
Abstract
In this review we examine the current understanding of how genetic deficits associated with neurodevelopmental disorders may impact synapse assembly. We then go on to discuss how the critical periods for these genetic deficits will shape the nature of future clinical interventions.
Similar content being viewed by others
INTRODUCTION
During development, the human central nervous system (CNS) carries out an extraordinary undertaking: it forms on the order of 1015 connections between ∼1012 neurons. Considering this huge bioengineering challenge, it may be surprising that for the majority of humans the end result is immensely successful at performing a wide range of tasks. These tasks range from monitoring of sensory inputs to motor control output, and to higher cognitive tasks such as memory storage and retrieval, and social interaction. Unfortunately, though, things do go wrong. This is underlined by the appearance of symptoms of neurodevelopmental disorders (NDDs) such as autism spectrum disorders (ASD) and intellectual disability that coincides with the period of massive synaptogenesis in the developing cortex during the first 2 years of an infant’s development (Huttenlocher and Dabholkar, 1997).
Here, we will use NDDs to collectively designate all disorders that impair the development of the CNS. These commonly include autism and ASDs, attention deficit-hyperactivity disorder (ADHD), intellectual disability (ID), Down’s syndrome, and Tourette’s syndrome (TS). In addition, a large body of evidence suggests that schizophrenia (SCZ) and some forms of bipolar disorder may have developmental roots. Some studies indicate a correlation between individuals destined to manifest SCZ as adults and delays in motor and cognitive milestones during childhood, reaching back to as early as 6 months of life (reviewed by Weinberger and Levitt, 2011). It is possible that some forms of bipolar disorder manifest themselves in children as pediatric mania; however, this is difficult to diagnose and is often misdiagnosed with ADHD and conduct disorder (Biederman et al, 2000). Thus, we consider any possible CNS disorder that may have roots during childhood as a NDD.
The temporal correlation between diagnosis of a subset of NDDs and the period of synapse assembly led to the hypothesis that the deficit underlying many cases of these disorders may reside at the synapse (Bourgeron, 2009; Zoghbi, 2003). Since formulation of the idea of a synaptic pathology (or ‘synaptopathy’) lying at the heart and being a common denominator for NDDs (Brose et al, 2010), many of the genes that have been implicated in these disorders by genetic studies have been examined either in the test tube, in tissue cultures, or in genetically modified organisms (primarily mice) with a possible synaptic etiology in mind. Astonishingly, a large proportion of the genes that have been identified in cases of NDDs have identified roles at the synapse either during development or in the adult (Aldinger et al, 2011; Peca et al, 2011b). Although not all of the studies argue for strictly developmental deficits, a few recent observations argue for shortfalls during synapse assembly and maturation in at least some forms of ID, ASD, and SCZ. Here we will examine the current understanding of the assembly of synapses and how mutations in some genes identified in NDDs affect this process.
THREE STEPS OF SYNAPSE ASSEMBLY
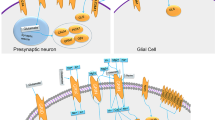
Synapse formation, as we currently understand it at the molecular and cellular level, can be dissected into three major steps: (1) contact formation, (2) transport of synaptic components within those neurites, and (3) immobilization of the synaptic components at the nascent synaptic contact sites (McAllister, 2007) (Figure 1). Genes implicated in NDDs have been found to be involved with all three aspects of synaptogenesis. In general, deficits in NDD susceptibility genes are uncommon, up to 2% of NDD cases for the more common variants (Toro et al, 2010), and collectively only account for between 10 and 20% of NDDs. However, identifying mutations that genuinely cause dysfunction of a gene, and thus implicating it in the etiology of NDDs, tells us much about the underlying biology of CNS and synapse development. Below, we present examples of genes with mutations and copy number variations (CNVs) that are both highly likely to result in protein function deficits or protein expression level differences, and for which studies clearly indicate an involvement in the three steps of synapse assembly (Table 1). This should not be understood as a comprehensive list, but serves as a demonstration of how genetic studies of the NDD population and basic research, primarily in mouse models, have helped further our understanding of synapse assembly and some aspects of NDDs. We then examine genes identified in syndromic forms of ASDs, and how the encoded proteins might function during synaptic development.

The three basic steps of synapse assembly. (1) First neuronal contact triggers a cascade of events through cell adhesion molecules such as Nlgns, Nrxns, and SynCAMs. Pre- and postsynaptic precursors need to be efficiently transported in axons and dendrites (2), by associating with motor proteins, such as Kinesin17, that ‘walk’ along microtubules. Neuronal contact must result in stopping, perhaps by means of kinases such as Cdk5, and maintenance of the synaptic material at the nascent synaptic site because of scaffolding molecules, such as SHANKs and CASK (3). This process eventually leads to the formation of a functional synaptic contact (4).
Neuronal Contact
Genes involved in neuronal cell adhesion and synaptic scaffolding are collectively the most prevalent genes found to date in genome-wide association studies (GWASs) of NDDs (Corvin, 2010; Kumar et al, 2011; O’Dushlaine et al, 2011). This suggests that cell adhesion events are critical for the correct assembly of synapses (Figure 1, top left). There are hundreds of genes encoding potential cell adhesion molecules (CAMs) expressed in the CNS; however, to date only a handful of them have been found to be able to induce synapse assembly (Bukalo and Dityatev, 2012; Washbourne et al, 2004). This has largely been defined by the expression of candidate CAMs in nonneuronal cells and their ability to induce either pre- or postsynaptic assembly in cocultured neurons (Biederer et al, 2002; Biederer and Scheiffele, 2007; Fu et al, 2003; Scheiffele et al, 2000; Siddiqui et al, 2010).
Neuroligins
The first family of CAMs that were shown to be sufficient to drive synapse assembly in vitro was the neuroligins (Scheiffele et al, 2000). These had been identified as ligands of the presynaptically localized α-latrotoxin receptors, the neurexins (Song et al, 1999). Indeed, subsequent studies demonstrated that binding of neuroligins (Nlgns) to the neurexins (Nrxns) resulted in assembly of presynaptic terminals in axons (Dean et al, 2003), and that binding of Nrxns to Nlgns drove postsynaptic differentiation in dendrites (Barrow et al, 2009; Nam and Chen, 2005). The in vitro studies suggest that Nlgn1 is key in triggering the recruitment of NMDA-type glutamate receptors (Barrow et al, 2009; Hoy et al, 2013), but this may be a late maturational step of synapse formation.
The neuroligins are encoded by five genes in humans (1, 2, 3, 4X, and 4Y), and mutations in NLGN1, 3, and 4X genes have been associated with autism (Sudhof, 2008) (Table 1). Whereas loss-of-function mutations have been found in patients with ASDs for NLGN3 and NLGN4X (Jamain et al, 2003; Laumonnier et al, 2004), an increase in copy number of the NLGN1 gene has been associated with ASD (Glessner et al, 2009; Szatmari et al, 2007) (Table 1). Analyses in mice have confirmed that loss-of-function mutations in Nlgn3 and Nlgn4 result in abnormal synaptic function and behavioral deficits (Ellegood et al, 2011; Jamain et al, 2008; Radyushkin et al, 2009; Tabuchi et al, 2007). It is important to note that studies performed with the same Nlgn3 and Nlgn4 mutations as characterized previously (Jamain et al, 2008; Tabuchi et al, 2007), but carried out in different labs, showed no significant effects on social behaviors or ultrasonic vocalizations (Chadman et al, 2008; Ey et al, 2012). These results open the possibility that genetic background and environmental effects may contribute to the subtle behavioral phenotypes. Since then, social deficits have been confirmed for the Nlgn3 knock-in mice, expressing a mutant form of Nlgn3 (R451C) identified in patients with autism, using multiple background strains of mice (Jaramillo et al, 2014). This result adds weight to the argument that mutations in these genes may be the cause of some cases of ASD in humans. Similarly, mice in which Nlgn1 was overexpressed, to mimic the copy number increase, also displayed anomalous synaptic characteristics (Dahlhaus et al, 2010; Hoy et al, 2013). Although overexpression of Nlgn1 resulted in deficits in learning and memory behavior, social behaviors tested were not affected (Dahlhaus et al, 2010; Hoy et al, 2013). In conclusion, the Nlgn family of genes has been clearly implicated in ASD etiology through genetic association and mouse studies.
Neurexins
The Nrxns are encoded by three genes in mammals; and all of them generate two forms of the protein from different start codons: the long α-Nrxns and the short β-Nrxns (Tabuchi and Sudhof, 2002). Approximately 0.5% of ASD cases harbor mutations in Nrxn 1α (NRXN1), underlining the importance of this gene in the etiology of NDDs (Reichelt et al, 2012). Analyses with loss-of-function mutations of the Nrxn genes have confirmed the role of α-Nrxns in shaping synapse function (Missler et al, 2003; Sons et al, 2006) and complex behaviors (Etherton et al, 2009; Laarakker et al, 2012), including social behaviors (Grayton et al, 2013). Deletion of all three α-Nrxn genes results in death of newly born mouse pups because of reduced synaptic transmission (Missler et al, 2003).
Further study of the extracellular domains of Nrxns demonstrated that they are relatively promiscuous, participating in molecular interactions with a suite of other synaptic proteins in addition to the Nlgns. These include LRRTMs, cerebellin, and the GABA A receptor. LRRTMs appear to act in a manner similar to Nlgns in postsynaptic assembly (de Wit et al, 2009; Ko et al, 2009; Linhoff et al, 2009; Wright and Washbourne, 2011). One important difference is that Nlgns primarily affect NMDA-type glutamate receptor recruitment and maintenance at synapses (Barrow et al, 2009; Hoy et al, 2013), whereas LRRTMs influence AMPA-type glutamate receptor presence at synapses (Soler-Llavina et al, 2011). Binding of Nrxns to cerebellin (Uemura et al, 2010), which in turn binds to glutamate receptor subunit δ2 (Matsuda et al, 2010), may be a specialization in the cerebellum to control synapse number between parallel fibers and Purkinje cells (Hirai et al, 2005). It is unclear how the cis interaction between Nrxns and the GABA A receptor contributes to synapse formation (Zhang et al, 2010). Presumably this interaction occurs in dendrites at an early stage during development, before Nrxns redistribute to axons (Barker et al, 2008). Thus, these studies suggest that Nrxns are key organizers of the pre- and postsynaptic specializations.
SynCAMs
Another family of molecules, identified by the ability to induce the formation of presynaptic terminals in coculture experiments, is the SynCAMs (Biederer et al, 2002). These proteins resemble nectins, in that they contain three extracellular immunoglobulin domains for homo- and heterophilic interaction. There are four SynCAM genes in humans (CADM1–4) (Biederer, 2006), one of which (CADM1) has been linked to ASD by virtue of two missense mutations that render SynCAM1 unable to be transported out of the endoplasmic reticulum to the plasma membrane (Zhiling et al, 2008). Knockout mice for SynCAM1 present a subtle reduction in synapse number (<10%), perhaps because of compensation by the other family members (Robbins et al, 2010). Examination of synapses by electron microscopy suggests an impact of SynCAM1 ablation on the size of both the presynaptic active zone and the postsynaptic density (Robbins et al, 2010). SynCAM1 suppresses long-term depression (LTD), as knockout mice show enhanced LTD, with no effect on long-term potentiation (Robbins et al, 2010). As a result, SynCAM1 KO mice exhibit enhanced spatial reference memory. The in vitro studies of SynCAM1 have suggested that the majority of this CAM function resides in its ability to modulate cytoskeletal dynamics during assembly of the presynaptic terminal (Stagi et al, 2010) and during formation of the postsynaptic spine (Cheadle and Biederer, 2012). Thus, SynCAM1 is clearly implicated in the development of fully functional synapses.
Mechanisms of CAM-triggered synapse assembly
To date, there appears to be a discrepancy between the sufficiency for CAMs to induce the formation of synaptic terminals in coculture systems and their necessity in the formation of synapses, especially in vivo. Expression of SynCAMs and Nlgns in nonneuronal cells and culturing of these cells with neurons suggests a potent ability of presynaptic SynCAMs and Nrxns in coordinating the recruitment of presynaptic components (Biederer et al, 2002; Scheiffele et al, 2000). But in vitro and in vivo studies of loss-of-function mutations of these genes have not shed light on how these molecules might perform this function (Robbins et al, 2010; Varoqueaux et al, 2006). This discrepancy may be because of compensation by other genes within the family, or by other CAM families. Alternatively, the effects of the loss of synaptic CAM function may only be discerned when there is competition between individual neurons, such that complete knockout does not show a difference, whereas if some neurons have reduced CAM expression they will exhibit reduced synapse numbers (Kwon et al, 2012).
Despite the lack of a strong effect on synapse formation by loss-of-function mutations, it was hypothesized that protein–protein interactions of the short intracellular tails of both Nrxns and SynCAMs might be the trigger for the accumulation of presynaptic components. Specifically, it was assumed that the presynaptic MAGUK protein CASK, which can bind to the terminal PDZ domain of both Nrxns and SynCAMs, might be the presynaptic organizer. This view was recently challenged by experiments in which the PDZ domain was switched for one that cannot bind to CASK, yet still resulted in functional presynaptic assembly (Gokce and Sudhof, 2013). Indeed, expression of a mutant form of Nrxn in which the entire intracellular tail was eliminated was still sufficient for presynaptic assembly to occur, including CASK recruitment (Gokce and Sudhof, 2013). These experiments suggest that Nrxns, and based on the similarity of the intracellular regions of perhaps also SynCAMs, function through an as yet unidentified transmembrane receptor to induce presynaptic assembly.
In contrast, the intracellular regions of Nlgns appear to play a central role in coordinating postsynaptic assembly and maturation. Protein–protein interactions of the intracellular tails of Nlgn1 help traffic NMDA receptors to synapses (Barrow et al, 2009) and mediate the switch in NMDA receptor type (from NR2B to NR2A containing) that occurs at synapses during development (Hoy et al, 2013). In addition, novel interaction domains within the C-terminus of Nlgns have been identified that are critical for both NMDA and AMPA receptor recruitment to synapses (Etherton et al, 2011; Shipman et al, 2012). Thus, whereas recruitment and maintenance of presynaptic components is not mediated by intracellular interactions of Nrxns and SynCAMs, it appears that protein–protein interactions within the C-terminal tail of Nlgns has a profound effect on postsynaptic assembly.
Axonal and Dendritic Transport of Synaptic Components
The vast majority of synaptic components are synthesized within the cell body. Thus, for the assembly of synapses to occur, synaptic precursor material must be transported within dendrites and axons to nascent synaptic sites (see Figure 1, top right). Transport of synaptic precursors to synapses involves motor proteins, dynamins, and kinesins that walk along microtubule tracks in both dendrites and axons (Goldstein et al, 2008; Hirokawa et al, 2010). Synaptic vesicle precursors and active zone precursors are transported in a microtubule-dependent manner in both antero- and retrograde directions (Ahmari et al, 2000; Bury and Sabo, 2011; Sabo and McAllister, 2003). Similarly, NMDA and AMPA receptors are transported along microtubules in both directions in dendrites (Washbourne et al, 2002). Although it is possible that some transmembrane synaptic components diffuse within the plasma membrane to synaptic sites (Groc et al, 2009), it is unlikely that this mechanism would deliver enough receptors, especially during peak synaptogenesis. Supporting a role for microtubule-based transport in synapse assembly, genes that encode axonal or dendritic motor proteins, microtubule-organizing proteins or regulators of microtubule transport have been uncovered as candidate genes in NDDs.
MAPT
As microtubules are the rails along which long-distance axonal and dendritic transport is achieved, one would expect mutations that affect microtubule stability or organization to result in NDDs. Indeed, rearrangements of human chromosome 17 that affect the microtubule-associated protein Tau (MAPT) have been identified in patients with learning disability and developmental delay (Koolen et al, 2006; Rovelet-Lecrux et al, 2012; Shaw-Smith et al, 2006). Although MAPT has been more commonly associated with neurodegenerative disorders, because of its presence in neurofibrillary tangles (Goedert et al, 2006), this gene plays an active role during development. MAPT promotes microtubule assembly and stability and may act as a linker protein between the microtubules and the plasma membrane. Thus, a reduction in MAPT can affect neuronal migration and deficits in dendritic arbor elaboration and synapse number (Sapir et al, 2012).
Kinesin17
A nonsense mutation in Kinesin17 gene (KIF17) was found in a case of schizophrenia (Awadalla et al, 2010; Tarabeux et al, 2010). The mutation resulted in truncation of the protein before the tail domain, the region important for interaction with the transport cargo (Goldstein et al, 2008). Similar truncations result in dominant negative motor proteins (Setou et al, 2002), explaining how such a mutation could result in deficits even when present in the heterozygous state (Tarabeux et al, 2010). Kinesin17 is an interesting candidate with regard to NDDs, as it was originally identified as a motor protein for NMDA receptor transport (Kayadjanian et al, 2007; Setou et al, 2000). Thus, overexpression of Kinesin17 results in enhanced spatial learning and memory in transgenic rodents (Wong et al, 2002). The ability to release the NMDA receptor-containing cargo is critical, as mutations that block phosphorylation of serines in the tail domain result in reduced synaptic NMDA receptor localization and deficient spatial learning and memory (Yin et al, 2012).
Gsk-3β
Mutations affecting splicing and expression levels of the glycogen synthase kinase 3β gene (GSK3B) are associated with schizophrenia (Blasi et al, 2013). Many of the roles of this signaling enzyme in neural function remain unclear. However, with regard to synapse formation, Gsk-3β has been implicated in the transport of amyloid precursor protein (APP) in axons, and in the wnt signaling pathway. Although the role of APP outside of the pathway to Alzheimer’s disease remains elusive, interactome studies suggest that APP can bind synaptotagmin-1, the calcium sensor for synaptic vesicle release (Kohli et al, 2012) and the axonal motor protein Kinesin-1 (Kamal et al, 2001). However, there has been some controversy as to whether APP is important for the transport of these components in axons (Lazarov et al, 2005). Gsk-3β phosphorylates APP, reducing its axonal transport (Takashima et al, 1995), suggesting that Gsk-3β may play a generalized role in regulating transport of synaptic components to synapses. In another potential synaptic function, Gsk-3β regulates wnt signaling at vertebrate synapses and the invertebrate neuromuscular junction to modulate presynaptic microtubules during synapse formation (Salinas, 2005). Gsk-3β plays a role in this pathway by phosphorylating microtubule-associated protein 1B (MAP1B), resulting in more dynamic microtubules (Goold et al, 1999). In addition, Gsk-3β plays a role in the mobilization of postsynaptic density protein-95 (PSD-95) (Nelson et al, 2013), suggesting a wide variety of roles for this kinase in shaping the synapse.
Recruitment and Maintenance of Synaptic Components at Nascent Synaptic Sites
Once synaptic precursors reach the nascent synaptic site, they need to stop, reorganize, and be maintained at the developing synapse (see Figure 1, bottom left). Kinases may regulate synaptic precursor stopping at the synapses, whereas scaffolding molecules will lock synaptic components into a network of interactions to hold them at the mature synapse (Figure 1, bottom right).
SHANKs
Mutations and microdeletions in the SHANK genes have been found among individuals with ASDs (Awadalla et al, 2010; Berkel et al, 2010; Guilmatre et al, 2014; Leblond et al, 2012; Lennertz et al, 2012; Sato et al, 2012). These genes encode scaffolding molecules, molecules whose sole job is to bind to other proteins inside the cell exclusively at synapses (Grabrucker et al, 2011). Studies of knockout mice for SHANKs 1, 2, and 3 suggest that reduced levels of these SHANK proteins result in synaptic deficits (Bozdagi et al, 2010; Sala et al, 2001; Wang et al, 2011) with consequences for cognitive and social behavior (Bozdagi et al, 2010; Peca et al, 2011a; Silverman et al, 2011; Wang et al, 2011; Wohr et al, 2011; Won et al, 2012). SHANKs are interacting partners of Nlgns and form a link between the transsynaptic CAMs and important synaptic signaling molecules inside the postsynaptic terminal including NMDA receptors (Petralia et al, 2005). Indeed, SHANK2 deficits can be reverted in mice by restoring NMDA receptor function (Won et al, 2012). The wealth of mutational information from the ASD population and the strong indications from mouse experiments point toward a significant contribution of SHANK gene deficits underlying ASD etiology.
CDK5
Although scaffolding molecules such as SHANKs can explain the maintenance of a variety of components at synapses, it is less clear how these components might initially be recruited to a nascent synaptic site. An inkling into mechanisms of recruitment can be gained by examining cyclin-dependent kinase 5 (CDK5). The gene for the regulatory subunit of CDK5 (CDK5R1) has been linked to nonsyndromic intellectual disability (Venturin et al, 2006) and dysregulation of CDK5 expression has been found in the brains of patients with schizophrenia (Engmann et al, 2011; Ramos-Miguel et al, 2012). CDK5 has been ascribed many neuronal functions ranging from neuronal migration to the regulation of synaptic strength (Cheung and Ip, 2007; Dhariwala and Rajadhyaksha, 2008; Smith and Tsai, 2002). However, a number of studies have specifically examined the role of CDK5 in transport of synaptic components to synapses. In the assembly of presynaptic terminals in C. elegans, CDK5 regulates the deposition of all presynaptic components (Park et al, 2011). In contrast, in vertebrates CDK5 specifically regulates the appearance of CASK (Samuels et al, 2007) and synapsin at synapses (Easley-Neal et al, 2013). In fact, CDK5 activity must be local to allow synapsin transport packets to stop at sites of nascent synapse formation (Easley-Neal et al, 2013). These studies suggest that kinases may play critical roles in regulating the recruitment of presynaptic components by generating a local STOP signal at sites of synapse formation. What exactly constitutes the STOP signal is unclear, but could include inhibition of the motor proteins (Yin et al, 2012) or uncoupling of the cargo from the motor proteins by phosphorylation (Samuels et al, 2007).
CASK
Mutations in the calcium/calmodulin-dependent serine kinase (CASK) gene have been identified in cases of X-linked optic atrophy, X-linked mental retardation, and microcephaly with disproportionate pontine cerebellar hypoplasia with associated intellectual disability (Dimitratos et al, 1998; Froyen et al, 2007; Hackett et al., 2009; Hayashi et al, 2008; Najm et al, 2008). CASK protein appears to have two very distinct functions (Hsueh, 2006). During embryonic development, ∼20% of the protein is present in the nucleus where it can interact with the transcription factor TBR1 (Hsueh et al, 2000). However, the protein also contains PDZ, SH3, and a guanylate kinase domain that are characteristic of membrane-associated guanylate kinase (MAGUK) proteins. MAGUK proteins, the most famous of which is perhaps PSD-95, are classic synaptic scaffolding proteins (Craven and Bredt, 1998). Indeed, CASK localizes at synapses and can interact with the intracellular tail of SynCAMs and Nrxns (Biederer et al, 2002; Hata et al, 1996). CASK associates with the α1B subunit of the voltage-gated calcium channel (VGCC) and this interaction determines VGCC localization at synapses (Maximov and Bezprozvanny, 2002; Maximov et al, 1999). As mentioned above, the synaptic localization is regulated by phosphorylation by Cdk5 (Samuels et al, 2007). Interestingly, from the perspective of synaptogenesis, CASK assembles with Mint1 and Veli to form a motor protein/cargo adaptor (Setou et al, 2000). This suggests that CASK may not only help maintain synaptic proteins, such as calcium channels at synapses, but also be involved in the transport of synaptic cargo to developing synapses (Figure 1, top right). Thus, mutations in CASK may contribute to neuronal and synaptic development at multiple levels; as a transcription factor during neuronal differentiation and migration, and during synaptic assembly and maturation.
SYNAPTIC FUNCTIONS OF SYNDROMIC NDD GENES
There are several syndromes that manifest with social deficits, bringing them into the autism spectrum. Three of these strictly mendelian syndromes, tuberous sclerosis, fragile X mental retardation, and Rett syndrome, are because of mutations in genes that have now been shown to have roles in synaptic function and potentially also synapse formation. Here we discuss how the gene products of the TSC1 and 2, FMR1, and MECP2 genes may be involved in synapse formation.
Tsc1 and Tsc2
Tuberous sclerosis (TSC) is an inherited disease manifested with nonmalignant growths in the brain, skin, and other tissues. These growths, often called tubers in the brain, arise because of imbalances in cell growth signals. Importantly, ∼60% of individuals with TSC also manifest deficits in intellectual ability and social behavior, placing TSC within the autism spectrum (Napolioni and Curatolo, 2008). The genes affected are the tumor suppressor genes Tsc1 and Tsc2. The encoded proteins, hamartin and tuberin, interact with Rheb GTPase thereby inactivating the mammalian target of rapamycin (mTOR) complex (Napolioni and Curatolo, 2008). The mTOR is a kinase that regulates not only cell growth, but also cell motility, transcription, and protein synthesis. Importantly, recent studies have uncovered the importance for mTOR signaling at the synapse, potentially explaining the association with neurodevelopmental disorders: mTOR acts as a regulator of activity-dependent transcription of synaptic plasticity genes (Gipson and Johnston, 2012; Troca-Marin et al, 2012). More recently, Tsc1 and Tsc2 have been shown to function antagonistically with another mendelian NDD gene, Fmr1 (Auerbach et al, 2011). Thus, Tsc1 and Tsc2 may be involved in the activity-dependent consolidation of synapses during development.
Fmr1
Fragile X mental retardation is the most common form of inherited intellectual disability and the most common single gene cause of autism. The disease arises because of chromosomal instabilities of the X chromosome (Xq27.3). The affected gene, FMR1, encodes for an mRNA binding protein that has been shown to regulate the transcription of synaptic mRNAs in an activity-dependent manner (McLennan et al, 2011). Examples of the genes that are regulated include those encoding glutamate receptor subunits and calcium-calmodulin kinase II (CaMKII). The chromosomal instabilities result in increased numbers of CGG repeats in the promoter region that in turn cause decreased expression levels of Mecp2 protein. This decrease in expression level results in synaptic deficits, including an increase in synaptic spine density and an increase in immature spine shapes in humans (Irwin et al, 2001) and mice (Greenough et al, 2001). In addition, knockout mice exhibit an increase in metabotropic glutamate receptor-dependent LTD (Huber et al, 2002; Martin and Huntsman, 2012). Interestingly, as the LTD effect was opposite to the LTD deficit observed in Tsc2 mice, crossing of the two mutations into the same mouse resulted in the effects canceling each other out (Auerbach et al, 2011). Thus, it has been hypothesized that Tsc2 and Fmr1 lie in parallel, antagonistic pathways that both control the synthesis of LTD-specific proteins at synapses (Auerbach et al, 2011). In another comparative study of knockout mice, it was shown that both Nlgn3 knockout mice and Fmr1-null mice share a similar phenotype, including perturbed metabotropic glutamate receptor-dependent LTD (Baudouin et al, 2012). These studies suggest that both syndromic and nonsyndromic forms of autism may share synaptic mechanisms. In addition, one can conclude that, in the case of FMR1 and Tsc2, mutations affect common pathways, although in different directions. In conclusion, with regard to synapse development, FMR1 appears to be critical in the pruning back of inappropriate synapses.
Mecp2
Rett syndrome is caused by mutations in the methyl-CpG binding protein 2 (MECP2) gene. This gene is located on the X chromosome in humans, leading to more severe defects in affected males than females. In fact, the majority of males die within the first 2 years after birth. The encoded protein has a DNA-binding domain that preferentially interacts with methylated DNA in chromatin. Based on this observation, it was assumed that the protein is a transcriptional repressor (Boyes and Bird, 1991); however, it has been also seen to drive the expression of some genes (Della Ragione et al, 2012). Importantly, it has been shown to affect the development of dendrites and synapses, regardless of when the gene is inactivated using a conditional mouse knockout model (Nguyen et al, 2012). Furthermore, it appears that the deficit may lie within glial cells. Coculturing of neurons with Mecp2-deficient astrocytes is sufficient to elicit the neuronal phenotype (Ballas et al, 2009). This suggests that Mecp2 may regulate the expression of secreted glial factors that contribute to neuronal development (see below).
GLIAL FACTORS CONTRIBUTING TO SYNAPSE ASSEMBLY
During early studies of the development of synapse formation in primary neuronal cultures, it was apparent that ‘feeder layers’ of glial cells, in particular astrocytes, were critical to drive synapse assembly between various populations of neurons (Banker, 1980; Kaech and Banker, 2006; Pfrieger and Barres, 1997). Since then, a number of factors released by astrocytes have been identified, including thrombospondins, glypicans, even cholesterol, and their mechanisms for driving synapse assembly and elimination have been examined (comprehensively reviewed by Clarke and Barres, 2013). As mentioned previously, mutations in Mecp2 (Ballas et al, 2009), and also FMR1 (Jacobs and Doering, 2010), result in astrocytes that do not maintain correct synaptic development, clearly implicating some of these factors in the etiology of ASD-linked disorders.
For this review, we will consider the glypican 6 (GPC6) gene in more detail, as it has been linked to ADHD and neuroticism (Calboli et al, 2010; Lesch et al, 2008), a character trait characterized by anxiety and depressed mood. Biochemical fractionation studies of astrocyte-conditioned media led to the identification of Gpc4 and Gpc6 as factors that could significantly increase the formation of synapses (Allen et al, 2012). Whereas astrocyte-secreted thrombospondins induce the formation of structural, but nonfunctional, synapses (Christopherson et al, 2005), Gpc4 and Gpc6 administration to neurons in culture results in the addition of functional synapses. Conversely, knockout mice for Gpc4 exhibit deficits in synapse formation (Allen et al, 2012). Together, these results argue for a significant role for this astrocyte-secreted factor in synaptic development. It will be interesting to discover the neuronal ligand that mediates the effects of Gpcs, thereby uncovering the molecular mechanism for astrocyte-induced synaptogenesis.
FUTURE DIRECTIONS AND CLINICAL IMPLICATIONS
In conclusion, genetic studies combined with studies primarily in mouse models of the candidate gene defects have contributed to our understanding of the underlying mechanisms of NDDs. Although much work is still needed, it is apparent that, at least for a subset of NDDs, deficits during development have a significant impact on synapse assembly and function, and consequently on behavior. Much work still needs to be carried out. The number of mouse models of candidate genes needs to be expanded to gain a more complete picture of the underlying molecular mechanisms of NDDs. Existing mouse models, or perhaps even zebrafish with high-throughput capacity (Tropepe and Sive, 2003), can be systematically tested for pharmacological rescue of behavioral deficits, thereby generating lead drug candidates for treatment studies. In addition, it will be important to study whether candidate gene function is absolutely necessary during development of the synapse or whether some aspects of synaptic function can be regained in the adult.
Although determining whether a genetic defect primarily affects development of synapses rather than mature synapse function may seem, at first glance, an esoteric endeavor, this difference has important implications for therapy. With the advent of viral-based gene therapy applications almost a realistic possibility in humans (Witt and Marks, 2011), it will be important to know where and also when an intervention is critical for a successful outcome. For example, if a synaptic deficit can be overcome in the adult, then this provides hope for many adults with this form of ASD. In contrast, when it is known that a developmental synaptic deficit is hardwired into the mature CNS, it advocates for an intervention as early as possible. Although the genetic critical periods are not the only deciding factors in the ability to treat a deficit, especially by pharmacological means, information on when and where a gene product’s function is necessary will certainly give insight into windows of opportunity.
To date, only a few of the NDD candidate genes have been studied with temporal requirements in mind. These kinds of experiments are difficult, requiring either the ‘rescue’ of deficits with viruses in a known location during development and in the adult, or the generation of conditional mutations, allowing for genetic rescue at given times. Such studies have been performed for Mecp2 and Nlgn3, and for Nlgn1 overexpression, but are necessary for more models of NDDs.
The most striking temporal rescue was demonstrated using a floxed STOP allele of the Mecp2 gene in mice. Re-expression of Mecp2 protein starting at 10 weeks after birth ameliorated neurological symptoms and promoted survival of male null mice many weeks beyond their null littermates that did not re-express Mecp2 (Guy et al, 2007). These experiments conclusively demonstrated that Rett-like defects in mice can be rectified in adult mice. Similarly, late expression (P30) of Nlgn3 in a null background rescued an ectopic synapse formation phenotype in the cerebellum (Baudouin et al, 2012). Nlgn3 knockout mice demonstrate a motor coordination phenotype that is rescued by re-expression during development. Amelioration of the motor coordination phenotype was not tested in the juvenile rescue experiment (Baudouin et al, 2012). It is also important to note that pharmacological intervention has been successful in improving synaptic and social deficits in adult mice. Shank2-deficient mice, which have the same microdeletion as seen in cases of ASD, manifest decreased NMDA receptor function, reduced social interaction, and reduced ultrasonic vocalization (Won et al, 2012). Treatment of adult mice with D-cycloserine, a partial NMDA receptor agonist, or CDPPB, a positive allosteric modulator of the metabotropic glutamate receptor 5 (mGluR5, which modulates NMDA receptor function), reversed the synaptic deficits and enhanced social interaction (Won et al, 2012).
To contrast these examples of juvenile/adult rescue of neuronal and neurological deficits, adult reduction of Neuroligin1 overexpression did not rescue synaptic or behavioral symptoms (Hoy et al, 2013). Neuroligin1 overexpression in the hippocampus results in increased spine head size and reduced learning and memory behavior. When overexpression was turned off at 2 months of age, spine heads remained larger in size and repeated testing with the novel object recognition test showed no improvement of memory for up to 4 weeks (Hoy et al, 2013). In contrast, 4 weeks of overexpression that began in the adult did lead to decreased novel object recognition. This study suggests that overexpression of Neuroligin1 is sufficient to hardwire synapses into a pathological state during development, and that this state cannot be reversed by reducing expression levels in the adult. It is, however, possible that a pharmacological intervention might be successful in improving the behavioral deficits.
Given the heterogeneity of results from these models, that is, that Neuroligin1 overexpression deficits cannot be rescued after development (Hoy et al, 2013) whereas MeCP2, Neuroligin3, and SHANK2 deletions can (Baudouin et al, 2012; Guy et al., 2007; Won et al, 2012), it is imperative that we widen these types of studies to other genetic models. It is tempting to hypothesize that overexpression can generally not be rescued, whereas lack of expression can. However, this general rule does not seem to be the case as the increased number of synapses driven by SynCAM1 overexpression during development can only be maintained by continuous SynCAM1 overexpression in the adult (Robbins et al, 2010). As rescue is not equally possible in developing and adult mice, it seems that each individual mutation may have a defined critical period and will have to be experimentally determined in each case.
With the advent of personalized medicine, we can expect to see prenatal genomic profiling to become increasingly routine. Genomic data will need to be supplemented with a basic understanding of (1) the genes involved in NDDs, (2) the parts of these genes that are essential for synaptic function, and (3) the critical periods for the function of each gene. It is difficult to extrapolate genetic critical periods from mouse studies to humans, and much work is necessary to confirm these trends in human patients. However, an integration of these studies with an individual’s genome sequence may eventually allow personalized interventions, such as gene therapy and/or pharmaceutical treatments for the most severe cases of NDDs.
FUNDING AND DISCLOSURE
The author declares no conflict of interest.
References
Ahmari SE, Buchanan J, Smith SJ (2000). Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci 3: 445–451.
Aldinger KA, Plummer JT, Qiu S, Levitt P (2011). SnapShot: genetics of autism. Neuron 72: 418–418 e411.
Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ et al (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486: 410–414.
Auerbach BD, Osterweil EK, Bear MF (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480: 63–68 In this study, crosses of Fmr1 and Tsc2 mutant mice to each other demonstrate antagonistic roles for these genes in metabotropic glutamate receptor-dependent LTD that cancel each other out.
Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR et al (2010). Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet 87: 316–324.
Ballas N, Lioy DT, Grunseich C, Mandel G (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci 12: 311–317.
Banker GA (1980). Trophic interactions between astroglial cells and hippocampal neurons in culture. Science 209: 809–810.
Barker AJ, Koch SM, Reed J, Barres BA, Ullian EM (2008). Developmental control of synaptic receptivity. J Neurosci 28: 8150–8160.
Barrow SL, Constable JR, Clark E, El-Sabeawy F, McAllister AK, Washbourne P (2009). Neuroligin1: a cell adhesion molecule that recruits PSD-95 and NMDA receptors by distinct mechanisms during synaptogenesis. Neural Dev 4: 17.
Baudouin SJ, Gaudias J, Gerharz S, Hatstatt L, Zhou K, Punnakkal P et al (2012). Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 338: 128–132 A comparison of two knockout mouse models demonstrates phenotypic similarities for Nlgn3 and Fmr1 ablations. This study also shows adult rescue of the synaptic Nlgn3 phenotype in the cerebellum.
Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U et al (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet 42: 489–491.
Biederer T (2006). Bioinformatic characterization of the SynCAM family of immunoglobulin-like domain-containing adhesion molecules. Genomics 87: 139–150.
Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET et al (2002). SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 297: 1525–1531.
Biederer T, Scheiffele P (2007). Mixed-culture assays for analyzing neuronal synapse formation. Nat Protoc 2: 670–676.
Biederman J, Mick E, Faraone SV, Spencer T, Wilens TE, Wozniak J (2000). Pediatric mania: a developmental subtype of bipolar disorder? Biol Psychiatry 48: 458–466.
Blasi G, Napolitano F, Ursini G, Di Giorgio A, Caforio G, Taurisano P et al (2013). Association of GSK-3beta genetic variation with GSK-3beta expression, prefrontal cortical thickness, prefrontal physiology, and schizophrenia. Am J Psychiatry 170: 868–876.
Bourgeron T (2009). A synaptic trek to autism. Curr Opin Neurobiol 19: 231–234.
Boyes J, Bird A (1991). DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell 64: 1123–1134.
Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N et al (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism 1: 15.
Brose N, O'Connor V, Skehel P (2010). Synaptopathy: dysfunction of synaptic function? Biochem Soc Trans 38: 443–444.
Bukalo O, Dityatev A (2012). Synaptic cell adhesion molecules. Adv Exp Med Biol 970: 97–128.
Bury LA, Sabo SL (2011). Coordinated trafficking of synaptic vesicle and active zone proteins before synapse formation. Neural Dev 6: 24.
Calboli FC, Tozzi F, Galwey NW, Antoniades A, Mooser V, Preisig M et al (2010). A genome-wide association study of neuroticism in a population-based sample. PLoS One 5: e11504.
Chadman KK, Gong S, Scattoni ML, Boltuck SE, Gandhy SU, Heintz N et al (2008). Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res 1: 147–158.
Cheadle L, Biederer T (2012). The novel synaptogenic protein Farp1 links postsynaptic cytoskeletal dynamics and transsynaptic organization. J Cell Biol 199: 985–1001.
Cheung ZH, Ip NY (2007). The roles of cyclin-dependent kinase 5 in dendrite and synapse development. Biotechnol J 2: 949–957.
Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A et al (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120: 421–433.
Clarke LE, Barres BA (2013). Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci 14: 311–321.
Corvin AP (2010). Neuronal cell adhesion genes: key players in risk for schizophrenia, bipolar disorder and other neurodevelopmental brain disorders? Cell Adh Migr 4: 511–514.
Craven SE, Bredt DS (1998). PDZ proteins organize synaptic signaling pathways. Cell 93: 495–498.
Dahlhaus R, Hines RM, Eadie BD, Kannangara TS, Hines DJ, Brown CE et al (2010). Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus 20: 305–322.
de Wit J, Sylwestrak E, O'Sullivan ML, Otto S, Tiglio K, Savas JN et al (2009). LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron 64: 799–806.
Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E et al (2003). Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci 6: 708–716.
Della Ragione F, Filosa S, Scalabri F, D’Esposito M (2012). MeCP2 as a genome-wide modulator: the renewal of an old story. Front Genet 3: 181.
Dhariwala FA, Rajadhyaksha MS (2008). An unusual member of the Cdk family: Cdk5. Cell Mol Neurobiol 28: 351–369.
Dimitratos SD, Stathakis DG, Nelson CA, Woods DF, Bryant PJ (1998). The location of human CASK at Xp11.4 identifies this gene as a candidate for X-linked optic atrophy. Genomics 51: 308–309.
Easley-Neal C, Fierro J Jr., Buchanan J, Washbourne P (2013). Late recruitment of synapsin to nascent synapses is regulated by Cdk5. Cell Rep 3: 1199–1212 This study demonstrated the necessity of local Cdk5 activity for the recruitment of synaptic precursors, Synapsin transport packets, to nascent synapses.
Ellegood J, Lerch JP, Henkelman RM (2011). Brain abnormalities in a Neuroligin3 R451C knockin mouse model associated with autism. Autism Res 4: 368–376.
Engmann O, Hortobagyi T, Pidsley R, Troakes C, Bernstein HG, Kreutz MR et al (2011). Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain 134: 2408–2421.
Etherton MR, Blaiss CA, Powell CM, Sudhof TC (2009). Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci USA 106: 17998–18003.
Etherton MR, Tabuchi K, Sharma M, Ko J, Sudhof TC (2011). An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J 30: 2908–2919.
Ey E, Yang M, Katz AM, Woldeyohannes L, Silverman JL, Leblond CS et al (2012). Absence of deficits in social behaviors and ultrasonic vocalizations in later generations of mice lacking neuroligin4. Genes Brain Behav 11: 928–941.
Froyen G, Van Esch H, Bauters M, Hollanders K, Frints SG, Vermeesch JR et al (2007). Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: important role for increased gene dosage of XLMR genes. Hum Mutat 28: 1034–1042.
Fu Z, Washbourne P, Ortinski P, Vicini S (2003). Functional excitatory synapses in HEK293 cells expressing neuroligin and glutamate receptors. J Neurophysiol 90: 3950–3957.
Gipson TT, Johnston MV (2012). Plasticity and mTOR: towards restoration of impaired synaptic plasticity in mTOR-related neurogenetic disorders. Neural Plast 2012: 486402.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S et al (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459: 569–573.
Goedert M, Klug A, Crowther RA (2006). Tau protein, the paired helical filament and Alzheimer's disease. J Alzheimers Dis 9: 195–207.
Gokce O, Sudhof TC (2013). Membrane-tethered monomeric neurexin LNS-domain triggers synapse formation. J Neurosci 33: 14617–14628 This study demonstrates that the previous assumption that an interaction of CASK with the PDZ motif of Nrxn triggers synaptic recruitment is flawed.
Goldstein AY, Wang X, Schwarz TL (2008). Axonal transport and the delivery of pre-synaptic components. Curr Opin Neurobiol 18: 495–503.
Goold RG, Owen R, Gordon-Weeks PR (1999). Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J Cell Sci 112 (Pt 19): 3373–3384.
Grabrucker AM, Schmeisser MJ, Schoen M, Boeckers TM (2011). Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol 21: 594–603.
Grayton HM, Missler M, Collier DA, Fernandes C (2013). Altered social behaviours in neurexin 1alpha knockout mice resemble core symptoms in neurodevelopmental disorders. PLoS One 8: e67114.
Greenough WT, Klintsova AY, Irwin SA, Galvez R, Bates KE, Weiler IJ (2001). Synaptic regulation of protein synthesis and the fragile X protein. Proc Natl Acad Sci USA 98: 7101–7106.
Groc L, Bard L, Choquet D (2009). Surface trafficking of N-methyl-D-aspartate receptors: physiological and pathological perspectives. Neuroscience 158: 4–18.
Guilmatre A, Huguet G, Delorme R, Bourgeron T (2014). The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol 74: 113–122.
Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315: 1143–1147 This study was the first ever demonstration of a genetic rescue of a NDD.
Hackett A, Tarpey PS, Licata A, Cox J, Whibley A, Boyle J et al (2009). CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur J Hum Genet 18: 544–552.
Hata Y, Butz S, Sudhof TC (1996). CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci 16: 2488–2494.
Hayashi S, Mizuno S, Migita O, Okuyama T, Makita Y, Hata A et al (2008). The CASK gene harbored in a deletion detected by array-CGH as a potential candidate for a gene causative of X-linked dominant mental retardation. Am J Med Genet A 146A: 2145–2151.
Hirai H, Pang Z, Bao D, Miyazaki T, Li L, Miura E et al (2005). Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat Neurosci 8: 1534–1541.
Hirokawa N, Niwa S, Tanaka Y (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68: 610–638.
Hoy JL, Haeger PA, Constable JR, Arias RJ, McCallum R, Kyweriga M et al (2013). Neuroligin1 drives synaptic and behavioral maturation through intracellular interactions. J Neurosci 33: 9364–9384 In this study, the Nlgn1 CNV is mimicked by overexpression in the mouse forebrain and shown to be sufficient to induce changes in the adult, but not necessary for maintenance of the deficit.
Hsueh YP (2006). The role of the MAGUK protein CASK in neural development and synaptic function. Curr Med Chem 13: 1915–1927.
Hsueh YP, Wang TF, Yang FC, Sheng M (2000). Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 404: 298–302.
Huber KM, Gallagher SM, Warren ST, Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99: 7746–7750.
Huttenlocher PR, Dabholkar AS (1997). Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol 387: 167–178.
Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP et al (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet 98: 161–167.
Jacobs S, Doering LC (2010). Astrocytes prevent abnormal neuronal development in the fragile X mouse. J Neurosci 30: 4508–4514.
Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC et al (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34: 27–29 This was the first demonstration that synaptic cell adhesion molecules are associated with ASD.
Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F et al (2008). Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci USA 105: 1710–1715.
Jaramillo TC, Liu S, Pettersen A, Birnbaum SG, Powell CM (2014). Autism-related neuroligin-3 mutation alters social behavior and spatial learning. Autism Res 7: 264–272.
Kaech S, Banker G (2006). Culturing hippocampal neurons. Nat Protoc 1: 2406–2415.
Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001). Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414: 643–648.
Kayadjanian N, Lee HS, Pina-Crespo J, Heinemann SF (2007). Localization of glutamate receptors to distal dendrites depends on subunit composition and the kinesin motor protein KIF17. Mol Cell Neurosci 34: 219–230.
Ko J, Fuccillo MV, Malenka RC, Sudhof TC (2009). LRRTM2 functions as a Neurexin ligand in promoting excitatory synapse formation. Neuron 64: 791–798.
Kohli BM, Pflieger D, Mueller LN, Carbonetti G, Aebersold R, Nitsch RM et al (2012). Interactome of the amyloid precursor protein APP in brain reveals a protein network involved in synaptic vesicle turnover and a close association with Synaptotagmin-1. J Proteome Res 11: 4075–4090.
Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R et al (2006). A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 38: 999–1001.
Kumar A, Swanwick CC, Johnson N, Menashe I, Basu SN, Bales ME et al (2011). A brain region-specific predictive gene map for autism derived by profiling a reference gene set. PLoS One 6: e28431.
Kwon HB, Kozorovitskiy Y, Oh WJ, Peixoto RT, Akhtar N, Saulnier JL et al (2012). Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nat Neurosci 15: 1667–1674.
Laarakker MC, Reinders NR, Bruining H, Ophoff RA, Kas MJ (2012). Sex-dependent novelty response in neurexin-1alpha mutant mice. PLoS One 7: e31503.
Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP et al (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet 74: 552–557.
Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR et al (2005). Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci 25: 2386–2395.
Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G et al (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet 8: e1002521.
Lennertz L, Wagner M, Wolwer W, Schuhmacher A, Frommann I, Berning J et al (2012). A promoter variant of SHANK1 affects auditory working memory in schizophrenia patients and in subjects clinically at risk for psychosis. Eur Arch Psychiatry Clin Neurosci 262: 117–124.
Lesch KP, Timmesfeld N, Renner TJ, Halperin R, Roser C, Nguyen TT et al (2008). Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J Neural Transm 115: 1573–1585.
Linhoff MW, Lauren J, Cassidy RM, Dobie FA, Takahashi H, Nygaard HB et al (2009). An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron 61: 734–749 This study took advantage of the cell adhesion molecule expression and coculture system by screening a cDNA library for molecules that induce presynaptic assembly, thereby identifying LRRTM proteins.
Martin BS, Huntsman MM (2012). Pathological plasticity in fragile X syndrome. Neural Plast 2012: 275630.
Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S et al (2010). Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science 328: 363–368.
Maximov A, Bezprozvanny I (2002). Synaptic targeting of N-type calcium channels in hippocampal neurons. J Neurosci 22: 6939–6952.
Maximov A, Sudhof TC, Bezprozvanny I (1999). Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem 274: 24453–24456.
McAllister AK (2007). Dynamic aspects of CNS synapse formation. Annu Rev Neurosci 30: 425–450.
McLennan Y, Polussa J, Tassone F, Hagerman R (2011). Fragile X syndrome. Curr Genomics 12: 216–224.
Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K et al (2003). Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423: 939–948.
Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J et al (2008). Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet 40: 1065–1067.
Nam CI, Chen L (2005). Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci USA 102: 6137–6142.
Napolioni V, Curatolo P (2008). Genetics and molecular biology of tuberous sclerosis complex. Curr Genomics 9: 475–487.
Nelson CD, Kim MJ, Hsin H, Chen Y, Sheng M (2013). Phosphorylation of threonine-19 of PSD-95 by GSK-3beta is required for PSD-95 mobilization and long-term depression. J Neurosci 33: 12122–12135.
Nguyen MV, Du F, Felice CA, Shan X, Nigam A, Mandel G et al (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J Neurosci 32: 10021–10034.
O’Dushlaine C, Kenny E, Heron E, Donohoe G, Gill M, Morris D et al (2011). Molecular pathways involved in neuronal cell adhesion and membrane scaffolding contribute to schizophrenia and bipolar disorder susceptibility. Mol Psychiatry 16: 286–292.
Park M, Watanabe S, Poon VY, Ou CY, Jorgensen EM, Shen K (2011). CYY-1/cyclin Y and CDK-5 differentially regulate synapse elimination and formation for rewiring neural circuits. Neuron 70: 742–757.
Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN et al (2011a). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472: 437–442.
Peca J, Ting J, Feng G (2011b). SnapShot: autism and the synapse. Cell 147: 706 706 e701.
Petralia RS, Sans N, Wang YX, Wenthold RJ (2005). Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci 29: 436–452.
Pfrieger FW, Barres BA (1997). Synaptic efficacy enhanced by glial cells in vitro. Science 277: 1684–1687.
Radyushkin K, Hammerschmidt K, Boretius S, Varoqueaux F, El-Kordi A, Ronnenberg A et al (2009). Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav 8: 416–425.
Ramos-Miguel A, Meana JJ, Garcia-Sevilla JA (2012). Cyclin-dependent kinase-5 and p35/p25 activators in schizophrenia and major depression prefrontal cortex: basal contents and effects of psychotropic medications. Int J Neuropsychopharmacol 16: 683–689.
Reichelt AC, Rodgers RJ, Clapcote SJ (2012). The role of neurexins in schizophrenia and autistic spectrum disorder. Neuropharmacology 62: 1519–1526.
Robbins EM, Krupp AJ, Perez de Arce K, Ghosh AK, Fogel AI, Boucard A et al (2010). SynCAM 1 adhesion dynamically regulates synapse number and impacts plasticity and learning. Neuron 68: 894–906 This study examined both the consequences of loss of function and overexpression of SynCAM1 on synapses and behavior in transgenic mice. It demonstrated that SynCAM1 overexpression is necessary to maintain an increased number of synapses.
Rovelet-Lecrux A, Hannequin D, Guillin O, Legallic S, Jurici S, Wallon D et al (2012). Frontotemporal dementia phenotype associated with MAPT gene duplication. J Alzheimers Dis 21: 897–902.
Sabo SL, McAllister AK (2003). Mobility and cycling of synaptic protein-containing vesicles in axonal growth cone filopodia. Nat Neurosci 6: 1264–1269.
Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31: 115–130.
Salinas PC (2005). Retrograde signalling at the synapse: a role for Wnt proteins. Biochem Soc Trans 33: 1295–1298.
Samuels BA, Hsueh YP, Shu T, Liang H, Tseng HC, Hong CJ et al (2007). Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron 56: 823–837.
Sapir T, Frotscher M, Levy T, Mandelkow EM, Reiner O (2012). Tau’s role in the developing brain: implications for intellectual disability. Hum Mol Genet 21: 1681–1692.
Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S et al (2012). SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet 90: 879–887.
Scheiffele P, Fan J, Choih J, Fetter R, Serafini T (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101: 657–669 This study was the first to demonstrate the sufficiency of a cell adhesion molecule, Nlgn1, for presynaptic assembly in a coculture system.
Setou M, Nakagawa T, Seog DH, Hirokawa N (2000). Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor- containing vesicle transport. Science 288: 1796–1802.
Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M et al (2002). Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature 417: 83–87.
Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S et al (2006). Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet 38: 1032–1037.
Shipman SL, Schnell E, Hirai T, Chen BS, Roche KW, Nicoll RA (2012). Functional dependence of neuroligin on a new non-PDZ intracellular domain. Nat Neurosci 14: 718–726.
Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM (2010). LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci 30: 7495–7506.
Silverman JL, Turner SM, Barkan CL, Tolu SS, Saxena R, Hung AY et al (2011). Sociability and motor functions in Shank1 mutant mice. Brain Res 1380: 120–137.
Smith DS, Tsai LH (2002). Cdk5 behind the wheel: a role in trafficking and transport? Trends Cell Biol 12: 28–36.
Soler-Llavina GJ, Fuccillo MV, Ko J, Sudhof TC, Malenka RC (2011). The neurexin ligands, neuroligins and leucine-rich repeat transmembrane proteins, perform convergent and divergent synaptic functions in vivo. Proc Natl Acad Sci USA 108: 16502–16509.
Song JY, Ichtchenko K, Sudhof TC, Brose N (1999). Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci USA 96: 1100–1105.
Sons MS, Busche N, Strenzke N, Moser T, Ernsberger U, Mooren FC et al (2006). alpha-Neurexins are required for efficient transmitter release and synaptic homeostasis at the mouse neuromuscular junction. Neuroscience 138: 433–446.
Stagi M, Fogel AI, Biederer T (2010). SynCAM 1 participates in axo-dendritic contact assembly and shapes neuronal growth cones. Proc Natl Acad Sci USA 107: 7568–7573.
Sudhof TC (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455: 903–911.
Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ et al (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 39: 319–328.
Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM et al (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318: 71–76.
Tabuchi K, Sudhof TC (2002). Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics 79: 849–859.
Takashima A, Yamaguchi H, Noguchi K, Michel G, Ishiguro K, Sato K et al (1995). Amyloid beta peptide induces cytoplasmic accumulation of amyloid protein precursor via tau protein kinase I/glycogen synthase kinase-3 beta in rat hippocampal neurons. Neurosci Lett 198: 83–86.
Tarabeux J, Champagne N, Brustein E, Hamdan FF, Gauthier J, Lapointe M et al (2010). De novo truncating mutation in Kinesin 17 associated with schizophrenia. Biol Psychiatry 68: 649–656.
Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F et al (2010). Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet 26: 363–372.
Troca-Marin JA, Alves-Sampaio A, Montesinos ML (2012). Deregulated mTOR-mediated translation in intellectual disability. Prog Neurobiol 96: 268–282.
Tropepe V, Sive HL (2003). Can zebrafish be used as a model to study the neurodevelopmental causes of autism? Genes Brain Behav 2: 268–281.
Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M et al (2010). Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 141: 1068–1079.
Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K et al (2006). Neuroligins determine synapse maturation and function. Neuron 51: 741–754.
Venturin M, Moncini S, Villa V, Russo S, Bonati MT, Larizza L et al (2006). Mutations and novel polymorphisms in coding regions and UTRs of CDK5R1 and OMG genes in patients with non-syndromic mental retardation. Neurogenetics 7: 59–66.
Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC et al (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet 20: 3093–3108.
Washbourne P, Bennett JE, McAllister AK (2002). Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci 5: 751–759.
Washbourne P, Dityatev A, Scheiffele P, Biederer T, Weiner JA, Christopherson KS et al (2004). Cell adhesion molecules in synapse formation. J Neurosci 24: 9244–9249.
Weinberger DR, Levitt P (2011). Neurodevelopmental origins of schizophrenia. In: Schizophrenia, 3rd edn, DR Weinberger, PJ Harrison (eds) Wiley-Blackwell Publishing: Oxford, UK. pp 393–412.
Witt J, Marks WJ Jr. (2011). An update on gene therapy in Parkinson’s disease. Curr Neurol Neurosci Rep 11: 362–370.
Wohr M, Roullet FI, Hung AY, Sheng M, Crawley JN (2011). Communication impairments in mice lacking Shank1: reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS One 6: e20631.
Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J et al (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486: 261–265 This study demonstrates the necessity of autism-associated gene Shank2 for social behavior, and shows pharmacological rescue of the phenotype using NMDA receptor partial agonist D-cycloserine.
Wong RW, Setou M, Teng J, Takei Y, Hirokawa N (2002). Overexpression of motor protein KIF17 enhances spatial and working memory in transgenic mice. Proc Natl Acad Sci USA 99: 14500–14505 This study established microtubule-based transport of a synaptic component, namely the NMDA receptor subunit NR2B, as playing a key role in defining synaptic function and behavior.
Wright GJ, Washbourne P (2011). Neurexins, neuroligins and LRRTMs: synaptic adhesion getting fishy. J Neurochem 117: 765–778.
Yin X, Feng X, Takei Y, Hirokawa N (2012). Regulation of NMDA receptor transport: a KIF17-cargo binding/releasing underlies synaptic plasticity and memory in vivo. J Neurosci 32: 5486–5499.
Zhang C, Atasoy D, Arac D, Yang X, Fucillo MV, Robison AJ et al (2010). Neurexins physically and functionally interact with GABA(A) receptors. Neuron 66: 403–416.
Zhiling Y, Fujita E, Tanabe Y, Yamagata T, Momoi T, Momoi MY (2008). Mutations in the gene encoding CADM1 are associated with autism spectrum disorder. Biochem Biophys Res Commun 377: 926–929.
Zoghbi HY (2003). Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302: 826–830.
Acknowledgements
The research in the author’s laboratory is supported by a grant from the National Institute of Neurological Disorders and Stroke (R01 NS065795).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Washbourne, P. Synapse Assembly and Neurodevelopmental Disorders. Neuropsychopharmacol 40, 4–15 (2015). https://doi.org/10.1038/npp.2014.163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.163
This article is cited by
-
Celf4 controls mRNA translation underlying synaptic development in the prenatal mammalian neocortex
Nature Communications (2023)
-
TAD evolutionary and functional characterization reveals diversity in mammalian TAD boundary properties and function
Nature Communications (2023)
-
Next-generation gene panel testing in adolescents and adults in a medical neuropsychiatric genetics clinic
neurogenetics (2021)
-
Live Neuron High-Content Screening Reveals Synaptotoxic Activity in Alzheimer Mouse Model Homogenates
Scientific Reports (2020)
-
Spaced training improves learning in Ts65Dn and Ube3a mouse models of intellectual disabilities
Translational Psychiatry (2019)
