dose-dependently blocked fear conditioning to this context (Figure 2c); there was a significant interaction between drug dose and stress treatment, F (3, 94)=17.236, p<0.0001; trend analysis showed a significant linear dose-dependent function of metyrapone in stressed animals, F (1, 94)=106.4, p<0.0001. The low freezing levels during the Day 5 test in the novel context showed that no groups generalized to this context (Figure 2d; p’s>0.05).
The critical behavioral data are the freezing levels during the Day 7 conditioning context test (Figure 2e); all animals received one footshock in this novel context 24 h earlier. A significant interaction between drug dose and stress treatment was found, F (3, 94)=2.957, p<0.05, and trend analysis indicated that pre-stress metyrapone dose-dependently decreased freezing in stressed animals in a linear manner, F (1, 94)=12.796, p<0.0005. However, metyrapone had no effect on freezing for unstressed animals (p>0.05).
These data demonstrate that the 15-shock stressor enhanced later fear conditioning to the single shock context (ie, SEFL occurred), and, importantly, that blocking CORT increases during the stressor subsequently blocked SEFL (Figure 2e). We also administered the 150 mg/kg dose of metyrapone immediately after the 15-shock stressor, before the single shock, or before the test and did not see an effect at any of those time points (data not shown), which supports our finding that CORT changes 7 days post-stressor were not reliably different. Moreover, CORT measurement in PTSD patients is typically taken long after the trauma, not immediately after, which may explain the inconsistencies in reported CORT levels in PTSD patients (Yehuda et al, 1990; Maes et al, 1998). This is not to say that CORT is not affected long-term by trauma, as we do see a disturbance in the circadian rhythm of basal CORT (Poulos et al, 2013). Therefore, increases in CORT specifically at the time of stress are critical for SEFL.
Co-Administration of Metyrapone and CORT Rescues SEFL
In order to confirm that metyrapone’s effect was mediated by its action on CORT, we determined whether co-administration of CORT restores sensitized fear after metyrapone injection. Additionally, we determined whether CORT administration in the absence of shock was sufficient to produce SEFL. A total of 116 experimentally naïve adult male Long Evans rats, approximately 300 g, were used in this study. Animals were given either a pre-training injection of 150 mg/kg metyrapone or vehicle in the rats’ housing area. Either 10 mg/kg CORT (Sigma-Aldrich) or vehicle (15% alcohol/85% saline) was injected 10 min prior to the 15-shock stressor. The rats then underwent the 7-day SEFL procedure. There were a total of eight groups, and n per group ranged between 12 and 17 (see Figure 2f).
A one-way ANOVA was performed to determine significant freezing differences during both context tests. A priori planned comparisons were also made to determine whether CORT alone could induce SEFL in unstressed animals, and if CORT can rescue SEFL from stressed animals that received metyrapone.
During the Day 4 stress context test, there was an overall difference between groups, F (7, 108)=36.355, p<0.0001 (Figure 2g). The freezing levels for stressed/vehicle animals (Group 0-0) were significantly higher than for unstressed/vehicle animals (Group 0-0), F (1, 108)=60.54, p<0.0001. The latter group showed similar freezing levels to unstressed controls, p>0.05. Stressed/vehicle rats froze significantly more than did stressed/metyrapone/vehicle animals (Group 150-0), F (1, 108)=47.43, p<0.0001, which showed similar freezing levels to both stressed/metyrapone/CORT animals (Group 150-10) and unstressed controls, p’s>0.05.
Like the first metyrapone experiment, no groups generalized to the second conditioning context on Day 5 (data not shown, p’s>0.05). On Day 7, there were reliable freezing differences between groups as analyzed by an overall one-way ANOVA, F (7,108)=6.619, p<0.0001 (Figure 2h). A priori planned comparisons indicated that the freezing levels of the stressed/vehicle animals were significantly higher than those of unstressed/vehicle animals receiving CORT (Group 0-10), F (1, 108)=11.891, p<0.001; this means that CORT alone does not induce sensitized fear. Although CORT without stress did not generate SEFL, CORT did restore SEFL in stressed rats that received metyrapone (Group 150-10). These animals froze significantly more than did their unstressed counterparts, F (1, 108)=7.297, p<0.01.
Whereas metyrapone prevented SEFL, CORT administration alone did not mimic the effect of stress (Figure 2h). However, SEFL was restored with CORT after metyrapone injection. Thus, changes in CORT are necessary but not sufficient for producing SEFL. This differs from the results from the Day 4 stress context test, where co-administration of CORT and metyrapone did not rescue freezing, showing a further dissociation of associative and nonassociative fear. Having metyrapone on board during the 15-shock stressor may have disrupted consolidation of the memory of the 15 shocks, but perhaps having enhanced CORT on board during the stressor creates enough of a state change to enhance later nonassociative freezing. CORT seems to have a permissive role critical to SEFL-inducing changes elsewhere. Additionally, CORT’s restoration of SEFL indicates that metyrapone’s effect is likely caused by its ability to block CORT synthesis.
BLA Inactivation Reduces SEFL
We next sought to find important brain regions for SEFL and targeted the BLA, a brain region crucial for fear conditioning and emotional learning in animals (Miserendino et al, 1990; Fanselow and Kim, 1994; Rogan et al, 1997; McKernan and Shinnick-Gallagher, 1997; Muller et al, 1997; Walker and Davis, 2002). Indeed, PTSD patients show enhanced amygdala activity during encoding and exposure to negative stimuli (Shin et al, 2006; Brohawn et al, 2010). Furthermore, the BLA has an important role in stress-induced fear responses (Kim et al, 2001; Adamec et al, 2005; Rodriguez Manzanares et al, 2005) and strikingly, animals with SEFL show long-lasting changes in gene expression in the BLA (Ponomarev et al, 2010).
Therefore, in order to determine the role of the BLA in SEFL, we inactivated the BLA using the GABAAR agonist muscimol (Sigma-Aldrich) either prior to or after the 15-shock stressor (see Figure 3a). All animals underwent surgery to implant guide cannulae into the BLA 10–14 days before the start of experiments. Briefly, rats were anesthetized (isoflurane: induction at 5%, maintenance 2.5%) before stereotaxic mounting (Kopf Instruments, Tujunga, CA). Two holes were drilled into the skull for implantation of 26-gauge guide cannulae (Plastics One, Roanoke, VA, USA) aimed bilaterally at the BLA; coordinates (from bregma) were: anterior/posterior −3.1 mm, medial/lateral +/− 5.2 mm, dorsal/ventral −7.6 mm. Guide cannulae were secured with dental acrylic cemented to anchoring skull screws.

Basolateral amygdala inactivation or intra-basolateral infusions of mifepristone reduces SEFL. (a) Experimental design for intra-amygdalar muscimol infusions. (b) Cannulae placement for amygdalar inactivation. (c) Freezing (mean+SEM) in the conditioning context on Day 3. **p<0.003 (one-way ANOVA, followed by planned contrasts). (d) Experimental design for mifepristone infusions. (e) Cannulae placement for mifepristone infusions. (f) Freezing (mean+SEM) in the conditioning context on Day 3. **p<0.001 (one-way ANOVA, followed by planned contrasts).
Animals were randomly assigned to one of three groups: pre-trauma muscimol animals received micro-infusions of muscimol 20 min before the 15 shocks, vehicle controls received ACSF 20 min before being placed in the stressor context without shocks, and post-trauma muscimol animals underwent the 15 shocks first, receiving micro-infusions of muscimol 45 min after the trauma. For infusions, 33-gauge injector cannulae that extend 1 mm below the guides were inserted. The drug was back-loaded via 33-gauge infusion cannulae into polyethylene tubing connected to 10 ml Hamilton micro-syringes (Hamilton Company, Reno, NV, USA), the infusion rate was 0.1 μl/min to reach a volume of 0.25 μl/side, delivered via a Harvard #22 syringe pump (Harvard Apparatus, South Natick, MA, USA). The animals then underwent a shortened 3-day SEFL procedure. Freezing in the conditioning context was recorded on Day 3. After exclusion of animals with misplaced cannulae groups consisted of: vehicle controls (n=4), pre-stressor muscimol (n=4), and post-stressor muscimol (n=4). A one-way ANOVA was performed to determine between-group differences for freezing during the context test.
Figure 3b depicts verification of correct bilateral cannulae placement on a schematic diagram. Figure 3c illustrates clear group differences during the context test in the conditioning context, confirmed statistically with a one-way ANOVA, F (2, 9)=8.17, p<0.009. A priori planned comparisons showed that post-stressor muscimol animals froze significantly more than did pre-stressor muscimol animals, F (1, 9)=13.5, p<0.003; however, pre-stressor muscimol animals did not show significantly different freezing levels from unstressed controls, p>0.05.
Pre-Stressor Intra-Amygdala Mifepristone Prevents SEFL
To determine whether CORT’s action was on the BLA, we infused the GR antagonist mifepristone directly into the BLA immediately prior to the 15-shock stressor (Figure 3d). Experimentally naïve adult male Long-Evans rats were used in this experiment. All animals underwent surgery to implant guide cannulae into the BLA 10–14 days before the start of experiments. Animals either received micro-infusions of mifepristone (Sigma-Aldrich) or vehicle (20% DMSO and 80% ACSF) 10 min before the 15 shocks, or were unstressed vehicle controls. The animals then underwent the shortened 3-day SEFL procedure. Freezing in the conditioning context was recorded on Day 3. After exclusion of animals with misplaced cannulae groups consisted of: stressed/mifepristone (n=5), stressed/ACSF (n=7), and unstressed/ACSF (n=5). A one-way ANOVA was performed to determine between-group differences for freezing during the context test.
Figure 3e shows verification of correct bilateral cannulae placement on a schematic diagram. Figure 3f shows freezing in the conditioning context test on Day 3. A one-way ANOVA confirmed statistically significant group differences in freezing, F (2, 14)=8.349, p<0.005. A priori planned comparisons showed that stressed/vehicle animals froze significantly more than did stressed/mifepristone animals, F (1, 14)=16.42, p<0.001; however, stressed/mifepristone animals did not show significantly different freezing levels from unstressed/vehicle controls, p>0.05. This indicates that CORT activation of GRs in the BLA is necessary for SEFL induction.
Summary
The experiments in this section elucidate the critical components of induction of stress-induced enhancement of fear. More specifically, a severe stressor initiates the HPA axis stress response to increase CORT levels. CORT acts centrally in the BLA, which must also be functional during the stressor, by binding to GRs. CORT synthesis blocked by metyrapone administration after the stressor did not affect fear sensitization, and CORT levels were not increased at any time point after the 15-shock stressor. The next section will address the underlying mechanisms of expression of enhanced fear.
STRESS-ENHANCED FEAR EXPRESSION
Next, to explore the potential mechanisms of SEFL expression, we looked for changes in glutamatergic receptor subunits in the BLA. Glutamatergic modifications in excitatory neurotransmission—in particular, NMDAR and AMPAR—within the amygdala participate in different components of fear learning, including acquisition, expression, and extinction (Miserendino et al, 1990; Fanselow and Kim, 1994; Walker and Davis, 2002). Increased excitatory neurotransmission in the BLA could also enhance fear conditioning (Fanselow et al, 1993).
Metyrapone Prevents a Post-Stressor GluA1 Increase
Rats received either 150 mg/kg metyrapone or vehicle 1 h before the 15-shock stressor (see Figure 4a). Two weeks after the test in the conditioning context, western blot analysis of BLA samples was performed in order to assess the relative abundance of AMPAR and NMDAR subunits in the BLA after SEFL and metyrapone treatments. Rats were sacrificed, 400-mm-thick coronal brain slices were made, from which the BLA was microdissected and immediately frozen at −80 °C. Tissue was then thawed and homogenized in ice cold buffer containing 1% SDS, 1 mM EDTA, and 10 mM Tris, pH 8.0. Protein concentrations were measured by the DC protein assay system (Bio-Rad, Hercules, CA). Samples (15 μg/lane) were loaded in 10- or 15-lane pre-cast 4 to 20% gradient SDS-polyacrylamide gels (Bio-Rad, Hercules, CA), and separated under reducing conditions using the Bio-Rad Mini–Protean 3 Cell system. Proteins were transferred to PVDF membranes (Immun-Blot PVDF membrane, 0.2 mm) by wet transfer (Bio-Rad, Hercules, CA). Blots were probed with anti-peptide GluA1 (C-terminus epitope- AB1504; 1:1000 dilution; EMD Millipore, Temecula, CA), anti-peptide GluA2 (rabbit polyclonal, AB1768-I; 1:6000 dilution; Millipore), and GluN1 (aa 834–938; 1:1000 dilution; Upstate: EMD Millipore) antibodies, followed by HRP-conjugated secondary (Goat anti-rabbit; EMD Millipore) antibody (1:2000 dilution), bands were detected by GE ECL prime or ECL2 Western blot detection kit (GE Healthcare Biosciences, Pittsburgh, PA) and images were captured using the LAS-3000 digital imaging system (Fujifilm, Tokyo, Japan) or developed onto film (GE Healthcare, Biosciences). An anti-peptide glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotech, Santa Cruz, CA) antibody (1:2000 dilution) was used as a loading control. Bands corresponding to the appropriate subunit were analyzed, and optical density measurements were compared by densitometry using ImageJ (NIH, Bethesda, MD).

Increases in GluA1 after SEFL treatment are prevented by metyrapone. (a) Experimental design. (b, d, f) Representative western blot images of GluA1, GluA2, GluN1, and GAPDH from the BLA of stressed and unstressed rats receiving vehicle or 150 mg/kg metyrapone. (c) Mean GluA1: GAPDH OD ratios (+SEM). Main effect of drug, p<0.005, **p<0.01 (one-way ANOVA, followed by a priori planned comparisons). (e) Mean GluA2: GAPDH OD ratios (+SEM). (g) Mean GluN1: GAPDH OD ratios (+SEM).
For the mean GluA1: GAPDH optical density ratios (+SEM), there was a main effect of drug F (1, 32)=9.011, p<0.005, but not of stress or stress by drug interaction (Figure 4b and c). A priori planned comparisons indicated that although stressed/vehicle animals showed a significantly increased amount of GluA1 compared with stressed/metyrapone animals, F (1, 32)=8.03, p<0.01, there were no GluA1 protein differences between stressed/metyrapone and unstressed controls, as well as between stressed and unstressed animals that both received metyrapone, p’s>0.05. There were no significant main effects of stress, drug, or a stress by drug interaction for GluA2 (Figure 4d and e), nor for GluN1 protein levels (Figure 4f and g) in the BLA, p’s>0.05.
Interestingly, besides preventing SEFL, metyrapone prevented the elevation in GluA1 and kept its expression level at that of unstressed controls. By contrast, there were no differences in GluA2 or GluN1 levels in stressed or metyrapone-treated groups. Because facilitating glutamatergic activity at AMPAR enhances the rate of fear conditioning (Kim et al, 1993) and SEFL occurs predominantly by increasing the rate of fear conditioning (Fanselow et al, 1993), the long-term increase in GluA1 subunits is a highly plausible mechanism for the expression of SEFL.
Summary
The results from this experiment interestingly show that besides preventing SEFL, metyrapone prevented the elevation in GluA1 in the BLA after SEFL and returned its expression levels to that of unstressed controls. By contrast, there were no differences in GluA2 or GluN1 levels in stressed or metyrapone-treated groups. During learning, sensory input relating to the CS and US converge on the BLA where a CS-US association is encoded via long-term potentiation at BLA synapses, dependent upon glutamate receptor-regulated synaptic plasticity (Rogan et al, 1997; Kim and Jung, 2006). NMDAR are important for acquisition of fear, while AMPAR, specifically GluA1-containing AMPAR, are more critical for the expression of fear via these long-term potentiation mechanisms (Kim et al, 1993; Fanselow and Kim, 1994; Walker and Davis, 2002; Hubert et al, 2013). Therefore, an increase in AMPAR subunits but not NMDAR subunits 2 weeks after the stressor is consistent with these findings. Similar results have been found in the hippocampus, where surface increases in GluA1 but not GluA2 occurred after restraint stress (Whitehead et al, 2013). AMPAR lacking GluA2 have high relative calcium permeability, whereas permeability of AMPAR containing GluA2 is very low; the former enhances cell excitability (Hollmann et al, 1991). However, this long-term upregulation of GluA1 2 weeks after the stressor is inconsistent with prior results showing that the expression of calcium-permeable AMPARs peaks 24 h after conditioning and subsides by 1 week. This shows a dissociation between SEFL, which we view as non-associative sensitization, and Pavlovian fear conditioning (Jarome et al, 2012). We postulate that this increased excitability may lead to enhanced plasticity in response to subsequent mild stressors.
FUTURE RESEARCH DIRECTIONS
The results from these studies discussed in this review elucidate novel neurobiological mechanisms underlying sensitized behavioral responses observed using the SEFL model in rats, with potential relevance to PTSD treatments in humans. Specifically, the collective findings show that that CORT acts on GRs in the BLA during the stressor to upregulate the GluA1 subunit of the AMPAR long-term, which elucidates novel mechanisms for the induction and the expression of SEFL.
The next logical step is to target this upregulation of GluA1 post-stressor. Metyrapone prevented SEFL only when given prior to stress, which severely limits its applicability as a potential treatment. Induction and maintenance of long-term potentiation requires the insertion of GluA1-containing AMPAR in the synapse. Although the process is induced by stress hormones (ie, glucocorticoids), maintenance of AMPAR at the synapse involve both protein kinase A-mediated phosphorylation and endosomal recycling (Kessels and Malinow, 2009; Hanley, 2010; Whitehead et al, 2013). Preventing the upregulation of GluA1 after the 15-shock stressor or perhaps blocking this synaptic AMPAR maintenance may prevent SEFL from developing following a stressor.
Moreover, it is critical to parse the nonassociative and associative components of SEFL and treat each with different methods. Associative fear toward the actual stress context is critical for survival, as it would leave the reaction of evading a truly dangerous situation intact. If such associative fear to the trauma context is contributing to negative symptomatology, however, then exposure treatment may still be necessary. Fortunately, behavioral neuroscience is also leading to developments that enhance exposure therapy (Davis et al, 2006; Monfils et al, 2009; Zelikowsky et al, 2014). Hence, the findings of these experiments suggest a potential for developing novel treatments for PTSD.
In conclusion, these findings have outlined a clear set of mechanisms that underlie the induction and expression of fear sensitization caused by an acute traumatic stressor; that is, stress-induced CORT in the BLA, mediated through GRs, increases GluA1-containing AMPAR in the BLA long-term. Moreover, the brain circuitry and neurotransmitter systems that mediate Pavlovian fear conditioning also mediate SEFL. These studies are the first of its kind to show that stress-induced GluA1-containing AMPAR increases in the BLA not only are important for associative fear expression but also for the expression of SEFL. These long-lasting GluA1 increases may be targeted to produce an enduring reversal of this enhanced fear responding, which suggests hope for those suffering from severe PTSD.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Adamec R, Blundell J, Burton P (2005). Role of NMDA receptors in the lateralized potentiation of amygdala afferent and efferent neural transmission produced by predator stress. Physiol Behav 86: 75–91.
American Psychiatric Association (2013) Diagnostic and statistical manual V. American Psychiatric Press: Washington, DC.
Antoni FA (1986). Hypothalamic control of adrenocorticotropin secretion: advances since the discovery of 41-residue corticotropin-releasing Factor. Endocr Rev 7: 351–378.
Baker DG, West SA, Nicholson WE, Ekhator NN, Kaschow JW, Hill KK et al (1999). Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 156: 585–588.
Bonne O, Grillon C, Vythilingam M, Neumeister A, Charney DS (2004). Adaptive and maladaptive psychobiological responses to severe psychological stress: implications for the discovery of novel pharmacotherapy. Neurosci Biobehav Rev 28: 65–94.
Boscarino JA (1996). Posttraumatic stress disorder, exposure to combat, and lower plasma cortisol among Vietnam veterans: findings and clinical implications. J Consult Clin Psychol 64: 191–201. Showed that in veterans, cortisol was inversely related to combat exposure, with veterans exposed to heavy combat having the lowest concentrations.
Brady KT, Killeen TK, Brewerton T, Lucerini S (2000). Comorbidity of psychiatric disorders and posttraumatic stress disorder. J Clin Psychiat 61: 22–32.
Bremner JD, Krystal JH, Southwick SM, Charney DS (1995). Functional neuroanatomical correlates of the effects of stress on memory. J Trauma Stress 8: 527–533.
Bremner JD, Licinio J, Darnel A, Krystal JH, Owens MJ, Southwick SM et al (1997). Elevated CSF corticotropin releasing factor concentrations in posttraumatic stress disorder. Am J Psychiat 154: 624–629.
Brohawn KH, Offringa R, Pfaff DL, Hughes KC, Shin LM (2010). The neural correlates of emotional memory in posttraumatic stress disorder. Biol Psychiat 68: 1023–1030.
Carrasco GA, Van de Kar LD (2003). Neuroendocrine pharmacology of stress. Eur J Pharmacol 463: 235–272.
Charney DS (2004). Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am J Psychiat 161: 195–216.
Christopher M (2004). A broader view of trauma: a biopsychosocial-evolutionary view of the role of the traumatic stress response in the emergence of pathology and/or growth. Clin Psych Rev 24: 75–98.
Cordero MI, Venero C, Kruyt ND, Sandi C (2003). Prior exposure to single stress facilitating subsequent contextual fear conditioning in rats. Evidence for a role of corticosterone. Horm Behav 44: 338–345. Demonstrated that prior exposure to aversive stimulation is sufficient to facilitate context-dependent fear conditioning and that increased glucocorticoid release at training may underlie memory facilitating effects induced by prior stress experiences.
Davis M, Ressler K, Rothbaum BO, Richardson R (2006). Effects of D-cycloserine on extinction: translation from preclincical to clinical work. Biol Psychiat 60: 369–375. Clear outline of the role that the partial NMDA agonist D-cycloserine (DCS) plays in facilitating fear extinction.
De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M (1998). Brain corticosteroid receptor balance in health and disease. Endocr Rev 19: 269–301.
Dutton CE, Adams T, Bujarski S, Badour CL, Feldner MT (2013). Posttraumatic stress disorder and alcohol dependence: individual and combined associations with social network problems. J Anxiety Disord 28: 67–74.
Dykman RA, Ackerman PT, Newton JEO (1997). Posttraumatic stress disorder: a sensitization reaction. Integr Physiol Behav Sci 32: 9–18.
Eberly RE, Harness AR, Engdahl BE (1991). An adaptational view of trauma response as illustrated by the prisoner of war experience. J Trauma Stress 4: 363–380.
Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A (2009). Amygdala inhibitory circuits and the control of fear memory. Neuron 62: 757–771.
Fanselow MS (1991) The midbrain periaqueductal gray as a coordinator of action in response to fear and anxiety. In: Depaulis A, Bandler R (eds). The Midbrain Periaqueductal Grey Matter: Functional, anatomical and Immuno-histohistochemical Organization (NATO ASI Series). Plenum Publishing Corp: New York, pp 151–173.
Fanselow MS (1998). Pavlovian conditioning, negative feedback, and blocking: mechanisms that regulate association formation. Neuron 20: 625–627.
Fanselow MS (2010). From contextual fear to a dynamic view of memory systems. Trends Cogn Sci 14: 7–15.
Fanselow MS, Bolles RC (1979). Naloxone and shock elicited freezing in the rat. J Comp Physiol Psych Physiological Psychology 93: 736–744.
Fanselow MS, DeCola JP, Young SL (1993). Mechanisms responsible for reduced contextual conditioning with massed unsignaled unconditional stimuli. J Exp Psychol Anim Behav Process 19: 121–137.
Fanselow MS, Gale GD (2003). The amygdala, fear, and memory. Ann NY Acad Sci 985: 125–134.
Fanselow MS, Kim JJ (1994). Acquisition of contextual Pavlovian fear conditioning is blocked by application of an NMDA receptor antagonist D,L-2-amino-5 phosphonovaleric acid to the basolateral amygdala. Behav Neurosci 108: 210–212. Demonstrated that NMDA receptors in the amygdala are critical for the induction of fear in rodents.
Fanselow MS, LeDoux JE (1999). Why we think plasticity underlying pavlovian fear conditioning occurs in the basolateral amygdala. Neuron 23: 229–232.
Fanselow MS, Lester LS (1988) A functional behavioristic approach to aversively motivated behavior: predatory imminence as a determinant of the topography of defensive behavior. In: Bolles RC, Beecher MD (eds). Evolution and Learning. Erlbaum: Hillsdale, NJ, pp 185–211.
Fendt M, Fanselow MS (1999). The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev 23: 743–760.
Friedman MJ (1994). Neurobiological sensitization models of post-traumatic stress disorder: their possible relevance to multiple chemical sensitivity syndrome. Toxicol Ind Health 10: 449–462.
Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR et al (2004). Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci 24: 3810–3815. Outlines the importance of the basolateral amygdala in both recent and remote fear memory.
Godsil BP, Blackmore MA, Fanselow MS (2005). Modulation of an activity response with associative and nonassociative fear in the rat: a lighting differential influences the form of defensive behavior evoked after fear conditioning. Learn Behav 33: 454–463.
Godsil BP, Fanselow MS (2004). Light stimulus change evokes an activity response in the rat. Learn Behav 32: 299–310.
Goisman RM, Allsworth J, Rogers MP, Warshaw MG, Goldenberg I, Vasile RG et al (1998). Simple phobia as a comorbid anxiety disorder. Depress Anxiety 7: 105–112.
Groves PM, Thompson RF (1970). Habituation: a dual process theory. Psychol Rev 77: 419–450.
Hageman I, Anderson H, Jorgensen MB (2001). Post-traumatic stress disorder: a review of psychobiology and pharmacotherapy. Acta Psychiat Scand 104: 411–422.
Hanley JG (2010). Endosomal sorting of AMPA receptors in hippocampal neurons. Biochem Soc T 38: 460–465.
Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R et al (2010). Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 468: 270–276.
Hembree EA, Foa EB, Dorfan NM, Street GP, Kowalski J, Tu X (2004). Do patients drop out prematurely from exposure therapy for PTSD? J Trauma Stress 16: 555–562.
Hollmann M, Hartley M, Heinemann S (1991). Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science 252: 851–853.
Hubert GW, Li C, Rainnie DG, Muly EC (2013). Effects of stress on AMPA receptor distribution and function in the basolateral amygdala. Brain Struct Funct 219: 1169–1179. Suggested that the shift of GluR1-containing AMPA receptors from dendritic stores into spines may be in part responsible for the persistent behavioral alterations observed following severe stressors.
Jarome TJ, Kwapis JL, Werner CT, Parsons RG, Gafford GM, Helmstetter FJ (2012). The timing of multiple retrieval events can alter GluR1 phosphorylation ad the requirement of protein synthesis in fear memory reconsolidation. Learn Mem 19: 300–306.
Jasnow AM, Cooper MA, Huhman KL (2004). N-methyl-D-aspartate receptors in the amygdala are necessary for the acquisition and expression of conditioned defeat. Neuroscience 12: 625–634.
Johnson EO, Kamilaris TC, Chrousos GP, Gold PW (1992). Mechanisms of stress a dynamic overview of hormonal and behavioral homeostasis. Neurosci Biobehav Rev 16: 115–130.
Kamin LJ (1968) “Attention-Like” processes in classical conditioning. In: Jones MR (ed). Miami Symposium on the Prediction of Behavior: Aversive Stimulation. University of Miami Press: Miami, FL, pp 9–33.
Kessels HW, Malinow R (2009). Synaptic AMPA receptor plasticity and behavior. Neuron 61: 340–350. Clearly outlines how AMPAR trafficking underlies several experience-driven phenomena-from neuronal circuit formation to the modification of behavior.
Kim M, Campeau S, Falls WA, Davis M (1993). Infusion of the non-NMDA receptor antagonist CNQX into the amygdala blocks the expression of fear-potentiated startle. Behav Neural Biol 59: 5–8. Demonstrated how AMPA receptors mediate the expression of fear in rodents.
Kim JJ, Jung MW (2006). Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev 30: 188–202.
Kim JJ, Lee HJ, Han JS, Packard MG (2001). Amygdala is critical for stress-induced modulation of hippocampal long-term potentiation and learning. J Neurosci 21: 5222–5228.
Krikorian R, Layton BS (1998). Implicit memory in posttraumatic stress disorder with amnesia for the traumatic event. J Neuropsych Clin N 10: 359–362.
Lemieux AM, Coe CL (1995). Abuse-related posttraumatic stress disorder: evidence for chronic neuroendocrine activation in women. Psychosom Med 57: 105–115.
Long VA, Fanselow MS (2012). Stress-enhanced fear learning in rats is resistant to the effects of immediate massed extinction. Stress 15: 627–636. Demonstrated how stress-enhanced fear learning, a rodent model of post-traumatic stress disorder, cannot be ameliorated with extinction.
Maes M, Lin A, Bonaccorso S, van Hunsel F, Van Gastel A, Delmeire L et al (1998). Increased 24-hour urinary cortisol excretion in patients with post-traumatic stress disorder and patients with major depression, but not in patients with fibromyalgia. Acta Psychiat Scand 98: 328–335.
Makkar SR, Shirley QZ, Cranney J (2010). Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacology 35: 1625–1652.
Maren S (1998). Overtraining does not mitigate contextual fear conditioning deficits produced by neurotoxic lesions of the basolateral amygdala. J Neurosci 18: 3088–3097.
Mason JW, Giller EL, Kosten TR, Ostroff RB, Podd L (1986). Urinary freecortisol levels in posttraumatic stress disorder patients. J Nerve Ment Dis 174: 145–149. Found stable, low levels of cortisol in post-traumatic stress disorder patients.
McDannald MA, Galarce EM (2011). Measuring Pavlovian fear with conditioned freezing and conditioned suppression reveals different roles for the basolateral amygdala. Brain Res 1374: 82–89.
McEwen BS, Weiss JM (1970). The uptake and action of corticosterone: regional and subcellular studies on rat brain. Prog Brain Res 32: 200–212.
McKernan MG, Shinnick-Gallagher P (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607–611.
Meyer EM, Long VA, Fanselow MS, Spigelman I (2013). Stress increases voluntary alcohol intake, but does not alter established drinking habits in a rat model of posttraumatic stress disorder. ACER 37: 566–574.
Milad MR, Quirk GJ (2002). Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70–74. Among the first to suggest that consolidation of extinction learning potentiates infralimbic activity, which inhibits fear during subsequent encounters with fear stimuli.
Miserendino MJ, Sananes CB, Melia KR, Davis M (1990). Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature 345: 716–718.
Monfils MH, Cowansage KK, Klann E, LeDoux JE (2009). Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science 324: 951–955.
Muller J, Corodimas KP, Fridel Z, LeDoux JE (1997). Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav Neurosci 111: 683–691.
Munck A, Guyre PM, Holbrook NJ (1984). Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev 5: 25–44.
Orsini CA, Kim JH, Knapska E, Maren S (2011). Hippocampal and prefrontal projections to the basal amygdala mediate contextual regulation of fear after extinction. J Neurosci 31: 17269–17277.
Owens MJ, Nemeroff CB (1991). Physiology and pharmacology of corticotrophin-releasing factor. Pharmacol Rev 43: 425–473.
Pare D, Quirk GJ, LeDoux JE (2004). New vistas on amygdala networks in conditioned fear. J Neurophysiol 92: 1–9.
Peri T, Ben-Shankhar G, Orr SP, Shalev AY (2000). Psychophysiologic assessment of aversive conditioning in posttraumatic stress disorder. Biol Psychiat 47: 512–519.
Pitkanen A, Savander V, LeDoux JE (1997). Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci 20: 517–523.
Pitman RK, Orr SP (1990). Twenty-four hour urinary cortisol and catecholamine excretion in combat-related posttraumatic stress disorder. Biol Psychiat 27: 245–247.
Ponomarev I, Rau V, Eger EI, Harris RA, Fanselow MS (2010). Amygdala transcriptome and cellular mechanisms underlying stress-enhanced fear learning in a rat model of posttraumatic stress disorder. Neuropsychopharmacology 35: 1402–1411.
Post RM, Weiss SR, Smith M, Li H, McCann U (1997). Kindling versus quenching. Implications for the evolution and treatment of posttraumatic stress disorder. Ann NY Acad Sci 821: 285–295.
Poulos AM, Reger M, Mehta N, Zhuravka I, Sterlace SS, Gannam C et al (2013). Amnesia for early life stress does not preclude the adult development of posttraumatic stress disorder symptoms in rats. Biol Psychiat 76: 306–314. Found that traumatic experiences during development when the hippocampus is still offline can promote lifelong changes in symptoms and neuropathology associated with human PTSD, even if there is no explicit memory of the early trauma.
Poulos AM, Zhuravka I, Long V, Gannam C, Fanselow MS (2015). Sensitization of fear learning to mild unconditional stimuli in male and female rats. Behav Neurosci 129: 62–67.
Quirk GJ, Likhtik E, Guillaume Pelletier J, Pare D (2003). Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci 23: 8800–8807.
Rasmusson AM, Charney DS (1997). Animal models of relevance to PTSD. Ann NY Acad Sci 821: 332–351.
Rau V, De Cola JP, Fanselow MS (2005). Stress-induced enhancement of fear learning: an animal model of posttraumatic stress disorder. Neurosci Biobehav Rev 29: 1207–1223. Proposes a valid rodent model of post-traumatic stress disorder, in which a severe stressor sensitizes responding to subsequent mild fear conditioning.
Rau V, Fanselow MS (2009). Exposure to a stressor produces a long lasting enhancement of fear learning in rats. Stress 12: 125–133.
Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB et al (2000). Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biol Psychiat 47: 769–776.
Reichardt HM, Schutz G (1998). Glucocorticoid signaling–multiple variations of a common theme. Mol Cell Endocrinol 146: 1–6.
Rodriguez Manzanares PA, Isoardi NA, Carrer HF, Molina VA (2005). Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J Neurosci 25: 8725–8734.
Rogan MT, Stäubli UV, LeDoux JE (1997). Fear conditioning induces associative long-term potentiation in the amygdala. Nature 391: 604–607. Suggested that fear conditioning is dependent on LTP-processes in the amygdala that underlies the long-term associative plasticity that allows for maintenance of the conditioned fear memory.
Rosen JB, Schulkin J (1998). From normal fear to pathological anxiety. Psychol Rev 105: 325–350.
Santini E, Ge H, Ren K, Pena de Ortiz S, Quirk GJ (2004). Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci 24: 5704–5710. Showed that rats infused intraventricularly or into the medial prefrontal cortex (mPFC) with the protein synthesis inhibitor anisomycin extinguished fear normally within a session but were unable to recall extinction the following day; extinction training also increased c-Fos levels in the mPFC.
Sapolsky RM (2000). Stress hormones: good and bad. Neurobiol Dis 7: 540–542.
Sapolsky RM, Romero M, Munck AU (2000). How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 21: 55–89.
Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Grundemann J et al (2014). Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81: 428–437.
Shin LM, Rauch SL, Pitman RK (2006). Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann NY Acad Sci 1071: 67–79.
Stander VA, Thomsen CJ, Highfill-McRoy RM (2014). Etiology of depression comorbidity in combat-related PTSD: a review of the literature. Clin Psychol Rev 34: 87–98.
Thompson BL, Erickson K, Schulkin J, Rosen JB (2004). Corticosterone facilitates retention of contextually conditioned fear and increases CRH mRNA expression in the amygdala. Behav Brain Res 149: 209–215. Suggested that repeated administration of CORT given before fear conditioning facilitates the acquisition of emotional memory, whereas CORT given after consolidation does not increase emotional memory.
Vale W, Spiess J, Rivier C, Rivier J (1981). Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and β-endorphin. Science 213: 1394–1397.
van Dam D, Ehring T, Vedel E, Emmelkamp PM (2013). Trauma-focused treatment for posttraumatic stress disorder combined with CBT for severe substance use disorder: a randomized controlled trial. BMC Psychiatry 13: 172.
Waddell J, Morris RW, Bouton ME (2006). Effects of the bed nucleus of the stria terminalis lesions on conditioned anxiety: aversive conditioning with long-duration conditional stimulus and reinstatement of extinguished fear. Behav Neurosci 120: 324–336.
Walker DL, Davis M (2002). The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmocol Biochem Behav 71: 379–392.
Walker DL, Toufexis DJ, Davis M (2003). Role of the bed nucleus of the stria terminalis versus the amygdala in fear, stress, and anxiety. Eur J Pharmacol 463: 199–216.
Whitehead G, Jo J, Hogg EL, Piers T, Kim DH, Seaton G et al (2013). Acute stress causes rapid synaptic insertion of Ca2+-permeable AMPA receptors to facilitate long-term potentiation in the hippocampus. Brain 136: 3753–3765.
Yehuda R (1997). Sensitization of the hypothalamic-pituitary-adrenal axis in posttraumatic stress disorder. Ann NY Acad Sci 821: 57–75.
Yehuda R (2001). Biology of posttraumatic stress disorder. J Clin Psychiat 62: 41–46.
Yehuda R, Kahana B, Binder-Brynes K, Southwick SM, Mason JW, Giller EL (1995). Low urinary cortisol excretion in Holocaust survivors posttraumatic stress disorder. Am J Psychiat 152: 982–986.
Yehuda R, Southwick SM, Nussbaum G, Wahby V, Giller EL, Mason JW (1990). Low urinary cortisol excretion in patients with posttraumatic stress disorder. J Nerve Ment Dis 78: 366–369.
Zelikowsky M, Hast TA, Bennett RZ, Merjanian M, Nocera NA, Ponnusamy R et al (2014). Cholinergic blockade frees fear extinction from its contextual dependency. Biol Psychiat 73: 345–352. Discovered that disrupting contextual processing during extinction with the cholinergic antagonist scopolamine blocked subsequent fear renewal.
Acknowledgements
We would like to thank Andrew Poulos, Andrey Mazarati, Sarah Sterlace, Sujean Oh, Halina Yee, Brian Chiang, Sarah Moore, Zachary Pennington, and Ginny Li for assistance with the behavioral and biochemical aspects of the study. Funding and Disclosure: This study was supported by NIH grants MH62122 and RC1MH0088184 to MSF, T32 grant 5 DA024635-03 to JNP, and F31AA021037 to EMM. Additional support was provided by a NARSAD Distinguished Investigator Award #18667 to MSF and by the Staglin Music Festival Center for Brain and Behavioral Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Perusini, J., Meyer, E., Long, V. et al. Induction and Expression of Fear Sensitization Caused by Acute Traumatic Stress. Neuropsychopharmacol 41, 45–57 (2016). https://doi.org/10.1038/npp.2015.224
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.224
This article is cited by
-
Examining sex differences in responses to footshock stress and the role of the metabotropic glutamate receptor 5: an [18F]FPEB and positron emission tomography study in rats
Neuropsychopharmacology (2023)
-
A prolonged stress rat model recapitulates some PTSD-like changes in sleep and neuronal connectivity
Communications Biology (2023)
-
Dopaminergic and cholinergic modulation of the amygdala is altered in female mice with oestrogen receptor β deprivation
Scientific Reports (2023)
-
Dose mediates the protracted effects of adolescent THC exposure on reward and stress reactivity in males relevant to perturbation of the basolateral amygdala transcriptome
Molecular Psychiatry (2023)
-
Refinement of the stress-enhanced fear learning model of post-traumatic stress disorder: a behavioral and molecular analysis
Lab Animal (2022)
