
Abstract
Fear to predictable threat and anxiety to unpredictable threat reflect distinct processes mediated by different brain structures, the central nucleus of the amygdala and the bed nucleus of the stria terminalis (BNST), respectively. This study tested the hypothesis that the corticotropin-releasing factor (CRF1) antagonist GSK561679 differentially reduces anxiety but increases fear in humans. A total of 31 healthy females received each of four treatments: placebo, 50 mg GSK561679 (low-GSK), 400 mg GSK561679 (high-GSK), and 1 mg alprazolam in a crossover design. Participants were exposed to three conditions during each of the four treatments. The three conditions included one in which predictable aversive shocks were signaled by a cue, a second during which shocks were administered unpredictably, and a third condition without shock. Fear and anxiety were assessed using the acoustic startle reflex. High-GSK had no effect on startle potentiation during unpredictable threat (anxiety) but increased startle potentiation during the predictable condition (fear). Low-GSK did not affect startle potentiation across conditions. Consistent with previous findings, alprazolam reduced startle potentiation during unpredictable threat but not during predictable threat. The increased fear by high-GSK replicates animal findings and suggests a lift of the inhibitory effect of the BNST on the amygdala by the CRF1 antagonist.
Similar content being viewed by others
INTRODUCTION
The involvement of corticotropin-releasing factor (CRF) receptors in anxiety and aversive states is well established (Buwalda, 1997; Liang et al, 1992). CRF1 receptors are centrally expressed in brain regions mediating these states, such as the extended amygdala (Griebel and Holsboer, 2012; Valdez, 2006). Clinically, CRF dysregulation occurs in mood and anxiety disorders (Baker et al, 1999; Ishitobi et al, 2012; Keck et al, 2008; Sautter et al, 2003). Such research has generated interest in CRF1 receptor antagonists as potential treatments for mood and anxiety disorders. Nevertheless, inconsistent evidence of efficacy in major depression (Binneman et al, 2008; Holsboer and Ising, 2008) and generalized anxiety disorder (Coric et al, 2010) led to a reconsideration. Specifically, CRF1 antagonists may reduce responses to acute rather than chronic stress (Koob and Zorrilla, 2012). However, minimal research has examined this issue in humans (Bailey et al, 2011; Binneman et al, 2008), and the current study fills this gap by examining the effects of a CRF1 antagonist GSK561679 (Verucerfont) on startle, a validated, cross-species marker, in healthy individuals (Grillon, 2008).
Research on startle in rodents implicates CRF1 in aversive states (Griebel and Holsboer, 2012; Koob, 2008; Valdez, 2006). Although startle is potentiated by aversive states, distinct neurocircuitry mediates potentiation in response to distinct types of threats (see Davis et al, 2010 for a review). Predictable threats evoke a short-duration startle potentiation mediated by the medial portion of the central nucleus of the amygdala (mCeA), whereas unpredictable threats evoke a longer-duration startle potentiation mediated by the bed nucleus of the stria terminalis (BNST) (Davis et al, 2010). Thus, mCeA and BNST support distinct aversive states, akin to fear, a defensive response to an explicit threat, and anxiety, a more sustained state of apprehension about uncertain future threat, respectively (Davis et al, 2010). In addition, the BNST, although not necessary for fear expression, has inhibitory influence on the mCeA (Campeau et al, 1997; Haufler et al, 2013) and on fear expression (Kim et al, 2013; Meloni et al, 2006; Walker et al, 2009b).
This study examined the effect of GSK561679 on an experimental model of fear and anxiety using the startle reflex. Fear was evoked by threat cues that predicted a shock, and anxiety was evoked by unpredictable shock (Schmitz and Grillon, 2012). Consistent with this animal literature, clinical studies using the experimental model dissociate a phasic fear response from a more sustained anxiety state in humans. Indeed, individuals with anxiety disorders display normal fear-potentiated startle to predictable threat but enhanced anxiety-potentiated startle to unpredictable threat (Grillon et al, 2008, 2009b). This latter response is reduced by antianxiety drugs such as the benzodiazepine alprazolam (Grillon et al, 2006) and the selective serotonin reuptake inhibitor citalopram (Grillon et al, 2009a). Thus, unpredictable shock evokes an anticipatory anxiety state that is increased in clinical anxiety and is reduced by anxiolytic treatments.
CRF1 receptors also differentially affect these two aversive states in rodents (Refojo et al, 2011; Sink et al, 2013). In this species, sustained anxiety is maintained by activation of CRF1 receptors and blocked by BNST infusion of CRH1 antagonists (Davis et al, 2010; Lee and Davis, 1997), whereas fear is either not affected or even enhanced by CRF1 antagonists (Meloni et al, 2006; Walker et al, 2009a, 2009b). This latter effect is consistent with an inhibitory role of the BNST on fear output circuit (Campeau et al, 1997; Haufler et al, 2013; Kim et al, 2013).
This study (ClinicalTrial.gov identifier, NCT01059227) extends this work to humans by comparing the effects on fear-potentiated and anxiety-potentiated startle across four medication conditions: high-dose GSK561679 (high-GSK), low-dose GSK561679 (low-GSK), placebo, and an active control (alprazolam) (Grillon et al, 2004). GSK561679 is an investigational drug for the treatment of mood and anxiety disorders. It is a safe, orally active, potent, and highly selective antagonist at the CRF1 receptor with good brain penetration and good in vitro and metabolic stability (Dunlop et al, 2014). GSK561679 has anxiolytic-like effects in the human threat test in marmosets (Fabio et al, 2008), but in humans it showed no efficacy in a major depression trial (Protocol no. CRS106139). Based on data in rodents, we expected GSK561679 to decrease anxiety-potentiated startle and to have either no effect or increase fear-potentiated startle (Walker et al, 2009a). Based on our previous work (Grillon et al, 2006), we also hypothesized that alprazolam would reduce anxiety-potentiated startle without affecting fear-potentiated startle.
MATERIALS AND METHODS
Participants
Only women were tested, as studies in animals showed reversible effects on sperm production (Dunlop et al, 2014). Subjects were paid healthy volunteers who gave written informed consent approved by the NIMH Human Investigation Review Board. Inclusion criteria included: (1) no past or current psychiatric disorders as per the Structured Clinical Interview for DSM-IV (First et al, 2002, 2) no history of a psychiatric disorder in any first-degree relatives, (3) no medical condition that interfered with the objectives of the study as established by a physician, (4) the use of two adequate means of birth control (see Supplementary Material for additional information), and (5) no use of illicit drugs or psychoactive medications as per history and confirmed by a negative urine screen. Participants met with a clinician before providing consent. In all, 39 subjects enrolled in the study and 31 (mean=30.4 years, SD=6.1 years) completed all of the sessions. One was excluded for failure to adhere to the study protocol (eg, no alcohol in the hours following a session), two decided not to participate in the study after providing written informed consent, one moved out of the state and did not want to come back for the last testing session, one used drug after the screening but before the first testing and was excluded from the study, one experienced nose bleed and heavy menses after low-GSK, one vomited after high-GSK, and one withdrew after personal problems unrelated to the study. Their mean score on the trait portions of the Spielberger State/Trait Anxiety Inventory (Spielberger, 1983) was 28.2 (SD=4.8). Body mass index of the study completers ranged from 18 to 33 (mean 24.2, SD 3.5).
Drugs
A double-blind, crossover design was implemented with each subject being exposed to each treatment—placebo, 1 mg alprazolam, 50 mg low-GSK, and 400 mg high-GSK—on four separate sessions. The treatments were given as identical-appearing capsules in two doses. The first, either one of the GSK561679 compounds or placebo, was given 3 h before testing. The second, either alprazolam or placebo, was given 1 h before testing. These timings were based on the pharmacokinetic of these drugs (Greenblatt and Wright, 1993; Tellew et al, 2010). Treatments were given following a Latin square design.
Procedure
The procedure was similar to that of our previous psychopharmacology studies examining responses to predictable and unpredictable shocks (Grillon et al, 2009a; Schmitz and Grillon, 2012). Subjects meeting eligibility criteria were invited to participate in an additional screening to examine their startle reactivity and tolerance of the shock. Participants underwent a startle assessment procedure with nine startle stimuli presented every 18–25 s. Following startle assessment, a shock workup procedure was initiated to deliver shocks on the nondominant wrist at a level that was highly unpleasant. Subjects were enrolled in the study to start on a later day if they showed a robust startle response and tolerated shocks between 3 and 5 μAmp.
Subjects then participated in four identical testing sessions separated by 6–20 days. A timeline of events is shown in Table 1. At the beginning of each testing day, the study was initiated only after verifying negative pregnancy and drug tests. During the procedure, subjects sat in a comfortable medical reclining chair. Psychophysiological recording electrodes were set up and a new baseline startle assessment (startle habituation no. 1) and new shock workup were initiated, identical to that of the first visit. Following the shock workup, the first drug ingestion took place. Two hours later, after a light meal, the second drug was administered. Approximately 50 min after the second drug ingestion, a second startle assessment (startle habituation no. 2) was performed. After 10 min, 3 h after the first drug administration, the threat experiment was initiated.
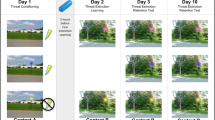
We used a test procedure with three 150-s conditions: No-threat (N), Predictable (P) threat, and Unpredictable (U) threat (NPU threat test; see Figure 1 and Schmitz and Grillon, 2012 for detailed explanations). The NPU verbal threat test can detect anxiolytic and anxiogenic effects of established and novel compounds (Grillon et al, 2006, 2007, 2009a, 2013). In addition, verbal threat is a reliable and replicable way of studying fear-potentiated startle in repeated designs for drug studies (Klumpers et al, 2010). In each condition, an 8-s cue was presented four times. The cues consisted of differently colored geometric shapes for the different conditions (eg, blue triangle for N, red square for P, and green square for U). The cues signaled a shock only in the P condition; they had no signal value in the N or U conditions.

Schematic of the threat experiment. There were three conditions—no-shock (N), predictable shock (P), and unpredictable shock (U)—presented in two orders, each including three N, two P, and two U in each of the two orders (UNPNPNU as shown or PNUNUNP). Each N, P, and U condition contained four 8-s cues of different colors and geometric shapes (for illustration purposes, the cues are squares in N, circles in P, and triangles in U). In each P condition, a shock (indicated by  ) was randomly associated with one of the four threat cues; it was administered 7.5 s after its onset. In each U condition, a shock was administered randomly in the absence of the cues. In the N condition, no shock was administered. Startle stimuli (indicated by ↑) were delivered in the presence and in the absence of the cue (ie, during intertrial intervals).
) was randomly associated with one of the four threat cues; it was administered 7.5 s after its onset. In each U condition, a shock was administered randomly in the absence of the cues. In the N condition, no shock was administered. Startle stimuli (indicated by ↑) were delivered in the presence and in the absence of the cue (ie, during intertrial intervals).
Participants received precise instructions with regard to risk of shock in each condition, including the contingency between shocks and cues in P and U. Instructions were also shown on a computer monitor throughout the experiment displaying the following information: ‘no shock’ (N), ‘shock only during red square’ (P), or ‘shock at any time’ (U). In each N, P, and U condition, six acoustic startle stimuli were delivered: (1) three during intertrial intervals (ITIs) (ie, in the absence of cues): one at 15–52 s, a second at 53–96 s, and a third at 97–140 s after the beginning of a condition; and (2) one during three of the four cues, 5–7 s after cue onset.
The threat experiment consisted of two threat series with a 5–10-min rest between threat series. Each series started with the delivery of four startle stimuli (pre-threat startle) to reduce initial startle reactivity and consisted of three N, two P, and two U conditions in one of the following two orders: P N U N U N P or U N P N P N U. Each participant received both orders, with one-half of the participants starting with P and the other one-half starting with U. One shock was administered in each individual P and U condition for a total of four shocks in P and four shocks in U. In each P, the shock was randomly associated with one of the four threat cues, being administered 7.5 s after the onset, ie, 500 ms before the termination, of that cue. In each U, the shock was given either 7 or 10 s after the termination of a cue. No startle stimuli followed a shock by <10 s.
During downtime periods when subjects were not tested, they did not have to answer questionnaires or rating scales. They were in a quiet environment, free to work on their studies, read, or watch TV.
Questionnaires and Analogue Scales
After each threat block, subjects retrospectively rated their anxiety level in the presence and absence of the cue in each condition (N, P, and U) on an analogue scale ranging from 0 (not at all fearful/anxious) to 10 (extremely fearful/anxious).
In addition, the state subscale of the State/Trait Anxiety Inventory (Spielberger, 1983) and visual analogue scales (VAS) of subjective sedation (Bond and Lader, 1974) were administered twice. Subjects used this latter questionnaire to rate how they presently felt on scales that assess physical and mental sedation (eg, ‘alert’/‘drowsy’, ‘lethargic’/‘energetic’). These ratings provided summary scores for mental and physical sedation/alertness. Greater sedation was reflected in higher scores. Ratings were made before drug ingestion and between the two threat series (see Table 1). Finally, immediately after the last recording, subjects rated the level of shock pain experienced during testing on a VAS ranging from 0 (not at all painful) to 10 (extremely painful).
Stimuli and Physiological Responses
Stimulation and recording were controlled by a commercial system (Contact Precision Instruments, London, UK). The acoustic startle stimulus was a 40-ms, 103-dB burst of white noise presented through headphones. The eyeblink reflex was recorded with electrodes placed under the left eye. The electromyographic signal was amplified with bandwidth set to 30–500 Hz and digitized at a rate of 1000 Hz.
Side Effects
In addition to the above questionnaires, potential side effects were assessed with a 21-item instrument of clinician-read, subject-endorsed rating of physical and mental symptoms (eg, somnolence, nausea, dizziness, fatigue, headache, anxiety) using a 4-point scale of 0 (not present) to 3 (extremely) for each item.
Data Analysis
The electromyographic eyeblink was rectified and smoothed with a 20-point moving average. Peak amplitude of the startle/blink reflex was determined in the 20–100-ms time frame after stimulus onset relative to a 50-ms prestimulus baseline and averaged within each condition. Data were analyzed using repeated measures analyses of variance (rANOVAs). The α was set at 0.05 for all statistical tests. Greenhouse–Geisser corrections (GG-ɛ) were used for main effects and interactions involving factors with more than two levels.
The effect of treatment on baseline startle reactivity was examined using the startle raw magnitude scores. Subsequently, the raw scores were standardized into T-scores within each participant as we have done in the past (Grillon et al, 2006, 2009a, 2013) in order to control for interindividual differences in startle reactivity.
RESULTS
Drug Effect on Baseline Startle (Raw Scores)
Baseline startle refers to startle measures in a context without a threat manipulation (ie, habituation procedures and N condition). First, startle magnitude during the habituation procedures of before and after drug ingestion (Table 2) were analyzed using a Time (before and after) × Treatment (placebo, alprazolam, low-GSK, and high-GSK) rANOVA. Startle habituated from the predrug to the postdrug tests (F(1, 30)=9.30, p=0.004), but this effect was not affected by treatment (Time × Treatment: F(1, 30)=0.44, NS). In addition, the Treatment main effect was not significant (F(1, 30)=1.80, NS).
Second, baseline startle magnitude of the N condition was analyzed using a one-way Treatment (placebo, alprazolam, low-GSK, and high-GSK) rANOVA. Startle magnitude differed among treatments (F(3, 90)=4.6, p=0.004) that was because of smaller startle magnitude during alprazolam (t(30)=3.2, p=0.003) and high-GSK (t(30)=3.1, p=0.003) compared with placebo (Table 2).
Fear-Potentiated Startle and Anxiety-Potentiated Startle (T-Scores)
The startle data appear in Table 3 (top). As in our previous studies and consistent with our a priori hypotheses (see, eg, Grillon et al, 2006, 2009a, 2013), we examined fear and anxiety separately. Fear-potentiated startle was operationally defined as the increased startle magnitude from ITI to the threat cue in P and anxiety-potentiated startle was operationally defined as the increased ITI startle magnitude from N to U. Fear-potentiated startle was analyzed using a two-way Stimulus Type (ITI, cue) × Treatment (placebo, alprazolam, low-GSK, and high-GSK) rANOVA. Anxiety-potentiated startle was analyzed using a two-way Condition (N and U) × Treatment (placebo, alprazolam, low-GSK, and low-GSK) rANOVA.
Fear-potentiated startle
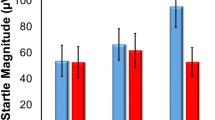
As expected, startle was potentiated by the threat cue (fear-potentiated startle), ie, startle magnitude was greater during the threat cue than during the ITI in P (F(3, 90)=102.1, p=0.00001; Figure 2 (top)).

(Top) Fear-potentiated startle (difference startle magnitude between threat cue and ITI in the predictable (P) condition). (Bottom) Anxiety-potentiated startle (difference ITI startle magnitude between the unpredictable (U) and the predictable (P) condition). *p<0.05, **p<0.01, ***p<0.001.
Fear-potentiated startle was affected by the treatment (Stimulus Type × Treatment: F(3, 90)=3.1, p=0.032), reflecting greater fear-potentiated startle during high-GSK compared with placebo (F(1, 30)=5.8, p=0.02) or alprazolam (F(1, 30)=5.4, p=0.02), but not low-GSK (F(1, 30)=1.8, NS). In addition, fear-potentiated startle did not differ between placebo and alprazolam (F(1, 30)=0.8, NS) or low-GSK (F(1, 30)=1.6, NS). Thus, as shown in Figure 2 (top), high-GSK increased fear-potentiated startle relative to two of the other three drug conditions. This effect remained significant when Treatment Order was added as a factor in the rANOVA (F(1, 30)=5.6, p=0.02).
Anxiety-potentiated startle
As expected, ITI startle was greater during the U compared with the N condition (anxiety-potentiated startle: F(1, 30)=78.3, p=0.00001; Figure 2 (bottom)).
Critically, treatment significantly affected anxiety-potentiated startle (Condition × Treatment: F(3, 90)=3.8, p=0.02). Specifically, alprazolam reduced anxiety-potentiated startle compared with placebo (F(1, 30)=5.0, p=0.03), low-GSK (F(1, 30)=10.7, p=0.002), and high-GSK561679 (F(1, 30)=16.3, p=0.0003). In addition, anxiety-potentiated startle did not differ between placebo and low-GSK (F(1, 30)=0.006, NS) or high-GSK (F(1, 30)=0.07, NS). Thus, as shown in Figure 2 (bottom), anxiety-potentiated startle was reduced by alprazolam, but was not affected by low-GSK or high-GSK. This alprazolam effect remained significant when Treatment Order was added as a factor in the rANOVA (F(1, 30)=5.2, p=0.03).
Retrospective Rating of Fear and Anxiety
The retrospective ratings appear in Table 3 (bottom). The ratings were analyzed using the same approach as with the startle data. Retrospective fear was calculated as the difference between ratings during the threat cue and during ITI in P, and anxiety was calculated as the difference ratings during ITI U minus N. Fear rating was greater during the threat cue compared with ITI in P (F(3, 90)=87.6, p<0.0001), but this effect was not affected by the treatment (Stimulus Type × Treatment: F(3, 90)=0.6, NS). Anxiety rating was greater during the U compared with the N condition (F(3, 90)=203.2, p<0.00001), but this effect was not affected by the treatment (Condition × Treatment: F(3, 90)=1.9, NS).
State Anxiety and Mental and Physical Sedation
Spielberger state anxiety ratings and sedation ratings were collected at baseline before treatment and between the two threat test series (Table 4). The scores were analyzed using a Time (before treatment, between threat series) × Treatment (placebo, alprazolam, low-GSK, and high-GSK) rANOVA. State anxiety significantly increased from baseline (mean=24.9, SEM=1.1) to after threat series-1 (mean=28.9, SEM=1) (Time: (F(1, 29)=19.0, p=0.00001). This increase did not differ among treatments (Time by Treatment: F(3, 87)=0.75, NS).
Sedation differed significantly between Time 1 and Time 2 (Time: F(1, 27)=332, p<0.00001), and this effect was modulated by Drug (Treatment × Time: F(3, 81)=8.7, p<0.0001). Sedation increased disproportionally during alprazolam relative to the other drug conditions (all (T1 vs T2), p<0.01).
Shock Rating
Shock ratings at the end of the experiment were analyzed with a one-way Treatment (4) rANOVA. Ratings did not differ significantly among treatments (placebo 6.1 (0.27); alprazolam 5.8 (0.28); low-GSK 5.9 (0.26); and high-GSK 6.0 (0.33); F(1, 30)=1.0, NS).
Safety Data
The treatments were well tolerated with very few side effects. Each item used to assess side effects was entered into a one-way Treatment (4) ANOVA. There was a treatment effect for (1) somnolence (F(3, 90)=8.1, p=0.0001) because of greater somnolence during alprazolam compared with placebo (t(30)=2.3, p=0.03), and lower somnolence during high-GSK compared with placebo (t(30)=2.1, p=0.04), and (2) fatigue (F(3, 90)=8.1, p=0.0001) because of greater fatigue during alprazolam compared with low-GSK (t(30)=2.9, p=0.005) and high-GSK (t(30)=3.2, p=0.002). Other side effects were mild and did not differ in severity between treatments.
DISCUSSION
This randomized control trial examined the effects of a CRF1 antagonist on fear- and anxiety-potentiated startle (Davis et al, 1997). We failed to demonstrate the hypothesized anxiolytic effect of GSK561679 on anxiety-potentiated startle, but we did show that high-GSK increased fear-potentiated startle. In addition, we replicated our prior report that alprazolam reduced anxiety-potentiated startle without affecting fear-potentiated startle (Grillon et al, 2006). The significance of these findings is discussed below.
Despite a host of studies demonstrating the antistress and antianxiety effect of CRF1 antagonists in animal models (see Griebel and Holsboer, 2012; Valdez, 2006 for reviews), clinical efficacy is not established. Studies in humans are scarce. Two studies reported no efficacy in major depression (compound GSK561679, clinical trial CRS106139; compound CP-316-311; Binneman et al, 2008), and one study in social anxiety disorder was completed with undisclosed results (NCT00555139). In contrast, positive results were reported in healthy individuals with R317573 in a proof-of-concept study using 7.5% CO2 to induce anxiety (Bailey et al, 2011). One possibility is that GSK561679 is anxiolytic in some tests but not in the NPU test.
There is substantial evidence that the NPU threat test can detect anxiolytic effects. This study replicated our previous reports (Grillon et al, 2006) that alprazolam reduces anxiety-potentiated startle but not fear-potentiated startle. Others studies have also found no effect of benzodiazepines on fear-potentiated startle (Baas et al, 2002; Scaife et al, 2005). We have also reported that 2-week treatment with the SSRI citalopram reduces anxiety-potentiated startle (Grillon et al, 2009a). In addition, anxiety-potentiated startle is increased in anxiety disorders (Grillon et al, 2008, 2009b), suggesting that the NPU test successfully models aversive states relevant to pathological anxiety. Moreover, there is some specificity in these medication effects: the fact that alprazolam consistently reduces anxiety-potentiated startle without affecting fear-potentiated startle indicates that different aversive states are differently sensitive to clinically effective anxiolytic treatments. This is consistent with the observation that infusion of CRF agonist in different brain areas affects distinct types of anxiety-like behavior in rodents (Bijlsma et al, 2011). Our data suggest that GSK561679 affects fear-like but not anxiety-like defensive responses.
The absence of anxiety-like effects might be considered in light of the suggestion that CRF1 antagonists have a nonlinear effect on anxiety-potentiated startle (Walker et al, 2009a, 2009b), raising the possibility that, despite using two dose levels, we missed the correct dose to observe an effect on anxiety-potentiated startle. In addition, it is still conceivable that CRF1 antagonists act on different components of anxiety than those modeled by NPU. Distinct BNST regions coordinate the modulation of independent defensive responses (eg, risk avoidance, respiration, startle) (Davis et al, 2010; Kim et al, 2013). These regions and their behavioral output may be differently sensitive to CRH antagonists.
Although high-GSK did not affect anxiety-potentiated startle, it did increase fear-potentiated startle to the threat cue. In rodents, CRF1 antagonists’ fear-enhancing effect has also been reported along with their anxiolytic effect (Walker et al, 2009a). In a review of their work, Walker et al (2009b) described several such experiments that showed that a CRF1 antagonist can increase cued-fear-potentiated startle, cued-fear-potentiated startle being the equivalent of the fear-potentiated startle to the threat cue in P of the present study. In addition, assuming that CRH antagonists act on the BNST, these results are consistent with an emerging literature showing that the BNST inhibits the mCeA (Haufler et al, 2013; Meloni et al, 2006) and fear-potentiated startle (Meloni et al, 2006). Taken together, these data suggest that the BNST displays regional specificity, exerting opposite influence on fear (inhibition) and anxiety (facilitation). One possibility therefore is that high-GSK was sufficient to inhibit BNST activity, leading to a disinhibition of mCeA (thus indirectly increasing fear), but was not sufficient to inhibit BNST neurons involved in the behavioral expression of anxiety.
High-GSK reduced baseline startle, raising the possibility that the increased fear-potentiated startle was an artifact of the effect of treatment on baseline startle. This is unlikely. First, alprazolam reduced baseline startle to a similar extent without noticeable effect on fear-potentiated startle. In addition, alprazolam reduced anxiety-potentiated startle. It is unclear how a reduction in startle reactivity could lead to both an increase and a decrease in startle potentiation. Second, in a prior study we showed that diphenhydramine, a sedative without anxiolytic properties, reduced baseline without significant effect on startle potentiation (Grillon et al, 2006).
The reducing effect of high-GSK on baseline startle magnitude was unexpected and we do not have a good explanation for this effect. We see three main possible explanations for this effect: sedation, contextual anxiety, and faster habituation. Startle is sensitive to sedative drugs such as alprazolam and diphenhydramine (Grillon et al, 2006). It is unlikely that the reduced baseline startle by high-GSK was because of sedation for two reasons. First, basic studies and clinical trials show that GSK561679 is not sedative (Dunlop et al, 2014). Second, we found that high-GSK had no subjective sedative effect or any significant side effects in the present study. Startle is enhanced by threatening contexts. For example, placing the shock electrodes increases startle (Grillon and Ameli, 1998a). One possibility is that our ‘baseline’ startle assessment during the threat experiment, ie, ITI startle during N, was not a good baseline startle because it was affected by the threatening context. If this were the case, then high-GSK would be anxiolytic for contextual anxiety. Although this is a possibility that may deserve to be investigated, we believe this explanation to be unlikely. The reduction in startle reactivity with high-GSK was of a relative large magnitude that would imply a high level of contextual anxiety. Such a level of contextual anxiety does not seem to be compatible with the relatively small contextual anxiety usually found in healthy controls (Grillon and Ameli, 1998a; Grillon et al, 1998b). Startle habituates rapidly with repeated stimulation. There was a clear reduction in overall startle reactivity in the placebo condition. This raises the possibility that GSK561679 speeds up habituation, a possibility that should be tested in future studies (see Supplementary Material for additional information on the effect of treatment on startle habituation).
The strengths and limitation of this study must be considered when interpreting these findings. Regarding strengths, we relied on a well-established cross-species experimental model of fear and anxiety, showing that these two facets of defensive responses can be functionally dissociated. Most importantly, the neural correlates of the behavioral responses tested with this experimental model have been well characterized, and permit us to infer neural mechanisms underlying findings of this study. In addition, this model has been shown to be sensitive to drugs that are used to treat anxious patients (Grillon et al, 2006, 2009a). Noteworthy, we replicated here our previous finding with alprazolam. Finally, we used a within-subject design that permitted us to avoid issues with potentially large interindividual differences in fear and anxiety that could mask drug effects. As for limitations, we tested only females, hence raising questions as to the generalization of the findings and the potential role of hormonal changes and oral contraception on the findings. A preliminary analysis showed no effect of oral contraception on fear-potentiated startle (see Supplementary Material). Our experimental model assesses normal adaptive responses to threat on healthy individuals. It is possible that CRH antagonists work on pathological states and not normal defensive responses. For example, CRH antagonists may work on anxiety associated with drug addiction (Zorrilla et al, 2014), suggesting that GSK561679 may be anxiolytic on chronic anxiety states. However, CRH antagonists show efficacy in animal models of adaptive defensive responses (Davis et al, 2010). Another limitation was that the effect of GSK561679 was found on potentiated startle but not on the retrospective fear/anxiety data. Reports of dissociation between objective and subjective measures are frequent in drug studies (Harmer et al, 2003; Kemp and Nathan, 2004), and we have found such a dissociation in all our psychopharmacological studies using the NPU threat test (Grillon et al, 2006, 2009a, 2011, 2013). The most likely reason for the differential effect of the GSS561679 on startle and subjective reports in this study is that the former measure was used to probe anxiety online, whereas the subjective anxiety measures were retrospective. Subtle differences in responding might have been affected by the passage of time and by the complexity of the design. Finally, it is highly likely that startle potentiation and subjective reports reflect the influence of different structures, subjective report being more cortically mediated than startle.
In sum, acute doses of the CRF1 antagonist GSK561679 did not exhibit an anxiolytic effect on anxiety-potentiated startle. On the other hand, the finding that GSK561679 increased fear-potentiated startle permits us to infer the following mechanism. CRF1 antagonist would lift the inhibitory control of BNST on CeA that is mediated by CRF1 action (Haufler et al, 2013; Kim et al, 2013; Walker et al, 2009b). Future basic research in animals and neuroimaging studies in humans should attempt to identify neural mechanisms underlying this effect as it may help deepen our insight into the neurobiology of fear and anxiety.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Baas JM, Grillon C, Bocker KB, Brack AA, CA III Morgan, Kenemans JL et al (2002). Benzodiazepines have no effect on fear-potentiated startle in humans. Psychopharmacology (Berl) 161: 233–247.
Bailey J, Papadopoulos A, Diaper A, Phillips S, Schmidt M, van der Ark P et al (2011). Preliminary evidence of anxiolytic effects of the CRF(1) receptor antagonist R317573 in the 7.5% CO(2) proof-of-concept experimental model of human anxiety. J Psychopharmacol 25: 1199–1206.
Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK et al (1999). Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 156: 585–588.
Bijlsma EY, van Leeuwen MLF, Westphal KGC, Olivier B, Groenink L (2011). Local repeated corticotropin-releasing factor infusion exacerbates anxiety- and fear-related behavior: differential involvement of the basolateral amygdala and medial prefrontal cortex. Neuroscience 173: 82–92.
Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T (2008). A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry 165: 617–620.
Bond A, Lader M (1974). The use of analogue scales in rating subjective feelings. Brit J Med Psychol 47: 211–218.
Buwalda B (1997). Physiological and behavioral effects of chronic intracerebroventricular infusion of corticotropin-releasing factor in the rat. Psychoneuroendocrinology 22: 297–309.
Campeau S, Falls WA, Cullinan WE, Helmreich DL, Davis M, Watson SJ (1997). Elicitation and reduction of fear: behavioural and neuroendocrine indices and brain induction of the immediate-early gene c-fos. Neuroscience 78: 1087–1104.
Coric V, Howard HF, Dan AO, Anantha S, Joseph P, Randy CD et al (2010). Multicenter, randomized, double-blind, active comparator and placebo-controlled trial of a corticotropin-releasing factor receptor-1 antagonist in generalized anxiety disorder. Depress Anxiety 27: 417–425.
Davis M, Walker DL, Lee Y (1997). Roles of the amygdala and bed nucleus of the stria terminalis in fear and anxiety measured with the acoustic startle reflex. Possible relevance to PTSD. Ann NY Acad Sci 821: 305–331.
Davis M, Walker DL, Miles L, Grillon C (2010). Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology 35: 105–135.
Dunlop B, Rothbaum B, Binder E, Duncan E, Harvey P, Jovanovic T et al (2014). Evaluation of a corticotropin releasing hormone type 1 receptor antagonist in women with posttraumatic stress disorder: study protocol for a randomized controlled trial. Trials 15: 240.
Fabio RD, St-Denis Y, Sabbatini FM, Andreotti D, Arban R, Bernasconi G et al (2008). Synthesis and pharmacological characterization of novel drug like corticotropin-releasing factor 1 antagonists. J Med Chem 51: 7370–7379.
First MB, Spitzer RL, Gibbon M, Williams JBW (2002) Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Non-patient Edition. (SCID-I/NP). Biometrics Research, New York State Psychiatric Institute: New York.
Greenblatt DJ, Wright CE (1993). Clinical pharmacokinetics of alprazolam. Clin Pharmacokinet 24: 453–471.
Griebel G, Holsboer F (2012). Neuropeptide receptor ligands as drugs for psychiatric diseases: the end of the beginning? Nat Rev Drug Discov 11: 462–478.
Grillon C (2008). Models and mechanisms of anxiety: evidence from startle studies. Psychopharmacology 199: 421–437.
Grillon C, Ameli R (1998a). Effects of threat of shock, shock electrode placement, and darkness on startle. Int J Psychophysiol 28: 223–231.
Grillon C, Baas JMP, Pine DS, Lissek S, Lawley M, Ellis V et al (2006). The benzodiazepine alprazolam dissociates contextual fear from cued fear in humans as assessed by fear-potentiated startle. Biol Psychiatry 60: 760–766.
Grillon C, Baas JP, Lissek S, Smith K, Milstein J (2004). Anxious responses to predictable and unpredictable aversive events. Behav Neurosci 118: 916–924.
Grillon C, Chavis C, Covington MS, Pine DS (2009a). Two-week treatment with citalopram reduces contextual anxiety but not cued fear. Neuropsychopharmacol 34: 964–971.
Grillon C, Heller R, Hirschhorn E, Kling MA, Pine DS, Schulkin J et al (2011). Acute hydrocortisone treatment increases anxiety but not fear in healthy volunteers: a fear-potentiated startle study. Biol Psychiatry 69: 549–555.
Grillon C, Krimsky M, Charney DR, Vytal K, Ernst M, Cornwell B (2013). Oxytocin increases anxiety to unpredictable threat. Mol Psychiatry 18: 958–960.
Grillon C, Levenson J, Pine DS (2007). A single dose of the selective serotonin reuptake inhibitor citalopram exacerbates anxiety in humans: a fear-potentiated startle study. Neuropsychopharmacology 32: 225–231.
Grillon C, Lissek S, Rabin S, McDowell D, Dvir S, Pine DS (2008). Increased anxiety during anticipation of unpredictable but not predictable aversive stimuli as a psychophysiologic marker of panic disorder. Am J Psychiatry 165: 898–904.
Grillon C, Morgan CA, Davis M, Southwick SM (1998b). Effects of experimental context and explicit threat cues on acoustic startle in Vietnam veterans with posttraumatic stress disorder. Biol Psychiatry 44: 1027–1036.
Grillon C, Pine DS, Lissek S, Rabin S, Bonne O, Vythilingam M (2009b). Increased anxiety during anticipation of unpredictable aversive stimuli in posttraumatic stress disorder but not in generalized anxiety disorder. Biol Psychiatry 66: 47–53.
Harmer CJ, Rogers RD, Tunbridge E, Cowen PJ, Goodwin GM (2003). Tryptophan depletion decreases the recognition of fear in female volunteers. Psychopharmacology 167: 411–417.
Haufler D, Nagy F, Pare D (2013). Neuronal correlates of fear conditioning in the bed nucleus of the stria terminalis. Learn Mem 20: 633–641.
Holsboer F, Ising M (2008). Central CRH system in depression and anxiety—evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol 583: 350–357.
Ishitobi Y, Nakayama S, Yamaguchi K, Kanehisa M, Higuma H, Maruyama Y et al (2012). Association of CRHR1 and CRHR2 with major depressive disorder and panic disorder in a Japanese population. Am J Med Genet B 159B: 429–436.
Keck ME, Kern N, Erhardt A, Unschuld PG, Ising M, Salyakina D et al (2008). Combined effects of exonic polymorphisms in CRHR1 and AVPR1B genes in a case/control study for panic disorder. Am J Med Genet B 147B: 1196–1204.
Kemp AH, Nathan PJ (2004). Acute augmentation of serotonin suppresses cardiovascular responses to emotional valence. Int J Neuropsychopharmacol 7: 65–70.
Kim S-Y, Adhikari A, Lee SY, Marshel JH, Kim CK, Mallory CS et al (2013). Diverging neural pathways assemble a behavioural state from separable features in anxiety. Nature 496: 219–223.
Klumpers F, van Gerven J, Prinssen E, Niklson I, Roesch F, Riedel W et al (2010). Method development studies for repeatedly measuring anxiolytic drug effects in healthy humans. J Psychopharmacol 24: 657–666.
Koob GF (2008). A role for brain stress systems in addiction. Neuron 59: 11–34.
Koob GF, Zorrilla EP (2012). Update on corticotropin-releasing factor pharmacotherapy for psychiatric disorders: a revisionist view. Neuropsychopharmacology 37: 308–309.
Lee Y, Davis M (1997). Role of the hippocampus, the bed nucleus of the stria terminalis, and the amygdala in the excitatory effect of corticotropin-releasing hormone on the acoustic startle reflex. J Neurosci 17: 6434–6446.
Liang KC, Melia KR, Miserendino MJD, Falls WA, Campeau S, Davis M (1992). Corticotropin-releasing factor: long-lasting facilitation of the acoustic startle reflex. J Neurosci 12: 2303–2312.
Meloni EG, Jackson A, Gerety LP, Cohen BM, Carlezon WA Jr. (2006). Role of the bed nucleus of the btria terminalis (BST) in the expression of conditioned fear. Ann NY Acad Sci 1071: 538–541.
Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM et al (2011). Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science 333: 1903–1907.
Sautter FJ, Bissette G, Wiley J, Manguno-Mire G, Schoenbachler B, Myers L et al (2003). Corticotropin-releasing factor in posttraumatic stress disorder (PTSD) with secondary psychotic symptoms, nonpsychotic PTSD, and healthy control subjects. Biol Psychiatry 54: 1382–1388.
Scaife JJC, Langley RRW, Bradshaw CCM, Szabadi EE (2005). Diazepam suppresses the acquisition but not the expression of ‘fear-potentiation’ of the acoustic startle response in man. J Psychopharmacol 19: 347–356.
Schmitz A, Grillon C (2012). Assessing fear and anxiety in humans using threat of predictable and unpredictable aversive events (the NPU-threat test). Nat Protoc 7: 527–532.
Sink KS, Walker DL, Freeman SM, Flandreau EI, Ressler KJ, Davis M (2013). Effects of continuously enhanced corticotropin releasing factor expression within the bed nucleus of the stria terminalis on conditioned and unconditioned anxiety. Mol Psychiatry 18: 308–319.
Spielberger CD (1983) Manual for the State-Trait Anxiety Inventory. Consulting Psychologist Press: Palo Alto, CA.
Tellew JE, Lanier M, Moorjani M, Lin E, Luo Z, Slee DH et al (2010). Discovery of NBI-77860/GSK561679, a potent corticotropin-releasing factor (CRF1) receptor antagonist with improved pharmacokinetic properties. Bioorg Med Chem Lett 20: 7259–7264.
Valdez GR (2006). Development of CRF1 receptor antagonists as antidepressants and anxiolytics: progress to date. CNS Drugs 20: 887–896.
Walker D, Yang Y, Ratti E, Corsi M, Trist D, Davis M (2009a). Differential effects of the CRF-R1 antagonist GSK876008 on fear-potentiated, light- and CRF-enhanced startle suggest preferential involvement in sustained vs phasic threat responses. Neuropsychopharmacology 34: 1533–1542.
Walker DL, Miles L, Davis M, Miles LA (2009b). Selective participation of the bed nucleus of the stria terminalis and CRF in sustained anxiety-like versus phasic fear-like responses. Prog Neuropsychopharmacol Biol Psychiatry 33: 1291–1308.
Zorrilla EP, Logrip ML, Koob GF (2014). Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol 35: 234–244.
Acknowledgements
This research was supported by the Intramural Research Program of the National Institute of Mental Health (ZIA MH002798) and by the NIMH grant MH069056 to CG. We gratefully acknowledge the efforts of members of the Section of the Neurobiology of Fear and Anxiety Disorders and of the Clinical Center at the NIMH.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
PowerPoint slides
Rights and permissions
About this article

Cite this article
Grillon, C., Hale, E., Lieberman, L. et al. The CRH1 Antagonist GSK561679 Increases Human Fear But Not Anxiety as Assessed by Startle. Neuropsychopharmacol 40, 1064–1071 (2015). https://doi.org/10.1038/npp.2014.316
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.316
This article is cited by
-
Cross-species anxiety tests in psychiatry: pitfalls and promises
Molecular Psychiatry (2022)
-
The novel vasopressin receptor (V1aR) antagonist SRX246 reduces anxiety in an experimental model in humans: a randomized proof-of-concept study
Psychopharmacology (2021)
-
Modeling anxiety in healthy humans: a key intermediate bridge between basic and clinical sciences
Neuropsychopharmacology (2019)
-
Acute prazosin administration does not reduce stressor reactivity in healthy adults
Psychopharmacology (2019)
-
Oxytocin facilitates adaptive fear and attenuates anxiety responses in animal models and human studies—potential interaction with the corticotropin-releasing factor (CRF) system in the bed nucleus of the stria terminalis (BNST)
Cell and Tissue Research (2019)
