
Abstract
Humans liberally use ethanol for its facilitating effects on social interactions but its effects on central nervous system function remain underexplored. We have recently described that very low doses of ethanol abolish long-term potentiation (LTP)-like plasticity in human cortex, most likely through enhancement of tonic inhibition [Lücke et al, 2014, Neuropsychopharmacology 39:1508-18]. Here, we studied the effects of low-dose ethanol on long-term depression (LTD)-like plasticity. LTD-like plasticity was induced in human motor cortex by paired associative transcranial magnetic stimulation (PASLTD), and measured as decreases of motor evoked potential input-output curve (IO-curve). In addition, sedation was measured by decreases in saccade peak velocity (SPV). Ethanol in two low doses (EtOH<10mM, EtOH<20mM) was compared to single oral doses of alprazolam (APZ, 1mg) a classical benzodiazepine, and zolpidem (ZLP, 10 mg), a non-benzodiazepine hypnotic, in a double-blinded randomized placebo-controlled crossover design in ten healthy human subjects. EtOH<10mM and EtOH<20mM but not APZ or ZLP enhanced the PASLTD-induced LTD-like plasticity, while APZ and ZLP but not EtOH<10mM or EtOH<20mM decreased SPV. Non-sedating low doses of ethanol, easily reached during social drinking, enhance LTD-like plasticity in human cortex. This effect is most likely explained by the activation of extrasynaptic α4-subunit containing gamma-aminobutyric type A receptors by low-dose EtOH, resulting in increased tonic inhibition. Findings may stimulate cellular research on the role of tonic inhibition in regulating excitability and plasticity of cortical neuronal networks.
Similar content being viewed by others

Introduction
Humans liberally ingest ethanol (EtOH) at low doses during social drinking for its facilitating effects on personal interactions, but the effects of low doses of EtOH on central nervous system function remain underexplored. Cellular studies demonstrated that low-dose EtOH (≤30 mM) enhances tonic inhibition mediated by the extrasynaptic α4-subunit containing gamma-aminobutyric acid type A receptor (α4-GABAAR) (Hanchar et al, 2006; Sundstrom-Poromaa et al, 2002; Wallner et al, 2003; Wei et al, 2004) although not all studies were able to replicate this finding (Borghese et al, 2006; Yamashita et al, 2006). In addition, low doses of EtOH inhibit N-methyl-D-aspartate receptor (NMDAR)-mediated currents (He et al, 2013; Lovinger et al, 1989, 1990; Weitlauf and Woodward, 2008).
We have recently reported that non-sedating very low doses of EtOH (<10mM) but not the classical benzodiazepine alprazolam (APZ), an allosteric positive modulator at synaptic α1-, α2-, α3- and α5-GABAARs, and zolpidem (ZLP), a non-benzodiazepine hypnotic with high affinity at the α1-GABAAR disrupt long-term potentiation (LTP)-like plasticity induced by paired associative transcranial magnetic stimulation (PASLTP) in human motor cortex (Lücke et al, 2014). Similar to LTP, long-term depression (LTD) is thought to play important roles in learning and memory formation (Collingridge et al, 2010), but LTD has been much less investigated compared to LTP. EtOH effects on LTD in slice preparations have been very variable, depending on the experimental model. EtOH increased (Hendricson et al, 2002) or decreased (Izumi et al, 2005) LTD in the hippocampus, led to a switch from LTP to LTD in the dorsal striatum (Yin et al, 2007), and decreased LTD in the cerebellum (Belmeguenai et al, 2008; Carta et al, 2006; He et al, 2013; Su et al, 2010). In addition, relatively scant evidence showed that enhancement of synaptic GABAAergic neurotransmission increased LTD (Choi et al, 2002), while its blockade by GABAAR antagonists disrupted LTD (Nishiyama et al, 2010; Watanabe et al, 2007; Zhu et al, 2013), or even resulted in an LTD to LTP shift (Hess and Donoghue, 1996).
At the systems level of human motor cortex, paired associative stimulation (PASLTD) can induce LTD-like plasticity, as measured by a decrease in motor evoked potential (MEP) amplitude that outlasts the PASLTD intervention (Huber et al, 2008; Müller et al, 2007; Rosenkranz et al, 2014; Wolters et al, 2003). Here we thought to investigate the effects of enhancing extrasynaptic GABAAergic inhibition (two low doses of EtOH: <10 mM, <20 mM) vs enhancing synaptic GABAAergic inhibition (single oral doses of 1mg of APZ, and 10 mg of ZLP) on PASLTD-induced LTD-like plasticity. We quantified α1-GABAAR-mediated sedation by measuring peak velocity of visually guided saccades (SPV) (de Visser et al, 2003; Lücke et al, 2014).
Given our previous findings on PASLTP-induced LTP-like plasticity (Lücke et al, 2014), we hypothesized drug-induced enhancement of PASLTD-induced LTD-like plasticity, predominately by low-dose EtOH but not or to a lesser extent by APZ and ZLP.
In accordance with the hypothesis, we show that low-dose EtOH but not APZ or ZLP enhanced the PASLTD-induced LTD-like plasticity, while APZ and ZLP but not low-dose EtOH decreased SPV. We discuss carefully that this double dissociation implicates increased tonic inhibition through activation of extrasynaptic α4-GABAARs by EtOH as the most likely mechanism for the enhancement of LTD-like plasticity. Findings may stimulate further cellular research on the role of tonic inhibition in regulating excitability and plasticity of cortical neuronal networks.
Materials and Methods
Subjects
Twelve healthy subjects (mean (±SD) age 24.6±3.95 years, range 19–31 years; body length, 181.5±7.89 cm, range 170–195 cm; body weight, 78.5±10.84 kg, range 60–99 kg; 3 female) participated in the study. Two subjects did not complete the study due to adverse effects under ZLP (nausea, vomiting). The presented data are therefore based on ten subjects. All subjects fulfilled the criteria according to a TMS safety questionnaire (Keel et al, 2001), had no history of neurological or psychiatric disease and were non-smokers as nicotine use alters PAS-induced LTD-like plasticity (Thirugnanasambandam et al, 2011). Subjects did not take any CNS-active drugs. This was verified by comprehensive urine analysis prior to each experimental session. An additional inclusion criterion for females was the use of a hormonal method of contraception to prevent hormonal fluctuations during menstrual cycle, which might affect motor cortical excitability (Smith et al, 2002; Smith et al, 1999). All subjects were right-handed as tested by the Edinburgh handedness questionnaire (Oldfield, 1971).
PAS-induced LTD-like plasticity is not expressed in all subjects. Therefore, to identify subjects suitable for this study, volunteers were screened in a session at least 1 week prior to the main study for a resting motor threshold (RMT) ≤50% of maximum stimulator output (Müller-Dahlhaus et al, 2008) and a significant PAS-induced decrease in MEP amplitude <1.0 (ratio of MEP amplitude post-PAS/pre-PAS) (Delvendahl et al, 2013; Voytovych et al, 2012).
All subjects gave their written informed consent prior to participation. The study was approved by the ethics committee of the Medical Faculty of the Goethe-University of Frankfurt and conformed to the latest version of the Declaration of Helsinki.
EMG Recordings
All motor evoked potential (MEP) measurements were conducted by surface electromyography (EMG) from the resting abductor pollicis brevis (APB) muscle of the dominant right hand using Ag-AgCl cup electrodes in a belly-tendon montage, while subjects were seated in a comfortable chair with their arms and hands lying relaxed on the armrests. EMG raw signals were amplified (Digitimer D360, Digitimer Ltd., Welwyn Garden City, UK), bandpass filtered (20 Hz - 2 kHz), digitized at a rate of 5kHz (CED Micro 1401, Cambridge Electronic design, Cambridge, UK) and stored on a laboratory computer. Spike2 v.3.05 software (Cambridge Electronic design, Cambridge, UK) was used for control of the experiment, data recording, and online and offline data analysis. All recordings were performed at rest. Target muscle relaxation was controlled by audio-visual feedback of the raw EMG at high gain (50 μV/Div).
Stimulation Procedures
Focal transcranial magnetic stimulation (TMS) was delivered to the hand area of the left primary motor cortex (M1) using a figure-of-eight stimulating coil (diameter of each wing, 70 mm) and a monophasic current waveform provided by a Magstim, 2002 magnetic stimulator (Magstim Company, Whitland, Wales, UK). The junction of the stimulating coil was positioned tangentially to the skull, with the handle pointing backwards approximately 45° away from the sagittal plane, to induce a current flow in the brain directed from lateral-posterior to medial-anterior. This way, the corticospinal system is excited predominantly trans-synaptically via horizontal cortico-cortical connections (Di Lazzaro et al, 2008). The coil was placed over the hot spot, defined as the optimal site over the left M1 to elicit consistently largest MEPs in the right APB at a slightly suprathreshold stimulation intensity. This coil position was marked on the scalp with a pen for correct placement of the coil throughout the experiment.
The individual resting motor threshold (RMT) was determined using a relative frequency method (Groppa et al, 2012), defined as the lowest stimulus intensity that elicited MEPs of ≥50 μV in at least five out of ten consecutive trials in the relaxed APB. In addition, for each subject the individual minimum stimulation intensity to elicit a peak-to-peak MEP amplitude of 1mV (SI1mV) was determined. Stimulation intensity is reported as a percentage of the maximum stimulator output (%MSO).
Motor cortical excitability was assessed measuring input-output curves of MEPs (IO-curves). Eleven stimulation intensities were applied at stimulus intensities of 0.5 to 1.5 × SI1mV in steps of 0.1 × SI1mV (Lücke et al, 2014; Rosenkranz et al, 2007a). Eight trials per stimulation intensity were recorded in randomized order to avoid hysteresis effects (Möller et al, 2009). The single-trial peak-to-peak MEP amplitudes were analyzed and then averaged according to stimulation intensity. MEP IO-curves rather than MEP amplitude at one single stimulus intensity were chosen as primary readout for drug-induced and PASLTD-induced changes in corticospinal excitability for the following reasons: (1) Previous studies demonstrated that PAS-induced LTD-like plasticity was expressed predominantly in the high-intensity range of the IO-curve (Ni et al, 2014; Rosenkranz et al, 2007a; Rosenkranz et al, 2007b). (2) Different parts of the IO-curve represent different physiological mechanisms. While the low-intensity part represents relatively direct excitation of the corticospinal neurons (I1-wave) the high-intensity part reflects more indirect excitation through interneurons (late I-waves) (Di Lazzaro et al, 2008). Therefore, differential drug effects on selected parts of the IO-curve after PASLTD would allow assignment of more specific mechanisms to the observed effects. (3) Finally, we wanted to be compatible with our previous research, where we also investigated IO-curves to study the effects of the same drugs as in the present study on PAS-induced LTP-like plasticity (Lücke et al, 2014).
Paired Associative Stimulation (PASLTD)
PASLTD was applied according to an established protocol (Jung and Ziemann, 2009; Müller et al, 2007; Voytovych et al, 2012) to induce LTD-like plasticity in M1. In brief, electrical stimulation of the right median nerve followed by focal single-pulse TMS of the contralateral M1 was applied 225 times at a rate of 0.25 Hz with an interstimulus interval (ISI) of 15.0±1.1 ms (mean (±SD)). ISI was calculated based on the individual N20 latency, measured by median nerve somatosensory-evoked potentials, minus 5 ms (N20-5 ms). Electrical stimulation was delivered through a bipolar electrode (constant current, pulse duration 1.0 ms) with an intensity of three times the perceptual sensory threshold. To control the attention directed to the stimulated hand during PAS (Stefan et al, 2004), subjects were instructed to count the number of flashes emitted from an LED fixed at the right wrist.
Saccadic Peak Velocity (SPV) Measurements
We measured visually guided saccadic peak velocity (SPV) (Velazquez-Perez et al, 2004) to assess drug-induced sedation. This is important, as we were concerned that sedation and a possible concomitant decrease in attention could have contributed to drug-related changes in PAS-induced LTD-like plasticity (Kamke et al, 2014). In addition, SPV is a behavioral marker of inhibition mediated through the α1-GABAAR (de Visser et al, 2003). Horizontal saccades were recorded by electronystagmography using Ag-AgCl surface electrodes over the outer canthus of each eye. Subjects were positioned in front of a screen (viewing distance, 0.9 m) and were instructed to make visually guided saccades in response to a white dot subtending an angle of view of 1° on a black background while the head was kept in straight position. The dot jumped at random intertrial intervals of 2–3 s (to limit anticipation of the next event) horizontally from one lateral edge to the opposite edge of the screen, subtending an angle of view of 40°. In each block 50 trials were recorded. Offline analysis was performed with MATLAB (version 6.1; Natick, MA) and customized software. Saccade onset and offset were identified manually, before applying a third-order polynomial fit to obtain SPV in °/s.
Pharmacokinetic Measurements
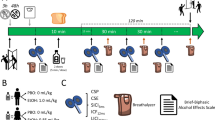
Pharmacokinetic analyses in blood and urine samples were performed at the Department of Forensic Toxicology, Goethe-University Frankfurt using chromatographic-mass spectrometry. Before each experiment, a urine drug screening was performed to test for CNS-active medical drugs and drugs of abuse. Blood plasma concentrations of the study medication were assessed immediately before (time point B1) and after (time point P1) PASLTD (Figure 1).

Timeline of experiments. Motor evoked potential input-output curves (IO-curve) and saccadic peak velocity (SPV) were measured at baseline (time point B0), post drug (time point B1) and post paired associative stimulation for induction of LTD-like plasticity (PASLTD, time point P1). The study medication was administered immediately (tablet) and 30 min (drink) after B0. Post drug measurements at B1 were performed 60 min after drink intake. PASLTD followed immediately after B1. Blood samples for measuring serum drug levels were taken at B1 and P1.
Experimental Design
We performed a randomized, double blind, double-dummy, placebo-controlled crossover study with five sessions (Figure 1). Study visits of a given participant were separated by at least 1 week. Experiments started with a urine drug test, followed by baseline (time point B0) measurements of SPV, RMT, SI1mV and MEP IO-curve. Study medication was administered as a tablet (immediately after the B0 measurements) and a drink (30 min later) (Table 1). Post drug measurements (SPV and IO-curve) were done 90 min after tablet intake and 60 min after drink intake (time point B1) to accommodate for the expected times of peak plasma concentrations of APZ (Greenblatt and Wright, 1993), ZLP (de Haas et al, 2010) and EtOH (Welling et al, 1977) in healthy young adults. Immediately before PASLTD a blood sample was taken. Five minutes after PASLTD (time point P1, Figure 1) SPV and IO-curve were measured and a second blood sample was taken. The stimulation intensities for measurement of the IO-curve were kept constant at all time points.
In each session a drink and a pill were administered in a double-dummy design according to the scheme in Table 1 to ensure blinding of the subjects. Tablets of APZ (1 mg) or ZLP (10 mg) were combined with a placebo drink (conditions 1 and 2 in Table 1). Ethanol was mixed with orange juice and bitter syrup to obfuscate the characteristic taste and administered in two dosages (see below) in combination with a placebo tablet (conditions 3 and 4 in Table 1). In the PBO session, the placebo tablet was given combined with the placebo drink (condition 5 in Table 1).
Alcohol dosages were calculated individually considering gender, body weight and body height (Ulrich et al, 1987; Widmark, 1932). Male subjects received ethanol doses of 0.29 g/kg and 0.55 g/kg, and female subjects 0.25 g/kg and 0.47 g/kg in the EtOH<10mM and EtOH<20mM conditions, respectively. These dosages were expected to result at time point B1 (i.e., 80 min after ingestion, and immediately prior to PASLTD, Figure 1) in plasma levels of approximately 0.2‰ (≈4.6 mM) for EtOH<10mM and 0.5‰ (≈11.6 mM) for EtOH<20mM (Table 2). Plasma levels at time point B1 were slightly but not significantly higher than those measured approximately 20 min later, at time point P1, in the previous study (Lücke et al, 2014). The naming of the lower-dose EtOH condition was accommodated in the present study from EtOH<5mM to EtOH<10mM, as the dose range exceeded 5 mM (Table 2).
To put the applied alcohol doses into the context of social drinking, a 70 kg male subject, for example, received 1.45 and 2.75 standard drink equivalents for the EtOH<10mM and EtOH<20mM conditions, respectively.
Statistics
All statistics were performed with SPSS Statistics software (IBM, v.22.0.0).
Drug effects on SPV were analyzed by a repeated measures analysis of variance (rmANOVA) with DRUG (5 levels: APZ, ZLP, EtOH<10mM, EtOH<20mM, PBO) and TIME (3 levels: B0, B1, P1) as within-subject factors.
Differences in RMT, SI1mV, and SPV at baseline (time point B0) between conditions were tested using rmANOVAs with DRUG (5 levels) as within-subject effect. Differences in IO-curve between conditions were tested at baseline (time point B0) and post-drug intake immediately before PASLTD (time point B1) using rmANOVAs with DRUG (5 levels) and SI (11 levels: 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4 and 1.5 × SI1mV) as within-subject effects. Drug effects on IO-curve (i.e., comparison of time point B1 vs B0) were analyzed by rmANOVA with the within-subject effects DRUG (5 levels), TIME (2 levels: B0, B1) and SI (11 levels). This analysis showed a significant DRUG*TIME*SI interaction that justified pairwise comparisons of PBO with all drugs in rmANOVAs with the effects of DRUG (2 levels: PBO vs drug of interest), TIME (2 levels) and SI (11 levels). In case of a significant triple interaction, post hoc comparisons tested the drug effect at each of the SIs using two-way rmANOVAs with the effects of DRUG and TIME, using Fisher’s Least Significant Difference test. The effects of drug on PASLTD-induced changes of the IO-curve (i.e., comparison of time point P1 vs B1) were similarly analyzed by performing an rmANOVA with the within-subject effects of DRUG (5 levels), TIME (2 levels: B1, P1) and SI (11 levels). Post hoc tests were analogously performed as above. MEP amplitudes recorded during PASLTD were averaged in bins of 25 consecutive trials (i.e., 9 bins in total for 225 trials). Drug effects on MEP amplitudes during the PASLTD intervention were then assessed by a two-way rmANOVA with the within-subject effects of DRUG (5 levels) and BIN (9 levels).
For all rmANOVAs, violation of sphericity was tested by Mauchly's test, and the Greenhouse-Geisser correction was applied when appropriate.
Two metrics were calculated that summarize PAS-induced changes of IO-curve in one single value: slope (Rosenkranz et al, 2007a; Rosenkranz et al, 2007b) and area under the IO-curve (AUIOC) (Lücke et al, 2014). Both metrics were determined for each subject, time point, and drug condition. Slope was defined as the linear regression of MEP amplitudes over the stimulus intensity range of the steepest part of the IO-curve (0.8–1.2 × SI1mV), while AUIOC was the sum of MEP amplitudes over the full IO-curve. These metrics did not reveal a significant LTD-like decrease in the PBO condition (slope at time point B1: 0.051±0.008 mV/%MSO, slope at P1: 0.046±0.011 mV/%MSO, P=0.44; AUIOC at B1: 15.02±2.19 mV, AUIOC at P1: 13.39±2.60 mV, P=0.15). These nil results were explained by the fact that the LTD-like decrease in MEP amplitude in the PBO condition was expressed only in the high-intensity range of the IO-curve (see Results, and previous studies (Ni et al, 2014)). As we wanted to analyze drug-induced changes (increases and decreases) of LTD-like plasticity, its expression in the PBO condition was a prerequisite. Therefore, slope and AUIOC were not used for any further statistical analyses.
Linear regression analyses were performed to test for correlations between the drug-induced changes in LTD-like plasticity and drug serum concentrations. For those SIs that showed a significant difference of LTD-like plasticity between DRUG and PBO, (MEPP1 – MEPB1)DRUG – (MEPP1 – MEPB1)PBO was calculated and related to drug serum concentration by calculating the Pearson coefficient.
In all tests, P≤0.05 was considered to be statistically significant. All values are given as means ±SEM, if not stated otherwise.
Results
The ten subjects who completed the study tolerated the study medications well without relevant adverse effects. None of the subjects had a positive urine drug screen prior to any of the sessions.
Drug Plasma Concentrations at Time Points B1 and P1
The blood concentrations of drugs measured before (time point B1) and after PASLTD (time point P1) are reported in Table 2. The blood concentrations of EtOH<10mM and EtOH<20mM were significantly lower at P1 compared to B1 (all P< 0.0001), due to fast metabolism of ethanol.
Drug Effects on Saccade Peak Velocity (SPV)
The baseline SPV was not different between drug conditions (rmANOVA, all P>0.05, Table 3). RmANOVA of the PBO condition showed no effect of TIME (3 levels) (F1.22,18=0.728, P=0.441), demonstrating that subjects performed a stable level for the period of the experiment (Figure 2). RmANOVA of all drug conditions and time points revealed significant effects of DRUG (F4,36=11.309, P< 0.0001), TIME (F2,18=8.588, P=0.004) and DRUG*TIME interaction (F8,72=2.293, P=0.034). Post hoc testing revealed that APZ caused a significant reduction of SPV at time points B1 (P=0.004) and P1 (P=0.012) compared to baseline (Figure 2). ZLP also caused a significant reduction at B1 (P<0.0001) and P1 (P=0.05) (Figure 2). In summary, APZ and ZLP caused similar sedative effects, due to their enhancement of neurotransmission through the α1-GABAAR. EtOH<10mM and EtOH<20mM did not change SPV (all P>0.1). There was no correlation between SPV and drug concentration in any of the drug conditions (all P>0.05).

Saccadic peak velocity (SPV) at time points B1 (post drug, white bars) and P1 (post PASLTD, black bars) normalized to SPV at baseline (B0) in the placebo (PBO), alprazolam (APZ), zolpidem (ZLP), and ethanol (EtOH<10mM, EtOH<20mM) conditions. All data are means±SEM (n=10). Asterisks indicate significant difference to baseline (P<0.05). Note that APZ and ZLP but not EtOH decrease SPV.
RMT and SI1mV at Baseline (Time Point B0)
RmANOVA did not show any differences of RMT (F4,36=1.698, P=0.17) or SI1mV (F4,36=1.816, P=0.15) at baseline between drug conditions (Table 3).
Drug Effects on IO-Curve (Comparison of Time Point B1 vs B0)
There was no difference in baseline IO-curves between drug conditions (F4,36=0.618, P=0.65, Figure 4a and d). In the PBO condition, there was no change in IO-curve pre (B0) versus post (B1) drug intake (TIME: F1,9=1.153, P=0.311; TIME*SI: F10,90=1.085, P=0.382), indicating stability of IO-curve recordings (Figure 3a).

(a) IO-curve (y-axes: MEP amplitude in mV, x-axes: stimulus intensity in multiples of SI1mV) at baseline (B0, black squared symbols) vs post drug (B1, blue diamonds), and (b) post drug (B1, blue diamonds) vs post PASLTD (P1, red circles) in the placebo (PBO) condition. Note that PBO had no effect on IO-curve when comparing time points B1 with B0, indicating reliability of IO-curve measurements. Also note that PASLTD resulted in a significant IO-curve depression at SIs of 1.3–1.5 × SI1mV (#P<0.05).
The rmANOVA with all drugs revealed a significant DRUG*SI (F40,360=1.772, P=0.004) and DRUG*TIME*SI (F40,360=1.708, P=0.006) interaction, suggesting a drug effect on IO-curve depending on stimulus intensity (Figure 4a–d). The significant triple-interaction justified pairwise comparisons of the PASLTD-induced changes in IO-curve between any of the drugs with PBO. The comparison of PBO vs APZ revealed significant effects of DRUG (F1,9=5.761, P=0.040), DRUG*SI (F10,90=4.392, P<0.0001) and DRUG*TIME*SI (F10,90=3.186, P=0.002). Post hoc two-way rmANOVAs with the effects of DRUG and TIME at the single SIs showed that APZ caused a significant MEP amplitude decrease at 1.4 × SI1mV (P=0.048, Figure 4a), but not at any of the other SIs.

(a-d) Drug effects (A: alprazolam (APZ), (b): zolpidem (ZLP), (c): ethanol < 10 mM (EtOH<10mM), (d): ethanol <20 mM (EtOH<20mM)) on IO-curve (y-axes, MEP amplitude in mV) at time point B1 (blue diamonds) vs B0 (black squares) compared to PBO (grey symbols). (e-h) Drug effects (e: APZ, f: ZLP, G: EtOH<10mM, h: EtOH<20mM) on PASLTD-induced change of IO-curve at time point P1 (red circles) vs B1 (blue diamonds) compared to PBO (grey symbols). X-axes: stimulus intensity (in multiples of SI1mV). Number signs indicate LTD-like plasticity in the PBO condition (#P<0.05), asterisks indicate significant difference of drug effects on IO-curve (B1 vs B0) or LTD-like plasticity (P1 vs B1) when compared to PBO (*P< 0.05). Note that APZ and ZLP but not EtOH depressed IO-curve, while both doses of EtOH but not APZ or ZLP increased PASLTD-induced LTD-like IO-curve depression.
The comparison of PBO vs ZLP revealed significant effects of DRUG (F1,9=5.207, P=0.048), DRUG*SI (F10,90=2.239, P=0.022) and DRUG*TIME*SI (F10,90=1.949, P=0.048). Post hoc two-way rmANOVAs with the effects of DRUG and TIME at the single SIs showed that ZLP caused a significant MEP amplitude decrease at 1.1 × SI1mV (P=0.016, Figure 4b), but not at any of the other SIs.
The comparison of PBO vs EtOH<10mM showed a significant DRUG*TIME*SI interaction (F10,90=2.127, P=0.030). However, post hoc two-way rmANOVAs with the effects of DRUG and TIME did not reveal a significant effect of EtOH<10mM at any of the single SIs (Figure 4c).
The comparison of PBO vs EtOH<20mM did not show a significant effect of DRUG or any of its interactions (Figure 4d).
In summary, APZ and ZLP resulted in marginal MEP amplitude decreases at single SIs while EtOH<10mM and EtOH<20mM did not change MEP amplitude. This is an important nil finding given the effects of EtOH on PASLTD-induced LTD-like plasticity (see below).
Drug Effects on MEP Amplitude During the PASLTD Intervention
MEP amplitudes recorded during the PASLTD-intervention did not differ between drug conditions, as demonstrated by a non-significant effect of DRUG (F4,32=0.347, P=0.84) and DRUG*BIN (F32,256=1.179, P=0.24) interaction. This is an important nil finding, indicating that drug condition did not alter the PASLTD intervention per se, while it did change the induction of plasticity by PASLTD (see below). Irrespective of drug condition, MEP amplitude increased over the course of PASLTD (BIN: F2.286,18.285=7.735, P=0.003) (Figure 5), similar to previous observations (Ilic et al, 2011).

MEP amplitudes measured during PASLTD intervention in the placebo (PBO), alprazolam (APZ), zolpidem (ZLP), EtOH<10mM and EtOH<20mM conditions. Each data point represents the mean±SEM of 25 consecutive trials of 10 subjects (i.e., 9 data points for 225 trials in total).
Drug Effects on PASLTD-Induced Changes of IO-Curve (Comparison of Time Point P1 vs B1)
At time point B1, there was no significant effect of DRUG (F4,36=5.886, P=0.06) but a significant DRUG*SI interaction (F40,360=2.250, P<0.001). Post hoc pair-wise comparisons of IO-curves at time point B1 between all study drugs and PBO revealed significant differences between APZ and PBO for DRUG (F1,9=7.420, P=0.023) and the DRUG*SI interaction (F10,90=7.119, P<0.001), and between ZLP and PBO for DRUG (F1,9=6.583, P=0.030) and the DRUG*SI interaction (F10,90=2.893, P=0.004). In contrast, there were no significant effects of DRUG or DRUG*SI interaction on IO-curves at time point B1 between EtOH<10mM or EtOH<20mM and PBO (all P > 0.15). The effects of APZ and ZLP were explained by a depression of the IO-curve (Figure 4). It is important to note that the lack of effects of ethanol on the IO-curve excludes a significant contribution of changes in corticospinal excitability on the enhancing effects of ethanol on PAS-induced LTD-like plasticity (see below).
In the PBO condition, there was no significant effect for TIME (F1,9=2.39, P=0.157), but the TIME*SI interaction on PASLTD-induced changes of IO-curve was significant (F10,90=3.849, P<0.0001). Post hoc testing revealed that PASLTD resulted in a decrease of IO-curve at P1 exclusively at high stimulation intensities of 130–150%SI1mV (all P<0.05, Figure 3b). This result is in line with previous studies, which showed that changes after PASLTD occurred predominantly in the high-intensity range of the IO-curve (Rosenkranz et al, 2007a; Rosenkranz et al, 2007b).
The rmANOVA including all drug conditions showed a significant effect of TIME (F1,9=11.562, P=0.008), TIME*SI (F10,90=5.228, P<0.0001) and DRUG*TIME*SI (F40,360=1.489, P=0.033), indicating that drugs modified the PASLTD-induced changes in IO-curve dependent on stimulation intensity (Figures 4e and h). The significant triple-interaction justified pairwise comparisons of the PASLTD-induced changes in IO curve between any of the drugs with PBO. The comparison of PBO vs APZ revealed significant effects of DRUG (F1,9=8.604, P=0.017), TIME (F1,9=13.159, P=0.006), DRUG*SI (F10,90=6.997, P<0.0001) and DRUG*TIME*SI (F10,90=2.890, P=0.004). However, post hoc two-way rmANOVAs with the effects of DRUG and TIME showed that APZ did not significantly alter the PASLTD effect in the PBO condition at any of the single SIs (Figure 4e).
The comparison of PBO vs ZLP showed a trend for DRUG (F1,9=5.038, P=0.051) and a significant DRUG*SI interaction (F10,90=2.101, P=0.032) but there were no effects of DRUG*TIME or DRUG*TIME*SI (Figure 4f). Therefore, ZLP did not significantly alter the PASLTD-induced IO-curve depression observed in the PBO condition.
The comparison of PBO vs EtOH<10mM revealed a significant interaction DRUG*TIME*SI (F10,90=2.371, P=0.015). Post hoc comparisons of the PASLTD effect at the single SIs showed that EtOH<10mM enhanced the MEP depression at 1.0 × SI1mV (P=0.032) and at 1.1 × SI1mV (P=0.033, Figure 4g).
The comparison of PBO vs EtOH<20mM revealed a significant interaction DRUG*TIME*SI (F10,90=1.90, P=0.05). Post hoc comparisons of the PASLTD effect at the single SIs showed that EtOH<20mM enhanced the MEP depression at 1.0 × SI1mV (P=0.033) and at 1.1 × SI1mV (P=0.032, Figure 4h).
In summary, EtOH<10mM and EtOH<20mM but not APZ or ZLP enhanced PASLTD-induced LTD-like plasticity in human M1. None of these effects correlated with serum ethanol levels at B1 or P1.
Discussion
The principal novel findings of this study were that EtOH<10mM and EtOH<20mM but not APZ or ZLP enhanced the PASLTD-induced LTD-like plasticity, while APZ and ZLP but not EtOH<10mM or EtOH<20mM decreased SPV, indexing α1-GABAAR-mediated sedation. The following paragraphs will discuss these findings in detail.
Drug Effects on Saccadic Peak Velocity (SPV)
We replicated our earlier findings that APZ and ZLP reduce SPV (Lücke et al, 2014; Premoli et al, 2014) while EtOH<10mM and EtOH<20mM had no significant effect (Lücke et al, 2014). SPV is an established sensitive electrophysiological surrogate marker of α1-GABAAR-mediated sedation (Blom et al, 1990; de Visser et al, 2003). In contrast to the classical benzodiazepine lorazepam, a selective agonist at α2-/α3-GABAARs did not decrease SPV unless high doses were given (de Haas et al, 2009). In addition, the largely selective α1-GABAAR agonist ZLP reduced SPV (de Haas et al, 2010), in accordance with our results. The nil results of EtOH on SPV are important because they demonstrate that EtOH<10mM and EtOH<20mM had no impact on α1-GABAAR-mediated neurotransmission and, therefore, sedation cannot be attributed to account for the enhancing effects of EtOH on LTD-like plasticity (see below). Our findings on a behavioral surrogate marker of α1-GABAAR-mediated inhibition are concordant with the notion that even high doses of EtOH (25–300 mM) had no effect on α1-GABAAR-mediated currents of acutely dissociated neurons from various brain regions (Criswell et al, 2003).
Drug Effects on IO-Curve
We demonstrated that APZ and ZLP but not EtOH<10mM and EtOH<20mM resulted in marginal IO-curve decreases at single stimulus intensities (Figure 4a and d). These data are largely in agreement with our previous study (Lücke et al, 2014). In particular, the present data replicate the nil effect of ethanol on IO-curve. This is also compatible with one earlier study where EtOH at a higher blood concentration (16.5±2.3 mM) than in the present study had no effect on IO-curve (Ziemann et al, 1995). This nil finding is of particular importance because it excluded changes in corticospinal excitability at the time of PASLTD that could have been responsible for the significant effects of EtOH on PASLTD-induced LTD-like plasticity (see below). Furthermore, EtOH has been implicated to inhibit NMDAR-mediated currents at doses of <25 mM (He et al, 2013; Lovinger et al, 1989, 1990; Weitlauf and Woodward, 2008). Since one previous study demonstrated a significant and dose-dependent IO-curve increase by ketamine, another NMDAR antagonist (Di Lazzaro et al, 2003), the present and previous (Lücke et al, 2014) nil findings of EtOH<10mM and EtOH<20mM on IO-curve make it unlikely that these low dosages of EtOH inhibited NMDARs to a significant extent.
In the previous study, we did not find significant IO-curve depression by APZ or ZLP (Lücke et al, 2014), but the effects in the present study were also only marginal, and significant only at single stimulation intensities. Different populations of subjects may have caused this slight discrepancy in otherwise identical experimental conditions. Also, the literature reported somewhat diverse results, with benzodiazepines such as lorazepam or diazepam having either no effect (Ilic et al, 2002; Ziemann et al, 1996) or producing a moderate IO-curve depression (Boroojerdi et al, 2001; Kimiskidis et al, 2006).
Drug Effects on PASLTD-Induced Changes of the IO-Curve
PASLTD-induced IO-curve depression is an established model of LTD-like plasticity at the systems level of human motor cortex (for review, (Müller-Dahlhaus et al, 2010)). Although this is not equivalent to LTD as studied at the cellular level, several properties suggest that PASLTD-induced corticospinal plasticity has overlapping mechanisms with LTD, and therefore has been termed LTD-like plasticity: it outlasts the PASLTD intervention by >30 min (Müller et al, 2007; Wolters et al, 2003), is associative as intervals between the electrical stimulus to median nerve and TMS of the contralateral M1 significantly different from N20-5 ms or 10 ms do not result in MEP depression (Wolters et al, 2003), is input specific, i.e. MEP depression occurs only in hand muscles supplied by the stimulated median nerve but not in those supplied by the ulnar nerve (Weise et al, 2006), and can be abolished by the NMDAR antagonist dextromethorphan and the voltage gated calcium channel blocker nimodipine (Wolters et al, 2003). In addition, epidural recordings from the cervical spinal cord and brain stem stimulation experiments verified that the PASLTD-induced MEP depression originates at the level of M1 rather than in the spinal cord (Di Lazzaro et al, 2009; Wolters et al, 2003). Finally, the neurobiological relevance of this form of LTD-like plasticity has been demonstrated by its homeostatic interactions with motor learning (Jung and Ziemann, 2009; Rosenkranz et al, 2007a; Ziemann et al, 2004), and its exaggerated expression in musicians (Rosenkranz et al, 2007b), focal hand dystonia (Weise et al, 2006), or after upper limb immobilization (Rosenkranz et al, 2014).
In agreement with previous studies (Rosenkranz et al, 2007a; Rosenkranz et al, 2007b), we found that the PASLTD-induced depression of the IO-curve in the PBO condition occurred specifically in the high-intensity range (1.3–1.5 × SI1mV, Figure 3b). This may reflect that the so called I1-wave caused by presumably direct transsynaptic activation of corticospinal neurons by TMS remained unchanged by PASLTD, while late I-waves (I2-I4-waves) caused by presumably indirect transsynaptic activation of corticospinal neurons through higher-order interneurons (for review, (Di Lazzaro et al, 2008)) were depressed by PASLTD (Di Lazzaro et al, 2009). This notion is supported by the finding that the low-intensity part of the IO-curve, not changed by PASLTD, is largely based on recruitment of the I1-wave if the induced current in M1 runs in posterior-to-anterior direction, while the high-intensity part of the IO-curve depressed by PASLTD is based on additional recruitment of late I-waves (Di Lazzaro et al, 1998a; Di Lazzaro et al, 1998b). The implication is that late I-waves but not the I1-wave are controlled by GABAAergic inhibition (Di Lazzaro et al, 2000; Di Lazzaro et al, 1998c). Therefore, we had speculated that increased GABAAergic neurotransmission by the drugs tested here would result in enhanced PASLTD-induced LTD-like IO-curve depression.
We found that EtOH<10mM and EtOH<20mM but not APZ or ZLP enhanced significantly PASLTD-induced LTD-like plasticity in human cortex at stimulus intensities of 1.0–1.1 × SI1mV (Figure 4e and h). The enhancing effect of EtOH on PASLTD-induced LTD-like IO-curve depression specifically at these mid-range intensities may be explained by the fact that late I-waves are just coming into play here (Di Lazzaro et al, 1998a; Di Lazzaro et al, 1998b), so that it may require the synergistic action of PASLTD and drug to result in MEP depression.
An important implication, also given the discussion above, is that the enhancement of the PASLTD-induced LTD-like plasticity by low-dose EtOH but not APZ or ZLP strongly suggests that this effect was mediated by increased tonic inhibition mediated by the extrasynaptic α4ß3δ-GABAAR. The double dissociation of the effects of EtOH vs APZ and ZLP on SPV (see above) vs PASLTD-induced LTD-like plasticity clearly speaks against a mechanism mediated by the α1-GABAAR. Modification of synaptic inhibition mediated through the α2-, α3- or α5-GABAAR is similarly unlikely because alprazolam exerts full positive allosteric modulation at the benzodiazepine-binding site of all of these receptors. Short-interval intracortical inhibition, a putative paired-pulse TMS marker of neurotransmission through the α2- and α3-GABAAR (Di Lazzaro et al, 2007; Di Lazzaro et al, 2006; Ziemann et al, 2014) has not been measured in the present study. This is a limitation, but previous studies demonstrated that classical benzodiazepines and ethanol increased short-interval intracortical inhibition, while zolpidem did not (Di Lazzaro et al, 2007; Di Lazzaro et al, 2006; Ziemann et al, 1995; Ziemann et al, 1996). Therefore, the uniform effects of benzodiazepines and ethanol on short-interval intracortical inhibition, i.e., α2- and α3-GABAAR mediated inhibition, would not explain their different effects on PAS-induced LTD-like plasticity observed in the present study. Finally a mechanism related to blockade of the NMDAR by EtOH was also unlikely to be involved because effective blockade of the NMDAR by dextromethorphan resulted in abolition rather than enhancement of PASLTD-induced LTD-like plasticity (Wolters et al, 2003).
These conclusions are compatible with the reported high sensitivity of the α4ß3δ-GABAAR to low-dose EtOH (Hanchar et al, 2006; Sundstrom-Poromaa et al, 2002; Wallner et al, 2003; Wei et al, 2004) even if this was not a unanimously supported finding in all studies (Borghese et al, 2006; Yamashita et al, 2006). The extrasynaptic α6ß3δ-GABAAR also exhibits high sensitivity to EtOH (Hanchar et al, 2006; Wallner et al, 2003; Wei et al, 2004) but is expressed exclusively in cerebellar granule cells, in contrast to the α4ß3δ-GABAAR that is widely distributed in the brain including neocortex (Pirker et al, 2000). As PASLTD-induced LTD-like plasticity occurs in M1 (Di Lazzaro et al, 2009; Wolters et al, 2003) we believe that the most likely candidate mechanism for the enhancement of PASLTD-induced LTD-like plasticity by low-dose EtOH is enhancement of extrasynaptic tonic inhibition through the α4ß3δ-GABAAR.
In our previous study we have reported a similarly selective suppression of LTP-like plasticity by EtOH<10mM and EtOH<20mM in the absence of significant effects by APZ or ZLP (Lücke et al, 2014). Together, these findings support the novel view that extrasynaptic tonic inhibition may constitute a key mechanism in regulating synaptic plasticity in human neocortex: enhancement of tonic inhibition leads to a rightward shift of a cell’s input-output relationship through a more hyperpolarized compared to normal state (Mitchell and Silver, 2003), a condition that favors LTD over LTP in neocortical circuits (Artola et al, 1990). Findings may stimulate cellular research on the role of tonic inhibition in regulating excitability and plasticity of cortical neuronal networks.
Behavioral Significance of the Present Findings
LTD of excitatory synaptic neurotransmission, besides LTP, is a leading candidate mechanism for memory and learning processes (Collingridge et al, 2010; Malenka and Bear, 2004). In addition, several studies have provided evidence that LTD in the nucleus accumbens and prefrontal cortex of rats plays a pivotal supportive role in drug addiction (Brebner et al, 2005; Van den Oever et al, 2008). It may be speculated that the enhancing effect of low-dose ethanol on LTD-like plasticity in the present study relates to detrimental effects of acute ethanol ingestion on memory formation and learning (Lister et al, 1991; Mattila et al, 1998; Zorumski et al, 2014). However, this remains to be tested in further experiments as memory or learning processes have not been studied here. Furthermore, the existence of any relation between enhancement of LTD by ethanol in primary motor cortex in the present study and LTD-dependent addictive behavior mediated by non-overlapping neurocircuits such as nucleus accumbens and prefrontal cortex (Collingridge et al, 2010; Noori et al, 2012) is entirely unclear. Therefore, whether the increase of PAS-induced LTD-like plasticity in motor cortex by low-dose ethanol provides a surrogate marker for the addictive potential of ethanol in given individuals would require further deliberate testing. On the other hand, it is of interest to note that several recent therapeutic trials applied high-frequency repetitive TMS to the dorsolateral prefrontal cortex of alcohol addicts and observed a decrease in spontaneous alcohol craving (Gorelick et al, 2014; Höppner et al, 2011; Mishra et al, 2010). High-frequency repetitive TMS typically leads to LTP-like plasticity (for review, (Ziemann et al, 2008)) so that these therapeutic effects may be explained by counteracting LTD-dependent mechanisms of addiction, in addition to other potential mechanisms, such as modulation of dopaminergic neurotransmission in addiction-related subcortical target areas of the dorsolateral prefrontal cortex (Feil and Zangen, 2010; Naim-Feil and Zangen, 2013).
Funding and Disclosure
The authors declare no conflict of interest.
References
Artola A, Brocher S, Singer W (1990). Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature 347: 69–72.
Belmeguenai A, Botta P, Weber JT, Carta M, De Ruiter M, De Zeeuw CI et al (2008). Alcohol impairs long-term depression at the cerebellar parallel fiber-Purkinje cell synapse. J Neurophysiol 100: 3167–3174.
Blom MW, Bartel PR, de Sommers K, Van der Meyden CH, Becker PJ (1990). The effects of alprazolam, quazepam and diazepam on saccadic eye movements, parameters of psychomotor function and the EEG. Fundam Clin Pharmacol 4: 653–661.
Borghese CM, Storustovu S, Ebert B, Herd MB, Belelli D, Lambert JJ et al (2006). The delta subunit of gamma-aminobutyric acid type A receptors does not confer sensitivity to low concentrations of ethanol. J Pharmacol Exp Ther 316: 1360–1368.
Boroojerdi B, Battaglia F, Muellbacher W, Cohen LG (2001). Mechanisms influencing stimulus-response properties of the human corticospinal system. Clin Neurophysiol 112: 931–937.
Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S et al (2005). Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310: 1340–1343.
Carta M, Mameli M, Valenzuela CF (2006). Alcohol potently modulates climbing fiber—>Purkinje neuron synapses: role of metabotropic glutamate receptors. J Neurosci 26: 1906–1912.
Choi S-Y, Morales B, Lee H-K, Kirkwood A (2002). Absence of Long-Term Depression in the Visual Cortex of Glutamic Acid Decarboxylase-65 Knock-Out Mice. J Neurosci 22: 5271–5276.
Collingridge GL, Peineau S, Howland JG, Wang YT (2010). Long-term depression in the CNS. Nat Rev Neurosci 11: 459–473.
Criswell HE, Ming Z, Griffith BL, Breese GR (2003). Comparison of effect of ethanol on N-methyl-D-aspartate- and GABA-gated currents from acutely dissociated neurons: absence of regional differences in sensitivity to ethanol. J Pharmacol Exp Ther 304: 192–199.
de Haas SL, Franson KL, Schmitt JA, Cohen AF, Fau JB, Dubruc C et al (2009). The pharmacokinetic and pharmacodynamic effects of SL65.1498, a GABA-A alpha2,3 selective agonist, in comparison with lorazepam in healthy volunteers. J Psychopharmacol 23: 625–632.
de Haas SL, Schoemaker RC, van Gerven JM, Hoever P, Cohen AF, Dingemanse J (2010). Pharmacokinetics, pharmacodynamics and the pharmacokinetic/ pharmacodynamic relationship of zolpidem in healthy subjects. J Psychopharmacol 24: 1619–1629.
de Visser SJ, van der Post JP, de Waal PP, Cornet F, Cohen AF, van Gerven JM (2003). Biomarkers for the effects of benzodiazepines in healthy volunteers. Br J Clin Pharmacol 55: 39–50.
Delvendahl I, Lindemann H, Heidegger T, Normann C, Ziemann U, Mall V (2013). Effects of lamotrigine on human motor cortex plasticity. Clin Neurophysiol 124: 148–153.
Di Lazzaro V, Dileone M, Profice P, Pilato F, Oliviero A, Mazzone P et al (2009). LTD-like plasticity induced by paired associative stimulation: direct evidence in humans. Exp Brain Res 194: 661–664.
Di Lazzaro V, Oliviero A, Meglio M, Cioni B, Tamburrini G, Tonali P et al (2000). Direct demonstration of the effect of lorazepam on the excitability of the human motor cortex. Clin Neurophysiol 111: 794–799.
Di Lazzaro V, Oliviero A, Profice P, Pennisi MA, Pilato F, Zito G et al (2003). Ketamine increases motor cortex excitability to transcranial magnetic stimulation. J Physiol 547: 485–496.
Di Lazzaro V, Oliviero A, Profice P, Saturno E, Pilato F, Insola A et al (1998a). Comparison of descending volleys evoked by transcranial magnetic and electric stimulation in conscious humans. Electroencephalogr Clin Neurophysiol 109: 397–401.
Di Lazzaro V, Pilato F, Dileone M, Profice P, Ranieri F, Ricci V et al (2007). Segregating two inhibitory circuits in human motor cortex at the level of GABAA receptor subtypes: A TMS study. Clin Neurophysiol 118: 2207–2214.
Di Lazzaro V, Pilato F, Dileone M, Ranieri F, Ricci V, Profice P et al (2006). GABAA receptor subtype specific enhancement of inhibition in human motor cortex. J Physiol 575: 721–726.
Di Lazzaro V, Restuccia D, Oliviero A, Profice P, Ferrara L, Insola A et al (1998b). Effects of voluntary contraction on descending volleys evoked by transcranial magnetic stimulation in conscious humans. J Physiol 508: 625–633.
Di Lazzaro V, Restuccia D, Oliviero A, Profice P, Ferrara L, Insola A et al (1998c). Magnetic transcranial stimulation at intensities below active motor threshold activates intracortical inhibitory circuits. Exp Brain Res 119: 265–268.
Di Lazzaro V, Ziemann U, Lemon RN (2008). State of the art: Physiology of transcranial motor cortex stimulation. Brain Stimulation 1: 345–362.
Feil J, Zangen A (2010). Brain stimulation in the study and treatment of addiction. Neurosci Biobehav Rev 34: 559–574.
Gorelick DA, Zangen A, George MS (2014). Transcranial magnetic stimulation in the treatment of substance addiction. Ann NY Acad Sci 1327: 79–93.
Greenblatt DJ, Wright CE (1993). Clinical pharmacokinetics of alprazolam. Therapeutic implications. Clin Pharmacokinet 24: 453–471.
Groppa S, Oliviero A, Eisen A, Quartarone A, Cohen LG, Mall V et al (2012). A practical guide to diagnostic transcranial magnetic stimulation: Report of an IFCN committee. Clin Neurophysiol 123: 858–882.
Hanchar HJ, Chutsrinopkun P, Meera P, Supavilai P, Sieghart W, Wallner M et al (2006). Ethanol potently and competitively inhibits binding of the alcohol antagonist Ro15-4513 to alpha4/6beta3delta GABAA receptors. Proc Natl Acad Sci USA 103: 8546–8551.
He Q, Titley H, Grasselli G, Piochon C, Hansel C (2013). Ethanol affects NMDA receptor signaling at climbing fiber-Purkinje cell synapses in mice and impairs cerebellar LTD. J Neurophysiol 109: 1333–1342.
Hendricson AW, Miao CL, Lippmann MJ, Morrisett RA (2002). Ifenprodil and ethanol enhance NMDA receptor-dependent long-term depression. J Pharmacol Exp Ther 301: 938–944.
Hess G, Donoghue JP (1996). Long-term depression of horizontal connections in rat motor cortex. Eur J Neurosci 8: 658–665.
Höppner J, Broese T, Wendler L, Berger C, Thome J (2011). Repetitive transcranial magnetic stimulation (rTMS) for treatment of alcohol dependence. World J Biol Psychiatry 12: 57–62.
Huber R, Määttä S, Esser SK, Sarasso S, Ferrarelli F, Watson A et al (2008). Measures of cortical plasticity after transcranial paired associative stimulation predict changes in electroencephalogram slow-wave activity during subsequent sleep. J Neurosci 28: 7911–7918.
Ilic NV, Milanovic S, Krstic J, Bajec DD, Grajic M, Ilic TV (2011). Homeostatic modulation of stimulation-dependent plasticity in human motor cortex. Physiol Res 60: S107–S112.
Ilic TV, Meintzschel F, Cleff U, Ruge D, Kessler KR, Ziemann U (2002). Short-interval paired-pulse inhibition and facilitation of human motor cortex: the dimension of stimulus intensity. J Physiol 545: 153–167.
Izumi Y, Nagashima K, Murayama K, Zorumski CF (2005). Acute effects of ethanol on hippocampal long-term potentiation and long-term depression are mediated by different mechanisms. Neuroscience 136: 509–517.
Jung P, Ziemann U (2009). Homeostatic and non-homeostatic modulation of learning in human motor cortex. J Neurosci 29: 5597–5604.
Kamke MR, Ryan AE, Sale MV, Campbell ME, Riek S, Carroll TJ et al (2014). Visual spatial attention has opposite effects on bidirectional plasticity in the human motor cortex. J Neurosci 34: 1475–1480.
Keel JC, Smith MJ, Wassermann EM (2001). A safety screening questionnaire for transcranial magnetic stimulation [letter]. Clin Neurophysiol 112: 720.
Kimiskidis VK, Papagiannopoulos S, Kazis DA, Sotirakoglou K, Vasiliadis G, Zara F et al (2006). Lorazepam-induced effects on silent period and corticomotor excitability. Exp Brain Res 173: 603–611.
Lister RG, Gorenstein C, Fisher-Flowers D, Weingartner HJ, Eckardt MJ (1991). Dissociation of the acute effects of alcohol on implicit and explicit memory processes. Neuropsychologia 29: 1205–1212.
Lovinger DM, White G, Weight FF (1989). Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243: 1721–1724.
Lovinger DM, White G, Weight FF (1990). NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci 10: 1372–1379.
Lücke C, Heidegger T, Röhner M, Toennes SW, Krivanekova L, Müller-Dahlhaus F et al (2014). Deleterious Effects of a Low Amount of Ethanol on LTP-Like Plasticity in Human Cortex. Neuropsychopharmacology 39: 1508–1518.
Malenka RC, Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44: 5–21.
Mattila MJ, Vanakoski J, Kalska H, Seppala T (1998). Effects of alcohol, zolpidem, and some other sedatives and hypnotics on human performance and memory. Pharmacol Biochem Behav 59: 917–923.
Mishra BR, Nizamie SH, Das B, Praharaj SK (2010). Efficacy of repetitive transcranial magnetic stimulation in alcohol dependence: a sham-controlled study. Addiction 105: 49–55.
Mitchell SJ, Silver RA (2003). Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron 38: 433–445.
Möller C, Arai N, Lücke J, Ziemann U (2009). Hysteresis effects on the input-output curve of motor evoked potentials. Clin Neurophysiol 120: 1003–1008.
Müller JF, Orekhov Y, Liu Y, Ziemann U (2007). Homeostatic plasticity in human motor cortex demonstrated by two consecutive sessions of paired associative stimulation. Eur J Neurosci 25: 3461–3468.
Müller-Dahlhaus F, Ziemann U, Classen J (2010). Plasticity resembling spike-timing dependent synaptic plasticity: the evidence in human cortex. Front Syn Neurosci 2: 1–11.
Müller-Dahlhaus JF, Orekhov Y, Liu Y, Ziemann U (2008). Interindividual variability and age-dependency of motor cortical plasticity induced by paired associative stimulation. Exp Brain Res 187: 467–475.
Naim-Feil J, Zangen A (2013). Addiction. Handb Clin Neurol 116: 613–630.
Ni Z, Gunraj C, Kailey P, Cash RF, Chen R (2014). Heterosynaptic modulation of motor cortical plasticity in human. J Neurosci 34: 7314–7321.
Nishiyama M, Togashi K, Aihara T, Hong K (2010). GABAergic activities control spike timing- and frequency-dependent long-term depression at hippocampal excitatory synapses. Front Synaptic Neurosci 2: 22.
Noori HR, Spanagel R, Hansson AC (2012). Neurocircuitry for modeling drug effects. Addict Biol 17 (5): 827–864.
Oldfield RC (1971). The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 9: 97–113.
Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G (2000). GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101: 815–850.
Premoli I, Castellanos N, Rivolta D, Belardinelli P, Bajo R, Zipser C et al (2014). TMS-EEG signatures of GABAergic neurotransmission in the human cortex. J Neurosci 34: 5603–5612.
Rosenkranz K, Kacar A, Rothwell JC (2007a). Differential modulation of motor cortical plasticity and excitability in early and late phases of human motor learning. J Neurosci 27: 12058–12066.
Rosenkranz K, Seibel J, Kacar A, Rothwell J (2014). Sensorimotor deprivation induces interdependent changes in excitability and plasticity of the human hand motor cortex. J Neurosci 34: 7375–7382.
Rosenkranz K, Williamon A, Rothwell JC (2007b). Motorcortical Excitability and Synaptic Plasticity Is Enhanced in Professional Musicians. J Neurosci 27: 5200–5206.
Smith MJ, Adams LF, Schmidt PJ, Rubinow DR, Wassermann EM (2002). Effects of ovarian hormones on human cortical excitability. Ann Neurol 51: 599–603.
Smith MJ, Keel JC, Greenberg BD, Adams LF, Schmidt PJ, Rubinow DA et al (1999). Menstrual cycle effects on cortical excitability. Neurology 53: 2069–2072.
Stefan K, Wycislo M, Classen J (2004). Modulation of associative human motor cortical plasticity by attention. J Neurophysiol 92: 66–72.
Su LD, Sun CL, Shen Y (2010). Ethanol acutely modulates mGluR1-dependent long-term depression in cerebellum. Alcohol Clin Exp Res 34: 1140–1145.
Sundstrom-Poromaa I, Smith DH, Gong QH, Sabado TN, Li X, Light A et al (2002). Hormonally regulated alpha(4)beta(2)delta GABA(A) receptors are a target for alcohol. Nat Neurosci 5: 721–722.
Thirugnanasambandam N, Grundey J, Adam K, Drees A, Skwirba AC, Lang N et al (2011). Nicotinergic Impact on Focal and Non-Focal Neuroplasticity Induced by Non-Invasive Brain Stimulation in Non-Smoking Humans. Neuropsychopharmacology 36: 879–886.
Ulrich L, Cramer Y, Zink P (1987). [Relevance of individual parameters in the calculation of blood alcohol levels in relation to the volume of intake]. Blutalkohol 24: 192–198.
Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN et al (2008). Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci 11: 1053–1058.
Velazquez-Perez L, Seifried C, Santos-Falcon N, Abele M, Ziemann U, Almaguer LE et al (2004). Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann Neurol 56: 444–447.
Voytovych H, Krivanekova L, Ziemann U (2012). Lithium: A switch from LTD- to LTP-like plasticity in human cortex. Neuropharmacology 63: 274–279.
Wallner M, Hanchar HJ, Olsen RW (2003). Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci USA 100: 15218–15223.
Watanabe K, Kamatani D, Hishida R, Kudoh M, Shibuki K (2007). Long-term depression induced by local tetanic stimulation in the rat auditory cortex. Brain Res 1166: 20–28.
Wei W, Faria LC, Mody I (2004). Low ethanol concentrations selectively augment the tonic inhibition mediated by delta subunit-containing GABAA receptors in hippocampal neurons. J Neurosci 24: 8379–8382.
Weise D, Schramm A, Stefan K, Wolters A, Reiners K, Naumann M et al (2006). The two sides of associative plasticity in writer's cramp. Brain 129: 2709–2721.
Weitlauf C, Woodward JJ (2008). Ethanol selectively attenuates NMDAR-mediated synaptic transmission in the prefrontal cortex. Alcohol Clin Exp Res 32: 690–698.
Welling PG, Lyons LL, Elliott R, Amidon GL (1977). Pharmacokinetics of alcohol following single low doses to fasted and nonfasted subjects. J Clin Pharmacol 17: 199–206.
Widmark E (1932) Die theoretischen Grundlagen und die praktische Verwendbarkeit der gerichtlich-medizinischen Alkoholbestimmung. Urban und Schwarzenberg: Berlin, Wien.
Wolters A, Sandbrink F, Schlottmann A, Kunesch E, Stefan K, Cohen LG et al (2003). A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. J Neurophysiol 89: 2339–2345.
Yamashita M, Marszalec W, Yeh JZ, Narahashi T (2006). Effects of ethanol on tonic GABA currents in cerebellar granule cells and mammalian cells recombinantly expressing GABA(A) receptors. J Pharmacol Exp Ther 319: 431–438.
Yin HH, Park BS, Adermark L, Lovinger DM (2007). Ethanol reverses the direction of long-term synaptic plasticity in the dorsomedial striatum. Eur J Neurosci 25: 3226–3232.
Zhu YY, Jing L, Duan TT, Yuan Q, Cao J, Zhou QX et al (2013). Patterned high-frequency stimulation induces a form of long-term depression dependent on GABAA and mACh receptors in the hippocampus. Neuroscience 250: 658–663.
Ziemann U, Ilic TV, Pauli C, Meintzschel F, Ruge D (2004). Learning modifies subsequent induction of LTP-like and LTD-like plasticity in human motor cortex. J Neurosci 24: 1666–1672.
Ziemann U, Lönnecker S, Paulus W (1995). Inhibition of human motor cortex by ethanol. A transcranial magnetic stimulation study. Brain 118: 1437–1446.
Ziemann U, Lönnecker S, Steinhoff BJ, Paulus W (1996). The effect of lorazepam on the motor cortical excitability in man. Exp Brain Res 109: 127–135.
Ziemann U, Paulus W, Nitsche MA, Pascual-Leone A, Byblow WD, Berardelli A et al (2008). Consensus: Motor cortex plasticity protocols. Brain Stimulation 1: 164–182.
Ziemann U, Reis J, Schwenkreis P, Rosanova M, Strafella A, Badawy R et al (2014). TMS and drugs revisited 2014. Clin Neurophysiol (doi:10.1016/j.clinph.2014.08.028).
Zorumski CF, Mennerick S, Izumi Y (2014). Acute and chronic effects of ethanol on learning-related synaptic plasticity. Alcohol 48: 1–17.
Acknowledgements
The authors declare that over the past three years the employment contract of AF has been supported by TEVA GmbH Deutschland, and UZ has received honoraria from Biogen Idec Deutschland GmbH, Bayer Vital GmbH, Medtronic and Servier for advisory work. These affiliations are unrelated to the work presented in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fuhl, A., Müller-Dahlhaus, F., Lücke, C. et al. Low Doses of Ethanol Enhance LTD-like Plasticity in Human Motor Cortex. Neuropsychopharmacol 40, 2969–2980 (2015). https://doi.org/10.1038/npp.2015.151
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.151
