
Abstract
Cortical interneurons, immunoreactive for neuronal nitric oxide synthase (nNOS) and the receptor NK1, express the functional activity marker Fos selectively during sleep. NREM sleep ‘pressure’ is hypothesized to accumulate during waking and to dissipate during sleep. We reported previously that the proportion of Fos+ cortical nNOS/NK1 neurons is correlated with established electrophysiological markers of sleep pressure. As these markers covary with the amount of NREM sleep, it remained unclear whether cortical nNOS/NK1 neurons are activated to the same degree throughout NREM sleep or whether the extent of their activation is related to the sleep pressure that accrued during the prior waking period. To distinguish between these possibilities, we used hypnotic medications to control the amount of NREM sleep in rats while we varied prior wake duration and the resultant sleep pressure. Drug administration was preceded by 6 h of sleep deprivation (SD) (‘high sleep pressure’) or undisturbed conditions (‘low sleep pressure’). We find that the proportion of Fos+ cortical nNOS/NK1 neurons was minimal when sleep pressure was low, irrespective of the amount of time spent in NREM sleep. In contrast, a large proportion of cortical nNOS/NK1 neurons was Fos+ when an equivalent amount of sleep was preceded by SD. We conclude that, although sleep is necessary for cortical nNOS/NK1 neuron activation, the proportion of cells activated is dependent upon prior wake duration.
Similar content being viewed by others
INTRODUCTION
Although the functions of sleep remain controversial, one of the strongest arguments for its fundamental importance is its homeostatic regulation. Sleep homeostasis refers to compensatory increases in sleep amount, sleep consolidation and/or sleep intensity that occur in response to a period of extended wakefulness (Borbely and Achermann, 2000). In mammals and birds, sleep intensity, usually assessed by measuring the spectral power of the EEG in the delta frequency range (0.5–4.5 Hz) during non-rapid eye movement sleep (NREM), is used as an index of the hypothetical sleep ‘pressure’ that has accumulated during wakefulness (Borbely and Achermann, 2000; Rattenborg et al, 2009).
Despite the widespread occurrence of sleep homeostasis among animal species, our understanding of the underlying mechanisms is incomplete. Current hypotheses implicate sleep factors such as adenosine or cytokines that accumulate during waking, and increase the propensity and depth of sleep (Coulon et al, 2012; Krueger et al, 2008; Landolt, 2008; Porkka-Heiskanen and Kalinchuk, 2011; Szymusiak and McGinty, 2008). Sleep factors are thought to act by inhibiting wake-promoting neurons (Porkka-Heiskanen and Kalinchuk, 2011; Rainnie et al, 1994), but may also act directly on the cerebral cortex (Clinton et al, 2011; Szymusiak, 2010). We have recently described a population of cortical GABAergic interneurons that is specifically activated during sleep (Gerashchenko et al, 2008; Pasumarthi et al, 2010). These neurons are identified by colocalized immunoreactivity for neuronal nitric oxide synthase (nNOS) and the substance P (SP) receptor NK1 (Dittrich et al, 2012). Activation of these neurons, assessed by immunoreactivity for the functional activity marker Fos, correlates with time spent in NREM sleep as well as with NREM delta power (Gerashchenko et al, 2008; Morairty et al, 2013). Prior wake duration ‘dose-dependently’ increased the proportion of Fos-labeled nNOS neurons when rats were subjected to 2, 4, or 6 h of sleep deprivation (SD) followed by a 2-h recovery sleep (RS) opportunity (Morairty et al, 2013). Based on these observations, we have suggested that cortical nNOS/NK1 neurons are inhibited by wakefulness and activated by sleep pressure (Kilduff et al, 2011).
In our previous studies, we increased sleep pressure by increasing the amount of prior wakefulness, which resulted in a compensatory increase in the amount of NREM sleep during RS. Therefore, we could not distinguish whether the time spent in NREM sleep or the magnitude of sleep pressure produced by prolonging wakefulness was the primary factor driving Fos expression in cortical nNOS/NK1 neurons. Fos expression typically reflects neuronal activity occurring during the prior 1–2 h to killing (Hoffman and Lyo, 2002; Zangenehpour and Chaudhuri, 2002). If cortical nNOS/NK1 neurons are uniformly activated throughout NREM sleep and inactive during wakefulness, Fos expression in these neurons should depend on the time spent in NREM during the 1–2 h before transcardial perfusion and thus would only indirectly correlate with measures of sleep pressure. Here, we sought to distinguish between these alternatives by dissociating the occurrence of NREM from the magnitude of sleep pressure using hypnotic medications to pharmacologically control NREM sleep duration under conditions in which the prior sleep/wake history was varied. To ensure that our results were not drug-specific, we utilized hypnotics with different mechanisms of action: the dual hypocretin/orexin receptor antagonist almorexant (ALM) and the GABAA receptor modulator zolpidem (ZOL). We find that, when time spent in NREM sleep is held constant, the proportion of cortical nNOS/NK1 cells activated is dependent upon prior sleep/wake history and that Fos expression in cortical nNOS/NK1 neurons reflects time kept awake (and, presumably, the accompanying sleep pressure) more robustly than any other parameter of NREM sleep.
MATERIALS AND METHODS
Animals
A total of 39 male Sprague–Dawley rats were studied. Animals were housed in separate cages in temperature-controlled recording chambers (20–24°C, 30–70% relative humidity) under a 12/12 light/dark cycle with food and water available ad libitum. The weights at experiment were 587 g±63 (mean±SD). All experimental procedures involving animals were approved by SRI International’s Institutional Animal Care and Use Committee and were in accordance with National Institute of Health (NIH) guidelines.
Surgical Procedures
Surgical procedures involved implantation of sterile telemetry transmitters (F40-EET, Data Sciences International, St Paul, MN, USA) as previously described (Morairty et al, 2013, 2008, 2012). Briefly, transmitters were placed intraperitoneally under isoflurane anesthesia. Biopotential leads were routed subcutaneously to the head and neck. EEG electrodes were placed epidurally 1.5 mm anterior to bregma and 1.5 mm lateral to midline, and 6 mm posterior to bregma and 4 mm lateral to midline on the right hemisphere. EMG leads were positioned bilaterally through the nuchal muscles.
Identification of Sleep/Wake States and Sleep/Wake Data Analyses
Behavioral state determinations and data analyses were conducted as previously described (Morairty et al, 2013, 2012). After at least 3-week postsurgical recovery, EEG and EMG were recorded via telemetry using DQ ART 4.1 software (Data Sciences International). Following completion of data collection, the EEG and EMG recordings were scored in 10 s epochs as waking, rapid eye movement sleep (REM), or NREM by expert scorers who examined the recordings visually using NeuroScore software (Data Sciences International). For calculation of bout durations, a bout was defined as consisting of a minimum of two consecutive epochs of a given state and ended with any single state change epoch. EEG spectra were analyzed with a fast Fourier transform algorithm using a Hanning Window without overlap (NeuroScore software, Data Sciences International) on all epochs without artifact. For comparisons of EEG spectra, average spectra of a specific state were normalized to the average spectra of the respective state during a 6 h baseline recording (Zeitgeber time 0–6, or ZT0-ZT6). For calculation of NREM EEG delta power (NRD), the mean of the power between 0.5–4.5 Hz of the averaged NREM spectra was calculated and normalized to the respective value of the 6 h baseline recording. NRD energy (NRDE) was calculated by multiplying the time (h) spent in NREM sleep by the normalized NRD power.
Detection of individual slow waves was adapted from (Vyazovskiy et al, 2007). Raw EEG was bandpass filtered (0.5–4.5 Hz) using the bandpassfilter.m function from the FieldTrip toolbox (http://www.ru.nl/neuroimaging/fieldtrip) in MATLAB (Mathworks, Natick, MA, USA). The first positive peak after a zero crossing was identified as a single slow wave. The slope was approximated as a straight line between that peak and the last negative peak preceding the zero crossing. All slopes from artifact-free NREM epochs were averaged for each rat. Slopes were normalized to the average NREM slopes from the respective baseline recordings.
SD Procedures
Animals were continuously observed while EEG and EMG were recorded and, when inactive and appeared to be entering sleep, cage tapping occurred. When necessary, an artist’s brush was used to stroke the fur or vibrissae. After ZOL, it was sometimes necessary to touch rats to keep them awake.
Experimental Protocol
The rats were assigned to six groups: (1) VEH with low sleep pressure (n=6); (2) VEH with high sleep pressure (n=7); (3) ZOL with low sleep pressure (n=6); (4) ZOL with high sleep pressure (n=7); (5) ALM with low sleep pressure (n=6); and (6) ALM with high sleep pressure (n=7). Dosing occurred at ZT12, 100 mg/kg p.o. in 2 ml/kg for both drugs. Perfusion occurred at ZT14 for VEH and ZOL groups and at ZT14.5 for ALM groups due to its longer latency to sleep onset (Black et al, 2013; Morairty et al, 2012, 2014). Rats in the high sleep pressure conditions were sleep deprived during the 6 h prior to dosing (Figure 1).

Experimental design. Sleep pressure increases during the active phase and decreases during the inactive phase. Rats were dosed at ZT12 (first vertical dashed line), when sleep pressure is lowest. We assume that sleep pressure increases slowly, if at all, during hypnotic-induced sleep; therefore, the corresponding curve remains low after dosing. To increase sleep pressure but keep the circadian conditions identical, rats in a second group were subjected to 6 h of SD starting at ZT6 and dosed at ZT12. Rats in both conditions were perfused 2 or 2.5 h after dosing (second vertical dashed line). Black and white bars indicate light conditions.
Immunohistochemical Procedures
Rats were killed with an overdose of euthanasia solution i.p. (SomnaSol, Butler-Schein, Dublin, OH, USA) and transcardially perfused with heparinized phosphate-buffered saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer. Brains were postfixed overnight in the same fixative and then immersed in 30% sucrose in phosphate-buffered saline until they sank. Coronal brain sections were cut at 40-μm thickness. Double immunohistochemistry for Fos and nNOS was performed on serial sections of rat brain as described previously (Gerashchenko et al, 2008; Pasumarthi et al, 2010). Sections were first incubated overnight with rabbit anti-Fos antibody (1:4000–5000, sc-52, Santa Cruz Biotechnology, Dallas, TX, USA), then with biotinylated donkey anti-rabbit antibody (1:500, Jackson ImmunoResearch, West Grove, PA, USA), followed by avidin-biotinylated horseradish peroxidase complex (1:200, PK-6100, Vector Laboratories, Burlingame, CA, USA), and nickel-enhanced 3,3′ diaminobenzidine (SK4100, Vector Laboratories) for a black reaction product. nNOS neurons were stained in the same sections by overnight incubation in rabbit-nNOS antibody (1:2000, 61–7000, Invitrogen, Camarillo, CA, USA), followed by biotinylated donkey anti-rabbit antibody and avidin-biotinylated horseradish peroxidase complex, and visualized with NovaRED (SK-4805, Vector Laboratories) for a red-brown reaction product.
Cell Counting
Single-labeled nNOS and double-labeled Fos+/nNOS cells were counted in one hemisection each at 1.4 mm anterior, 0.5 mm posterior, and 3.0 mm posterior to bregma (Paxinos et al, 1999). The percentage of nNOS neurons expressing Fos was calculated as described previously (Gerashchenko et al, 2008; Pasumarthi et al, 2010). Micrographs for publication were taken at × 200 magnification on a Leica DM 5000B microscope (Leica Microsystems, IL, USA) with a Microfire S99808 camera (Optronics, CA, USA) in Stereoinvestigator (MBF Bioscience, Williston, VT, USA). Adjustments of brightness, color or contrast were applied to the whole image and performed in Photoshop (Adobe Systems, San Jose, CA, USA).
Statistics
Statistical tests were performed using Excel (Microsoft, Redmond, WA, USA), MATLAB and R (R Foundation for Statistical Computing). For each studied variable, we used Mann–Whitney U-tests to test whether it significantly distinguished between the high sleep pressure and low sleep pressure groups within the same drug treatment condition. The NREM bout duration histograms (Figure 2) were tested with two-way permutation ANOVA (Manly, 2007) with 5000 iterations of the factors ‘bout duration’ and ‘sleep pressure’. If an interaction was found, the sleep pressure conditions were compared for each bout duration using Holm–Sidak-corrected t-tests. For comparison of EEG power spectra, we first performed two-way permutation ANOVA with 5000 iterations with factors ‘frequency bin’ and ‘sleep pressure’. If interactions were found, the source of the interaction was evaluated through bin-by-bin (0.122 Hz) uncorrected t-tests between the sleep pressure conditions. Only changes that affected a range of frequencies were considered potentially meaningful, whereas isolated bins with significant changes were ignored. To determine how strongly different physiological parameters distinguished the high vs low sleep pressure groups, we calculated Hedges’ g (difference of the means divided by pooled standard deviation) and the 95% confidence intervals as an effect size measure (Hedges and Olkin, 1985). As Hedges’ g is a parametric measure, data were first transformed to achieve a normal distribution. The percent time in NREM and the %Fos+/nNOS neurons were arcsine transformed. NREM bout durations, NRD, slow wave slopes (each normalized by respective baseline values), and NRDE were log10 transformed. Normal distributions after data transformation were verified visually using normal probability plots. To test whether Hedges’ g for %Fos/nNOS was significantly different from Hedges’ g for any other variable, g was first transferred to Fisher’s z (Borenstein et al, 2009). The z for %Fos/nNOS was then compared with z for every other variable (Meng et al, 1992) and the p-values were Holm–Sidak-corrected for multiple comparisons.

Sleep/wake parameters in the 6 experimental conditions. (a–c) Time spent awake between ZT6 and the time of transcardial perfusion. High sleep pressure groups were sleep deprived from ZT6-ZT12, low sleep pressure groups were left undisturbed so that the amount of sleep pressure differed. Lighting conditions are indicated below the panels. Dosing occurred at ZT12. (d–f) Time spent in wake, NREM, and REM during the 90 min before transcardial perfusion. After ZOL dosing, the time spent in any state did not differ between the sleep pressure conditions. Horizontal lines indicate group medians. *p<0.05, U-test. (g–i) NREM bout duration frequency histograms during the 90 min before transcardial perfusion following VEH (g), ALM (h) and ZOL (I) dosing. *p<0.05, Holm–Sidak test after significant interaction in permutation ANOVA. (j) Average NREM bout durations. *p<0.05, U-test.
RESULTS
Sleep Time can be Dissociated from Sleep/Wake History Using Hypnotics
After experimental manipulation of sleep pressure as illustrated in Figure 1, the VEH-dosed rats showed the expected influence of waking history on sleep propensity: whereas undisturbed rats showed an increased time spent awake beginning at lights off (ZT12), rats that were sleep deprived during the preceding 6 h showed strongly reduced wakefulness at the same time of day (Figure 2a). ALM decreased the time spent awake in both groups but, at the dose used, the sleep-deprived rats showed a stronger reduction of wakefulness than rats that were undisturbed for the 6 h preceding dosing, indicating an additive effect of ALM and sleep pressure (Figure 2b). In contrast, ZOL caused a strong reduction of wakefulness irrespective of the preceding wake history (Figure 2c). As we previously found that Fos expression in rat cortical nNOS neurons is dependent on NREM time during the 90 min preceding transcardial perfusion (Morairty et al, 2013), we focused on that time window for the following analyses. Figure 2d–f depicts the time each rat spent in wake, NREM, and REM during the 90 min immediately before transcardial perfusion. Whereas the time spent in wake, NREM and REM differed between the low and high sleep pressure groups treated with either VEH or ALM, these physiological parameters did not differ between the groups treated with ZOL, indicating a decoupling between prior sleep/wake history and vigilance states with ZOL treatment.
ZOL Disrupts Established Measures of Sleep Pressure
Given the results in Figure 2d–f, we evaluated whether the high and low sleep pressure groups could be distinguished after ALM and ZOL treatment using four established measures of sleep pressure: NREM bout duration, NRD, NRDE, and slow wave slopes. NREM bout duration frequency histograms were shifted towards longer bout durations in the sleep-deprived groups (Figure 2g–i). Although there was a significant interaction between the factors ‘bout duration’ and ‘sleep pressure’ following VEH (F5,55=6.74, p=0.007) and ALM (F5,55=4.60, p=0.0008), this interaction did not reach statistical significance for ZOL (F5,55=2.28, p=0.056). Accordingly, the average NREM bout durations were longer for sleep-deprived than undisturbed rats following VEH (p=0.002) and ALM (p=0.008), but not for ZOL (p=0.073; Figure 2j).
As expected, the NREM EEG power spectra showed an elevated power in the delta range in the high sleep pressure group for VEH-dosed rats (Figure 3b). The wake and NREM spectra for ALM-dosed rats resembled those of VEH-dosed rats, including the increased NREM delta power in the high sleep pressure group (Figure 3c and d). Following ZOL, wake and NREM spectra were strongly altered compared with the corresponding baseline recordings, as indicated by the deviations from the basal value 1 in Figure 3e and f. In contrast to VEH and ALM, neither a main effect of ‘sleep pressure’ nor an interaction of ‘frequency bin’ and ‘sleep pressure’ was found following ZOL for either wake or NREM spectra, indicating that spectral power after ZOL did not depend on prior sleep/wake history. (REM spectra are not shown because, in some groups, too little REM occurred to calculate representative spectra). Figure 3g and h depicts normalized NRD (0.5–4.5 Hz) and NRDE for each rat. Both measures significantly distinguished the high vs low sleep pressure groups following VEH (p=0.005 for NRD, p=0.005 for NRDE) as well as ALM (p=0.022 for NRD, p=0.001 for NRDE) treatment, whereas no difference was found following ZOL (p=1 for NRD, p=0.63 for NRDE).

Spectral analyses. Wake (a, c, e) and NREM (b, d, f) EEG power spectra for the 90 min before transcardial perfusion were normalized by the respective baselines (ZT0–6). Interactions of factors ‘frequency’ and ‘sleep pressure’ (permutation ANOVA) are indicated for each panel. The degrees of freedom are 491 and 5401 for all interactions. The p-values for post hoc uncorrected bin-by-bin t-tests are indicated below the spectra. Following VEH and ALM dosing, increased sleep pressure coincided with increased NREM delta power (arrows) whereas, after ZOL dosing, NREM delta power was increased irrespective of sleep pressure. (g) Average NREM delta power (NRD) during the 90 min before transcardial perfusion. (h) NREM delta energy (NRDE) during 90 min before transcardial perfusion. Both NRD and NRDE distinguished between the high and low sleep pressure groups following VEH and ALM but not following ZOL dosing. Horizontal lines indicate group medians. *p<0.05, U-test.
Finally, we measured the average slopes of individual EEG slow waves during NREM. As expected, the slow wave slopes were steeper for sleep-deprived rats than for undisturbed rats following VEH (Figure 4b). This difference was preserved after ALM (Figure 4c) but not after ZOL (Figure 4d). Consequently, the average slow wave slope was significantly greater in the high sleep pressure than in the low sleep pressure group following VEH (p=0.008) and ALM (p=0.001) but not following ZOL (p=0.366; Figure 4e).

Slopes of NREM EEG slow waves during the 90 min before transcardial perfusion. (a) The raw EEG trace (top) was bandpass filtered in the slow wave range 0.5–4.5 Hz (bottom). Positive (green) and negative peaks (red) were identified. A straight line between a negative and a positive peak encompassing a zero crossing was defined as the slope of the respective slow wave. (b, c, d) Average slow waves±SEM for the experimental groups. (e) Average NREM slow wave slopes. The slopes distinguished between the low and high sleep pressure groups following VEH and ALM but not following ZOL dosing. Horizontal lines indicate group medians. *p<0.05, U-test.
Fos Expression in nNOS Neurons Depends on Prior Sleep/Wake History and Resultant Sleep Pressure
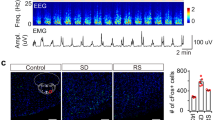
To determine whether the percentage of Fos+ cortical nNOS neurons depends on prior sleep/wake history or only on NREM time during the 90 min before transcardial perfusion, we performed double immunohistochemistry for Fos and nNOS. As depicted in Figure 5a–e, sleep-deprived rats showed higher levels of %Fos/nNOS than undisturbed rats irrespective of drug treatment. Consequently, %Fos/nNOS significantly distinguished between the high vs low sleep pressure conditions following VEH (p=0.001), ZOL (p=0.001) and ALM (p=0.001; Figure 5e). Notably, the separation between conditions was absolute after each drug treatment, i.e., there were no overlapping data points.

Fos expression in cortical nNOS neurons depends on sleep pressure. (a–d) Example micrographs of Fos/nNOS double immunohistochemistry. Following both hypnotics, nNOS neurons were single-labeled (arrows) in low sleep pressure conditions. (a, c) In the high sleep pressure conditions (b, d), many nNOS neurons were double-labeled for Fos (black triangles) irrespective of the drug treatment. Scale bar indicates 50 μm. (e) Proportion of Fos+ cortical nNOS neurons. Note that %Fos/nNOS completely separated all high sleep pressure groups from the respective low sleep pressure groups. Horizontal lines indicate group medians. *p<0.05, U-test. (f–h) Effect sizes for the difference between low and high sleep pressure groups using six different measures. Data for each of the six parameters listed on the abscissa were transformed to obtain normal distributions. For each variable, Hedges’ g±95% confidence interval was calculated as an effect size for the difference between the two groups following VEH (f), ALM (g), and ZOL (h) treatment. Following each drug treatment, the strongest effect was seen for %Fos/nNOS. * indicates significantly (p<0.05) smaller g than that for %Fos/nNOS after Holm–Sidak correction for multiple comparisons.
Fos/nNOS is the Best Indicator of Sleep/Wake History and Resultant Sleep Pressure
To determine which physiological measure was most closely related to prior sleep/wake history, we quantified the effect sizes (Hedges’ g) for the difference between the high vs low sleep pressure groups for each of the parameters measured in the present study (see Methods). For each drug treatment, Hedges’ g was greatest for %Fos/nNOS (Figure 5f and h). Following VEH, %Fos/nNOS separated the sleep pressure groups significantly better than bout duration, NRD, or NRDE (Figure 5f) and better than all parameters following ALM (Figure 5g).
DISCUSSION
These results demonstrate that the extent of activation of cortical nNOS/NK1 neurons is determined by prior sleep history. When sleep pressure is assumed to be low, cortical nNOS neurons are largely inactive (Figure 5a–e) even in the presence of high amounts of NREM sleep as illustrated by the ZOL group in Figure 2e.
%Fos/nNOS Depends on Sleep/Wake History
Using hypnotic treatment, we were able to dissociate time spent asleep during the 90 min before killing from the prior sleep/wake history. Following ZOL, neither time spent in wake, NREM, nor REM differed between rats that were sleep-deprived and rats that were left undisturbed for the preceding 6 h. Nonetheless, in ZOL-treated rats, the proportion of Fos+ cortical nNOS neurons was significantly greater in the high sleep pressure than the low sleep pressure group. We conclude that cortical nNOS neurons are not activated simply by the occurrence of NREM sleep, rather, the %Fos/nNOS depends on the magnitude of sleep pressure that has accumulated during the time preceding sleep onset. Together with our previous studies (Gerashchenko et al, 2008; Morairty et al, 2013), these results demonstrate that cortical nNOS neurons are responsive to homeostatic sleep drive.
We found that the proportion of Fos+ cortical nNOS neurons was a better indicator of prior sleep/wake history than total time spent in NREM, average NREM bout duration, NREM delta power, or the average slope of NREM slow waves (Figure 5f–h). Following ZOL, %Fos/nNOS was the only measure that significantly distinguished between the low and high sleep pressure groups. This observation makes it unlikely that Fos expression in cortical nNOS neurons is downstream of any of these variables (e.g., driven by slow wave activity), although experimental confirmation will depend on the ability to selectively manipulate cortical nNOS/NK1 neurons.
In agreement with our previous findings (Morairty et al, 2014), the hypnotic efficacy of ALM and ZOL was comparable at the doses used in the present study. Nonetheless, the same dose of ALM produced different amounts of sleep in the high sleep pressure and low sleep pressure groups. This result is consistent with the view that ALM removes a wake-inducing input—Hcrt tone—whereas ZOL actively inhibits neuronal activation. Nonspecific neuronal inhibition might bias the system towards sleep, whereas Hcrt antagonism might just impair the ability to stay awake in the presence of endogenous sleep pressure. A more detailed comparison will be needed to test if this is indeed a qualitative difference between the drugs or an effect of non-equivalent doses. Nonetheless, despite increased NREM sleep, ALM did not increase levels of Fos in cortical nNOS neurons in the low sleep pressure condition, which is consistent with the results obtained from the ZOL experiment.
nNOS/NK1 Neurons and NREM Delta Power
The finding that, following ZOL, NRD did not differ between sleep-deprived and undisturbed rats despite the pronounced difference in %Fos/nNOS between groups was surprising, as we have previously found that activation of these neurons may facilitate NRD (Morairty et al, 2013). Therefore, we had expected that increased Fos expression in cortical nNOS neurons would coincide with increased NRD in conditions when total NREM time did not differ. Given the pronounced effects of ZOL on the EEG, it is conceivable that ZOL masked the effects of cortical nNOS neuron activation on the NREM EEG. The low frequencies of the NREM power spectra were conspicuously increased following ZOL irrespective of sleep pressure (Figure 3d). As cortical nNOS neurons are GABAergic (Kubota et al, 2011), the GABAA modulator ZOL might act directly on the downstream targets of these neurons. This interpretation is in agreement with the finding that the sleep pressure-dependent modulation of the NREM EEG was not impaired by the Hcrt antagonist ALM, as sleep-deprived rats showed significantly elevated NRD. Based on Hedges’ g, the difference in NRD between high and low sleep pressure conditions was not smaller following ALM than VEH treatment (Figure 5).
Although the downstream targets of sleep-active cortical nNOS neurons have not yet been identified, these neurons are present in all cortical areas (Vincent and Kimura, 1992), form long-range cortico-cortical projections (Tomioka et al, 2005), and appear to be the origin of a dense nNOS-positive fiber network (Vincent and Kimura, 1992; Yousef et al, 2004) that is suited for producing a near simultaneous NO signal throughout a large cortical volume (Philippides et al, 2005). Thus, it seems likely that cortical nNOS neurons may exert a widespread effect on the cortex during sleep. This inference is supported by our recent finding that nNOS KO mice show deficits in the regulation of delta power and consolidation of NREM sleep (Morairty et al, 2013). However, direct and specific experimental manipulation of these neurons will be necessary to determine the specific effects on cortical activity patterns.
Regulation of nNOS/NK1 Neurons
Although NREM sleep appears to be a permissive state for the activation of cortical nNOS neurons, we demonstrated here that the magnitude of activation of these cells during NREM depends on prior sleep/wake history. The mechanism by which prior wake time is linked to the activation of cortical nNOS neurons is of great interest, as it could provide insight into how the accumulation of sleep pressure is tracked by the brain. A better understanding of this mechanism could prove relevant for facilitating restorative sleep or combating pathological sleepiness. The integration of time spent awake might occur at the level of the nNOS/NK1 neurons themselves. Locally accumulating sleep factors, such as adenosine and cytokines, might activate these neurons (Kilduff et al, 2011). Another such factor could be SP. mRNA levels of the gene coding for SP are increased in the cortex by SD (Martinowich et al, 2011). Cortical nNOS neurons co-express the SP receptor NK1 and are strongly and directly activated in vitro by SP (Dittrich et al, 2012).
Fos expression in cortical nNOS/NK1 neurons remains minimal as long as rats are kept awake, irrespective of accrued sleep pressure (Morairty et al, 2013). Therefore, if integration of sleep pressure indeed occurs at the level of cortical nNOS neurons, a wake-related inhibitory input must be assumed that prevents activation of these cells before sleep onset. This view is congruent with the model we have presented previously (Kilduff et al, 2011).
Alternatively, the integration of sleep pressure might occur upstream of cortical nNOS neurons. In this scenario, cortical nNOS neurons would receive activating input only during NREM, the magnitude of which depending on the sleep/wake history. In order to identify the mechanisms by which sleep pressure is linked to activation of cortical nNOS neurons, it will be critical to characterize the anatomical and neurochemical inputs to these cells.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Black SW, Morairty SR, Fisher SP, Chen TM, Warrier DR, Kilduff TS (2013). Almorexant promotes sleep and exacerbates cataplexy in a murine model of narcolepsy. Sleep 36: 325–336.
Borbely AA, Achermann P (2000). Sleep homeostasis and models of sleep regulation. In: Kryger MH, Roth T, Dement WC, (eds). Principles and Practice of Sleep Medicine 3rd edn. W.B. Saunders Company: Philadelphia, PA, USA. pp 377–390.
Borenstein M, Hedges LV, Higgins JPT, Rothstein HR (2009) Introduction to Meta-Analysis. Wiley: West Sussex.
Clinton JM, Davis CJ, Zielinski MR, Jewett KA, Krueger JM (2011). Biochemical regulation of sleep and sleep biomarkers. J Clin Sleep Med 7: S38–S42.
Coulon P, Budde T, Pape HC (2012). The sleep relay—the role of the thalamus in central and decentral sleep regulation. Pflugers Arch 463: 53–71.
Dittrich L, Heiss JE, Warrier DR, Perez XA, Quik M, Kilduff TS (2012). Cortical nNOS neurons co-express the NK1 receptor and are depolarized by Substance P in multiple mammalian species. Front Neural Circuits 6: 31.
Gerashchenko D, Wisor JP, Burns D, Reh RK, Shiromani PJ, Sakurai T et al (2008). Identification of a population of sleep-active cerebral cortex neurons. Proc Natl Acad Sci USA 105: 10227–10232.
Hedges LV, Olkin I (1985) Statistical Methods for Meta-Analysis. Academic Press: Orlando, FL, USA.
Hoffman GE, Lyo D (2002). Anatomical markers of activity in neuroendocrine systems: are we all 'fos-ed out'? J Neuroendocrinol 14: 259–268.
Kilduff TS, Cauli B, Gerashchenko D (2011). Activation of cortical interneurons during sleep: an anatomical link to homeostatic sleep regulation? Trends Neurosci 34: 10–19.
Krueger JM, Rector DM, Roy S, Van Dongen HP, Belenky G, Panksepp J (2008). Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci 9: 910–919.
Kubota Y, Shigematsu N, Karube F, Sekigawa A, Kato S, Yamaguchi N et al (2011). Selective coexpression of multiple chemical markers defines discrete populations of neocortical GABAergic neurons. Cereb Cortex 21: 1803–1817.
Landolt HP (2008). Sleep homeostasis: a role for adenosine in humans? Biochem Pharmacol 75: 2070–2079.
Manly BFJ (2007) Randomization, Bootstrap and Monte Carlo Methods in Biology 3rd edn. Chapman & Hall/CRC: Boca Raton, FL, USA.
Martinowich K, Schloesser RJ, Jimenez DV, Weinberger DR, Lu B (2011). Activity-dependent brain-derived neurotrophic factor expression regulates cortistatin-interneurons and sleep behavior. Mol Brain 4: 11.
Meng X-L, Rosenthal R, Rubin DB (1992). Comparing correlated correlation coefficients. Psychol Bull 111: 172–175.
Morairty SR, Dittrich L, Pasumarthi RK, Valladao D, Heiss JE, Gerashchenko D et al (2013). A role for cortical nNOS/NK1 neurons in coupling homeostatic sleep drive to EEG slow wave activity. Proc Natl Acad Sci USA 110: 20272–20277.
Morairty SR, Hedley L, Flores J, Martin R, Kilduff TS (2008). Selective 5HT2A and 5HT6 receptor antagonists promote sleep in rats. Sleep 31: 34–44.
Morairty SR, Revel FG, Malherbe P, Moreau JL, Valladao D, Wettstein JG et al (2012). Dual hypocretin receptor antagonism is more effective for sleep promotion than antagonism of either receptor alone. PLoS One 7: e39131.
Morairty SR, Wilk AJ, Lincoln WU, Neylan TC, Kilduff TS (2014). The hypocretin/orexin antagonist almorexant promotes sleep without impairment of performance in rats. Front Neurosci 8: 3.
Pasumarthi RK, Gerashchenko D, Kilduff TS (2010). Further characterization of sleep-active neuronal nitric oxide synthase neurons in the mouse brain. Neuroscience 169: 149–157.
Paxinos G, Kus L, Ashwell K, Watson C (1999) Chemoarchitectonic Atlas of the Rat Forebrain 1st edn. Academic Press: San Diego, CA, USA.
Philippides A, Ott SR, Husbands P, Lovick TA, O’Shea M (2005). Modeling cooperative volume signaling in a plexus of nitric-oxide-synthase-expressing neurons. J Neurosci 25: 6520–6532.
Porkka-Heiskanen T, Kalinchuk AV (2011). Adenosine, energy metabolism and sleep homeostasis. Sleep Med Rev 15: 123–135.
Rainnie DG, Grunze HC, McCarley RW, Greene RW (1994). Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science 263: 689–692.
Rattenborg NC, Martinez-Gonzalez D, Lesku JA (2009). Avian sleep homeostasis: convergent evolution of complex brains, cognition and sleep functions in mammals and birds. Neurosci Biobehav Rev 33: 253–270.
Szymusiak R (2010). Hypothalamic versus neocortical control of sleep. Curr Opin Pulm Med 16: 530–535.
Szymusiak R, McGinty D (2008). Hypothalamic regulation of sleep and arousal. Ann NY Acad Sci 1129: 275–286.
Tomioka R, Okamoto K, Furuta T, Fujiyama F, Iwasato T, Yanagawa Y et al (2005). Demonstration of long-range GABAergic connections distributed throughout the mouse neocortex. Eur J Neurosci 21: 1587–1600.
Vincent SR, Kimura H (1992). Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience 46: 755–784.
Vyazovskiy VV, Riedner BA, Cirelli C, Tononi G (2007). Sleep homeostasis and cortical synchronization: II. A local field potential study of sleep slow waves in the rat. Sleep 30: 1631–1642.
Yousef T, Neubacher U, Eysel UT, Volgushev M (2004). Nitric oxide synthase in rat visual cortex: an immunohistochemical study. Brain Res Brain Res Protoc 13: 57–67.
Zangenehpour S, Chaudhuri A (2002). Differential induction and decay curves of c-fos and zif268 revealed through dual activity maps. Brain Res Mol Brain Res 109: 221–225.
Acknowledgements
Research supported by NIH R01 HL59658, USAMRAA Grant DR080789P1, DFG fellowship DI 1718/1-1, and SRI International internal funds. We thank Alan Wilk, Michael Miller, and Webster Lincoln for assistance with the experiments. The authors declare no financial conflict of interests regarding the subject of this work. Over the past 3 years, SRM has received research support from F Hoffmann-LaRoche, Ltd., Sunovion Pharmaceuticals, Inc., and CHDI, Inc. Over the past 3 years, TSK has served as a consultant for NIH and Japanese Society for the Promotion of Science; made paid educational presentations for the benefit of APSS, LLC, the Physician’s Postgraduate Press and the University of Arkansas for Medical Sciences; and received research support from F Hoffmann-LaRoche, Ltd., Sunovion Pharmaceuticals, Inc., CHDI, Inc., and Inscopix, Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dittrich, L., Morairty, S., Warrier, D. et al. Homeostatic Sleep Pressure is the Primary Factor for Activation of Cortical nNOS/NK1 Neurons. Neuropsychopharmacol 40, 632–639 (2015). https://doi.org/10.1038/npp.2014.212
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.212
This article is cited by
-
Somatostatin neurons in prefrontal cortex initiate sleep-preparatory behavior and sleep via the preoptic and lateral hypothalamus
Nature Neuroscience (2023)
-
The natural Disc1-deletion present in several inbred mouse strains does not affect sleep
Scientific Reports (2017)
-
The Dual Hypocretin Receptor Antagonist Almorexant is Permissive for Activation of Wake-Promoting Systems
Neuropsychopharmacology (2016)
-
Nicht invasive Gehirnstimulation zur Beeinflussung von Wachheit und Schlaf
NeuroTransmitter (2016)
