
Abstract
Our understanding of the active role that astrocytes play in modulating neuronal function and behavior is rapidly expanding, but little is known about the role that astrocytes may play in drug-seeking behavior for commonly abused substances. Given that the nucleus accumbens is critically involved in substance abuse and motivation, we sought to determine whether nucleus accumbens astrocytes influence the motivation to self-administer ethanol following abstinence. We found that the packing density of astrocytes that were expressing glial fibrillary acidic protein increased in the nucleus accumbens core (NAcore) during abstinence from EtOH self-administration. No change was observed in the nucleus accumbens shell. This increased NAcore astrocyte density positively correlated with the motivation for ethanol. Astrocytes can communicate with one another and influence neuronal activity through gap-junction hemichannels. Because of this, the effect of blocking gap-junction hemichannels on the motivation for ethanol was examined. The motivation to self-administer ethanol after 3 weeks abstinence was increased following microinjection of gap-junction hemichannel blockers into the NAcore at doses that block both neuronal and astrocytic channels. In contrast, no effect was observed following microinjection of doses that are not thought to block astrocytic channels or following microinjection of either dose into the nucleus accumbens shell. Additionally, the motivation for sucrose after 3 weeks abstinence was unaffected by NAcore gap-junction hemichannel blockers. Next, Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) were selectively expressed in NAcore astrocytes to test the effect of astrocyte stimulation. DREADD activation increased cytosolic calcium in primary astrocytes, facilitated responding for rewarding brain stimulation, and reduced the motivation for ethanol after 3 weeks abstinence. This is the first work to modulate drug-seeking behavior with astrocyte-specific DREADDs. Taken together, our findings demonstrate that NAcore astrocytes can shape the motivation to self-administer ethanol; suggesting that the development of ligands which selectively stimulate astrocytes may be a successful strategy to abate ethanol-seeking behavior.
Similar content being viewed by others
INTRODUCTION
Addiction is associated with adaptive changes within discrete brain structures. Similar to neurons, astrocytes display temporally and regionally specific adaptations to abused substances. Astrocytic adaptations are commonly measured via immunoreactivity of the structural filament glial fibrillary acidic protein (GFAP) (Sofroniew and Vinters, 2010). For example, total GFAP expression and the number of cells expressing GFAP (GFAP+) increased in the nucleus accumbens core (NAcore) after repeated cocaine injections, but no change was observed in the dorsal striatum (Bowers and Kalivas, 2003). Additionally, rat strains that had self-administered more morphine expressed more GFAP in the ventral tegmental area (Beitner-Johnson et al, 1993). In the prefrontal cortex, prelimbic GFAP+ astrocyte density was lower in rats that exhibited higher ethanol (EtOH) preference (Miguel-Hidalgo, 2005) while dorsolateral astrocyte density was lower in EtOH-dependent humans (Miguel-Hidalgo et al, 2002). These studies illustrate that astrocytes respond to commonly abused substances in a region-specific manner.
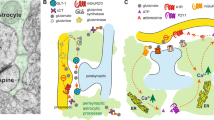
Astrocytes are the predominant mammalian brain cell type, but because they do not fire action potentials, the capacity of astrocytes to modulate complex behaviors was only recently appreciated. It is now known that astrocytes can release transmitter through exocytosis (Parpura et al, 1994), transporters (Szatkowski et al, 1990), gap-junction hemichannels (Ye et al, 2003), and ion channels following stimulation of G-protein-coupled receptors (Woo et al, 2012). Notably, manipulations of astrocytic transporters, which increased extracellular transmitter, reduced responding for rewarding brain stimulation (John et al, 2012) and blocked the reinstatement of cocaine-seeking behavior (Baker et al, 2003). Moreover, the reinstatement of cocaine-seeking behavior did not occur in transgenic mice expressing a dominant-negative SNARE domain in astrocytes (Turner et al, 2012). These mice exhibit deficient gliotransmission as well as disruptions in the expression and/or function of cell-surface proteins, including ion channels, transporters, and receptors. Together, these studies suggest that specific routes of astrocytic transmitter flux can differentially influence the seeking of drug and rewarding brain stimulation.
We sought to determine whether nucleus accumbens astrocytes can influence the motivation to self-administer EtOH after abstinence. The nucleus accumbens is comprised of at least two anatomically and functionally distinct subregions: the NAcore and the shell (NAshell; Zahm, 2000). The NAcore is implicated in the control of behavior by discrete cues, while the NAshell is implicated in the control of behavior by spatial/contextual information as well as primary reinforcement (Chaudhri et al, 2010; Pierce and Kumaresan, 2006). This study focused on the NAcore because activation of the human NAcore by drug-related stimuli is predictive of relapse risk (Heinz et al, 2009). Several studies have examined the ability of different stimuli to provoke drug-seeking behavior using the extinction-reinstatement paradigm. Alternatively, abstinence is imposed as humans do not typically undergo explicit extinction of drug-related stimuli and behaviors (Epstein et al, 2006). Our hypothesis was that NAcore astrocytes modulate the motivation to self-administer EtOH after abstinence. To test this hypothesis, we employed a loss- and gain-of-function approach and quantified motivation via breakpoint obtained on an EtOH-maintained progressive ratio reinforcement schedule (Richardson and Roberts, 1996). For loss-of-function studies, gap-junction hemichannel blockers were microinjected into the NAcore. The abundance of gap-junction hemichannels in astrocytes enable both extracellular (Hassinger et al, 1996) and intracellular (Dermietzel and Spray, 1993) communication through large networks of electrically coupled astrocytes. However, gap-junctions are found in many cell types, and their ligands are pharmacologically promiscuous. Therefore, we also performed gain-of-function studies using Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) that were selectively expressed in NAcore astrocytes. The impact of astrocytic DREADD activation on behavior was further probed with intracranial self-stimulation (ICSS).
MATERIALS AND METHODS
Subjects
Procedures were IACUC approved and compliant with the NIH Guide for the Care and Use of Laboratory Animals. Male rats (Harlan, Dublin VA: 225–250 g Han Wistar for EtOH; 250–290 g Sprague Dawley for ICSS) were individually housed (lights on 06:00 am) with ad libitum home cage food and water, unless indicated. Habituation and surgical recovery were 1 week each.
Reagents
In vivo: EtOH (20%, v/v, from 95%) and/or sucrose (2%, w/v) were diluted in water. 18-α-glycyrrhetinic acid (18α: 100 μM) and mefloquine (25 or 750 μM), both obtained from Sigma (St Louis, MO), were dissolved in 0.05% DMSO/PBS. Clozapine-N-oxide (CNO, obtained from NIDDK) was dissolved in 0.5% DMSO/saline. Cocaine (obtained from NIDA) was dissolved in saline. In vitro: AAV5 was diluted in serum-free media (SFM, 4 × 109 particles/ml), rhod-2 AM (Invitrogen, Grand Island, NY) was dissolved in 0.05% DMSO/pluronic in SFM. CNO and (S)-DHPG (Tocris, Bristol, UK) were dissolved in saline.
Vector
rAAV5/GFAP-HA-hm3D-IRES-mCitrine: HA-hM3D (Gαq) was inserted upstream of IRES-mCitrine.pcDNA3 and subcloned into pAAV5 containing the minimal human GFAP promoter with SalI and EcoRI. Purified virus was obtained pre-dialyzed (350 mM NaCl, 5% D-sorbitol in PBS, UNC Gene Therapy Center, Chapel Hill, NC) and microinjected at 4 × 1012 particles/ml, 1 μl/side over 10 min, and 7 min allowed for diffusion. Testing began 2 weeks later. The volume of transfected cells was determined with serial sections using the method of Cavalieri (Gundersen and Jensen, 1987; Stereologer, SRC, Tampa, FL).
Ca++ Measures
Primary astrocytes were prepared as previously described (Zou et al, 2011) and are detailed in Supplementary Methods. Cells were transfected (100 μl AAV5: Gαq-DREADD or GFP) and cytosolic calcium ([Ca++]i) measured 3–4 days later. On test day, media was changed to SFM (1 h, 37 °C, 5% CO2) and cells loaded with rhod-2 (3 μM, 15 min, 30 °C, 5% CO2), as previously described (Sun et al, 2013). After washing (SFM, 15 min), recordings were obtained (AxioObserver Z.1, AxioVision 4.6, Zeiss, Oberkochen, Germany). Specifically, rhod-2 was excited (180 ms) and emission sampled every second for a 30 s baseline and then every 10 s thereafter (20x PlanApo, CCD) for 12 min to avoid photobleaching. The relative cytosolic fluorescence of 24 individual cells was normalized to baseline and experiments performed in duplicate. Sequential treatments were CNO (1 nM, 12 min), SFM wash (30 min), re-baseline, and (S)-DHPG (2 μM, 12 min). Doses were based upon earlier reports (Ferguson et al, 2011; Toms and Roberts, 1999) and preliminary data (not shown).
Surgery
Stainless steel cannulae (26 gauge, AmazonSupply) and obturators (33 gauge, AmazonSupply) or a polyamide-insulated cathode (30 gauge, PlasticsOne, Roanoke, VA) were implanted under isoflurane using the following coordinates from bregma. NAcore: +1.2 AP, ±1.94 ML, −6.82 DV, 6 °θ; NAshell: +2.2 AP, ±2.35 ML, −6.25 DV, 8 °θ; medial forebrain bundle: −2.8 AP, +1.7 ML, and −8.8 DV from skull surface. The cannulae were angled to avoid the lateral ventricles. The anode was grounded to a skull screw.
Intermittent EtOH or Sucrose Access
Procedures were previously described (Hopf et al, 2010) and are detailed in Supplementary Methods. Subject handling continued during the 3 weeks abstinence period.
Operant EtOH Self-Administration
As per procedures adapted from prior reports (Bowers et al, 2008), rats were water deprived during shaping that began with two, 15 h, fixed ratio one sessions (FR1) for 10% sucrose (w/v) in 20% EtOH (v/v). After these sessions, 1–2 h home cage water was allowed. Next, rats were trained to respond on a FR3 for one to three, 1 h sessions per stage: (1) 10% sucrose/20% EtOH, 3–4 h home cage water; (2) 5% sucrose/20% EtOH, 5–6 h water; (3) 3% sucrose/20% EtOH, ad libitum home cage water; (4) 1.5% sucrose/20% EtOH, ad libitum water; (5) 8 weeks of contiguous daily sessions for 0% sucrose/20% EtOH and ad libitum home cage water. Subject handling continued during the 3 weeks abstinence period.
Breakpoint
Procedures for the determination of the cue-provoked and EtOH-maintained breakpoint were previously described (Bowers et al, 2008) and are detailed in Supplementary Methods. Breakpoint was defined as total active-lever depressions in the last completed ratio. Thus, lever depressions that did not yield reinforcer delivery were not included in breakpoint calculations. Sessions timed out after 15 min of inactivity, which generally occurred within 1 h. All drugs were administered in a counterbalanced fashion. Mefloquine (25–750 μM), 18-α (100 μM), or vehicle were microinjected (0.5 μl/side at 0.25 μl/min) with an additional 2 min allowed for diffusion. CNO (3 mg/kg) or vehicle was administered (1 ml/kg, i.p.), and experiments occurred 30 min later.
ICSS
Rats were trained to respond for ICSS as previously described (Bauer et al, 2013) and are detailed in Supplementary Methods. Test sessions occurred 30 min after CNO (1.5–6 mg/kg, i.p.) or immediately after cocaine (10 mg/kg, i.p.).
Histology
Tissue processing was adapted from prior reports (Long et al, 1998) and are detailed in Supplementary Methods. All groups were processed in parallel. Every 6th section was collected via systematic-uniform random sampling and incubated with anti-GFAP (1:15K, Dako, Carpinteria, CA; 4 °C, 15 h) prior to visualization (HRP and DAB, Vector Labs, Burlingame, CA) and then Nissl counterstained. GFAP+ cells were counted (blinded) using unbiased stereological principles (Long et al, 1998; Stereologer, Zeiss PlanApo 63x) and density obtained by dividing cell counts by the measured brain volume. Immunofluorescent sections were incubated (4 °C, 15 h) with Cy3-GFAP monoclonal (1:800, Sigma) or β-tubulin-III (1:600, Abcam and then 1:1000 AF594, Invitrogen) and Hoescht-33342 counterstained (1:10K, 8 min).
Statistical Analysis
Run rate was defined as active-lever depressions-1/interval duration. Stereology and breakpoint (defined above) were analyzed by either a one- or two-way ANOVA, and time course was analyzed by a repeated measures ANOVA. Treatment and/or virus were factors. The primary ICSS-dependent variable was reinforcement rate (stimulations/min) normalized as percent Maximum Control Rate that was analyzed by a two-way ANOVA with frequency and post-virus day or treatment as factors. Significant main effects were followed by a Bonferroni correction, Scheffé, Dunnet’s, or Holm–Sidak post-hoc, as appropriate. Significance was considered as p<0.05.
RESULTS
NAcore GFAP+ Astrocyte Density Increased after 3 Weeks Abstinence from Ethanol
There is considerable interest in identifying adaptations that develop during drug abstinence and which may drive relapse. Here, astrocyte populations in the NAcore and NAshell were examined after abstinence from EtOH self-administration. These brain regions are critically involved in EtOH relapse, and within these regions GFAP is restricted to astrocytes.
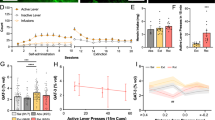
As individual animal models cannot recapitulate the entirety of alcohol use disorders (AUDs), astrocyte packing density was measured following abstinence from two distinct self-administration paradigms (Figure 1). Intermittent EtOH access (EtOH-IA) models some cardinal features of AUDs; eg, escalating consumption and preference (Hopf et al, 2010). Operant EtOH self-administration models the positive reinforcing and motivating effects of EtOH. Figure 1a illustrates experimental timelines of these models. We found that NAcore GFAP+ astrocyte density was sensitive to abstinence time and treatment (Figure 1b, Supplementary Figure S1A). More specifically, astrocyte density increased 3 weeks after either EtOH-IA or operant EtOH, but was unchanged 24 h after EtOH-IA. In contrast, NAcore astrocyte density decreased 3 weeks after intermittent sucrose (S-IA). Sucrose self-administration served as a control for EtOH. Because altered NAcore astrocyte density could reflect cell number or GFAP synthesis, GFAP protein expression was measured in crude NAcore homogenate 3 weeks after operant EtOH self-administration. No change was observed in GFAP expression (Supplementary Figure S1B), suggesting that astrocyte packing density increased.

NAcore GFAP+ astrocyte density increased after 3 weeks abstinence from EtOH. (a) Experimental timeline. Five treatment groups were analyzed: (i) age- and handling-matched naive, (ii) after 24 h abstinence from EtOH-IA, (iii) after 3 weeks abstinence from EtOH-IA, (iv) after 3 weeks abstinence from operant EtOH self-administration, and (v) after 3 weeks abstinence from S-IA. (b) A significant one-way ANOVA (F(4,25)=13.74, p=0.0001) followed by a Dunnett’s post-hoc indicated that NAcore astrocyte density increased after 3 weeks abstinence from either EtOH-IA or operant EtOH. Additionally, a significant decrease was observed after 3 weeks abstinence from S-IA. No change was observed in (c) non-GFAP+ cell density (p=0.17) or (d) region volume (p=0.92). (e) Following 3-week abstinence, the EtOH breakpoint positively correlated with NAcore astrocyte density for both the EtOH-IA (R2=0.87, p=0.01) and operant EtOH cohorts (R2=0.79, p=0.04). In contrast, no correlation was observed in the NAshell for either the EtOH-IA (p=0.89) or operant (p=0.10) cohorts. M, Monday, etc.; abst, 3 weeks abstinence; oper, operant. Data represent mean±SEM, *p<0.05.
In contrast to the NAcore, NAshell astrocyte density increased after either 24 h or 3 weeks abstinence from EtOH-IA (Supplementary Figure S1C). However, NAshell astrocyte density was unchanged 3 weeks after either operant EtOH or S-IA (Supplementary Figure S1C). Moreover, no changes were noted in non-GFAP+ cell density or region volume for the NAcore (Figure 1c and d) or NAshell (Supplementary Figure S1D and E).
Although GFAP is commonly used as an astrocytic marker following pharmacological insult or injury (Eng et al, 2000), GFAP antiserum does not label all astrocytes. To better understand the response of NAcore astrocytes during EtOH abstinence, we also measured aldehyde dehydrogenase 1L1 (ALDH1L1) and glutamine synthetase. ALDH1L1 appears to be expressed in all adult astrocytes, but not other cell types (Cahoy et al, 2008). Glutamine synthetase can protect neurons from excitotoxicity by taking up and converting excess ammonia and glutamate into glutamine. Glutamine synthetase antiserum labels all cells in the astrocyte family including astrocytes, Bergmann glia, Müller cells, tanycytes, and ependymal cells (Anlauf and Derouiche, 2013). Of these, only the astrocytes are found in the region analyzed. Neither the number of cells expressing ALDH1L1 (Supplementary Figure S1G) nor ALDH1L1 protein expression (Supplementary Figure S1H) changed in the NAcore following EtOH self-administration. This is in agreement with an immunoblotting study of schizophrenic brain samples, which found increased GFAP expression, but no change in ALDH1L1 expression compared to controls (Feresten et al, 2013). In contrast, Supplementary Figure S1I indicates that glutamine synthetase expression increased in the NAcore after 3 weeks abstinence from EtOH. These data support earlier work showing increased packing density of glutamine synthetase-positive astrocytes during abstinence from ethanol self-administration (Miguel-Hidalgo, 2006).
The Ethanol Breakpoint Correlated with NAcore GFAP+ Astrocyte Density
The NAcore is implicated in the motivational and cue-reactivity aspects of AUDs (Heinz et al, 2009). Therefore, we hypothesized that NAcore astrocytes contribute to the motivation to self-administer EtOH after 3 weeks abstinence. To begin evaluating this hypothesis, ethanol self-administration by formerly abstinent rats was provoked by a compound cue that was previously associated with EtOH availability (Bowers et al, 2008). The cue mimics EtOH-associated olfactory, auditory, and visual stimuli that contribute to the ‘first drink’ phenomenon. Similar stimuli can precipitate relapse in alcoholics (Ludwig et al, 1974). Figure 1e shows that the EtOH breakpoint positively correlated with NAcore astrocyte density. Because breakpoint is used as a proxy for motivation (Richardson and Roberts, 1996), we subsequently examined if NAcore astrocytes influence the motivation for EtOH.
Microinjection of Gap-Junction Hemichannel Blockers into the NAcore Increased the Motivation to Self-Administer Ethanol after 3 Weeks Abstinence
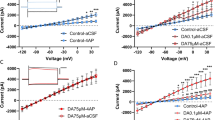
Figure 2a illustrates the experimental timeline. Astrocytes express a large number of gap-junction hemichannels, which are used for both extracellular (Hassinger et al, 1996) and intracellular (Dermietzel and Spray, 1993) communication. As the motivation to self-administer EtOH positively correlated with GFAP+ astrocyte number in the NAcore, we tested the hypothesis that NAcore astrocytes can influence EtOH self-administration by interfering with the capacity of these cells to communicate through gap-junction hemichannels. Given that many cell types express structurally similar gap-junction hemichannels, precise pharmacological manipulation is not currently possible. Accordingly, two gap-junction hemichannel blockers were examined in separate cohorts: mefloquine or 18-α. Mefloquine blocks neuronal gap-junction hemichannels at low doses, while 10- to 100-fold higher doses also target astrocytes (Cruikshank et al, 2004). NAcore neuronal gap-junction hemichannel blockade alone did not influence the EtOH breakpoint (Figure 2b). However, also targeting astrocytes with a 30-fold higher mefloquine dose increased the EtOH breakpoint. Additionally, microinjection of the relatively selective gap-junction hemichannel blocker 18-α (Davidson and Baumgarten, 1988) into the NAcore similarly increased the EtOH breakpoint (Figure 2b). Although 18-α is not cell-specific, it is intriguing that the EtOH breakpoint was increased to the same extent by both the high mefloquine dose and 18-α given that EtOH preference was similarly increased by microinjection of either an astrocyte-specific toxin or the same dose of 18-α (Miguel-Hidalgo et al, 2008) that was used here. Importantly, while increased active-lever responding was apparent throughout testing (Figure 2c), no differences were observed in response rates over the first several reinforcers earned (Figure 2d), inactive-lever responding (Supplementary Figure S2A), or latency to the first active-lever press (Supplementary Figure S2B). These data suggest that gap-junction hemichannel blockers did not produce obvious off-target motor effects during EtOH self-administration. Supplementary Table S1 summarizes blood EtOH levels after the test session.

NAcore gap-junction hemichannel blocker microinjection selectively increased the EtOH breakpoint. (a) Experimental timeline. (b) Microinjection of gap-junction hemichannel blockers into the NAcore increased the EtOH breakpoint (F(3,40)=5.56, p=0.003). A Scheffé post-hoc revealed that breakpoint was only affected by 18-α or mefloquine doses that target both neurons and astrocytes. Mefloquine doses that are thought to only target neurons were without effect. No effect of treatment order was found (p=0.61). (c) Cumulative active-lever responding during the test session. (d) A one-way repeated measures ANOVA did not indicate differential response rates across initial reinforcer deliveries (p=0.98). Microinjection of mefloquine into the (e) NAshell did not alter the EtOH breakpoint (p=0.75) or the (f) sucrose breakpoint when microinjected into the NAcore (p=0.86). Data represent mean±SEM, *p<0.05.
NAshell microinjection of either mefloquine dose did not affect the EtOH breakpoint (Figure 2e). Additionally, microinjection of the higher dose into the NAcore did not influence the breakpoint of sucrose-abstinent rats for sucrose (Figure 2f). Supplementary Figure S2C–E illustrates microinjection sites. These data partially support our hypothesis that NAcore astrocytes influence the motivation to self-administer EtOH after abstinence. To further test this hypothesis, we examined if NAcore astrocyte stimulation impacts responding for ‘rewarding’ brain stimulation.
Stimulating NAcore Astrocytes Increased ICSS Responding
Astrocytes predominantly respond to transmitter through G-protein-coupled receptors, which increase [Ca++]i (Porter and McCarthy, 1996). The most studied of these receptors are Gαq-coupled. Given that astrocytes cannot be pharmacologically isolated, we sought to determine whether Gαq-DREADDs could be used to stimulate astrocytes. DREADDs exhibit no basal activity, but are activated by the selective agonist CNO (Armbruster et al, 2007). Astrocyte-specific DREADDs were first tested in culture. Figure 3a illustrates the experimental timeline where a Ca++-sensitive dye was loaded into primary astrocytes that were expressing Gαq-DREADDs under the GFAP promoter (Figure 3b). Bath application of CNO increased [Ca++]i in primary astrocytes expressing Gαq-DREADD (Figure 3c), but not if GFP was expressed instead (Figure 3c). (S)-DHPG, a positive control and agonist of Gαq-coupled receptors, increased [Ca++]i in both cultures (Figure 3d). These in vitro data illustrate that activation of Gαq-DREADDs can mobilize primary astrocyte [Ca++]i to a similar extent as activation of endogenous Gαq-coupled receptors with (S)-DHPG. These [Ca++]i elevations are similar to what others have observed in primary astrocytes following (S)-DHPG application (Toms and Roberts, 1999). Finally, a full characterization of these astrocytic Gαq-DREADDs was recently published (Agulhon et al, 2013). Among these measures were [Ca++]i elevations that lasted tens of seconds following brief application of CNO to a slice preparation.

Activation of astrocytic Gαq-DREADDs increased [Ca++]i. (a) Experimental timeline. (b) Astrocytes expressing Gαq-DREADD and mCitrine or GFP were loaded with the Ca++-sensitive dye rhod-2. (c) After bath applying CNO, a one-way repeated measures ANOVA indicated that [Ca++]i increased in astrocytes that were expressing Gαq-DREADD (vector: F(1,46)=331.45, p=0.001; time: F(69,3174)=170.97, p<0.0001; interaction: F(69,3174)=150.02, p<0.0001). No change was observed in astrocytes expressing GFP. (d) Bath application of (S)-DHPG, a specific agonist of endogenous Gαq-coupled receptors, increased [Ca++]i regardless of whether Gαq-DREADD or GFP was expressed. Following (S)-DHPG application, a one-way repeated measures ANOVA revealed a small, but significant difference between transgenes (transgene: F(1,46)=6.67, p=0.01; time: F(69,3174)=284.47, p=0.001; interaction F(69,3174)=6.01, p=0.0001). Arrow, onset of drug treatment; treatment bar, duration of drug treatment. Data represent the mean of 24 cells normalized to baseline ±SEM.
Next, we tested the hypothesis that stimulation of astrocytic Gαq-DREADDs would alter responding for ICSS. ICSS quantitatively estimates the impact of reward-altering manipulations (Bauer et al, 2013). Figure 4a illustrates the experimental timeline. Gαq-DREADDs were selectively expressed in NAcore astrocytes extending ∼1 mm3 around the injection site (Figure 4b and c, Supplementary Figure S3A), which is less than half the estimated adult rat NAcore volume (Wissman et al, 2012). Importantly, manipulating any number of astrocytes can be consequential as an astrocyte can associate with up to 105 synapses and hundreds of dendrites (Bushong et al, 2002). A lack of chromatolysis (Supplementary Figure S3B) suggested that Gαq-DREADD expression did not cause overt toxicity.

Responding for ICSS was facilitated by activation of astrocytic Gαq-DREADDs that were expressed in the NAcore. (a) Experimental timeline. (b) Low-magnification confocal images showing that Gαq-DREADD was expressed in NAcore astrocytes. Note that similar expression was observed in Gαq-DREADD transgenic mice (Agulhon et al, 2013), and that GFAP antiserum detects a more limited number of astrocyte processes. White arrow, transfected astrocyte; gray arrow, non-transfected astrocyte. An example of the volume of transfected cells is shown in Supplementary Figure S3. (c) High-magnification confocal images chosen to show transfected astrocyte complexity and vector specificity. (d) A two-way ANOVA revealed that ICSS was unaffected by vehicle (p=0.17) or post-transfection time (p=0.29). (e) A moderate, but significant ICSS facilitation was observed following 3 mg/kg CNO (frequency, F(9,45)=50.07, P<0.0001; treatment, F(1,5)=13.55, p=0.01). % MCR, percent Maximum Control Rate. Data represent mean±SEM. Scale bar=50 μm.
While ICSS baseline was not affected by either Gαq-DREADD expression or vehicle (Figure 4d), CNO significantly facilitated ICSS (Figure 4e) at a dose previously used to activate neuronal DREADDs (Ferguson et al, 2011). CNO did not alter ICSS if Gαq-DREADD was not expressed or other CNO doses were used (Supplementary Figure S3C–E). As ICSS facilitation occurred within a timeframe (1.5 h) where CNO back-conversion to clozapine has not been shown (Guettier et al, 2009), CNO and astrocyte Gαq-DREADD expression appear to be required. Importantly, motoric capacity appeared unaltered because responding was similar across treatments at both the lowest and highest frequencies tested. Finally, ICSS was strongly facilitated by the positive control cocaine regardless of Gαq-DREADD expression (Supplementary Figure S3F). These results suggest that NAcore astrocytes can modulate sensitivity to rewarding stimulation. Accordingly, we examined if stimulation of astrocytic Gαq-DREADDs that were expressed in the NAcore shapes the motivation to self-administer EtOH after 3 weeks abstinence.
Stimulating NAcore Astrocytes Decreased the Motivation to Self-Administer Ethanol after 3 Weeks Abstinence
Figure 5a illustrates the experimental timeline. The same CNO dose found to facilitate ICSS (Figure 4e) was also found to decrease the EtOH breakpoint in rats that had Gαq-DREADDs expressed in NAcore astrocytes (Figure 5b). CNO had no effect if GFP was expressed instead (Figure 5b). Although Gαq-DREADD-expressing rats exhibited muted responding after CNO administration (Figure 5c), motor depressant effects were not apparent as response rates over the first several reinforcers earned (Figure 5d), inactive-lever responding (Figure 5e), and latency to the first active-lever depression (Supplementary Figure S4A) were similar. Supplementary Figure S4B and C illustrates cannulae placements.

The motivation to self-administer ethanol after 3 weeks abstinence was decreased by activation of astrocytic Gαq-DREADDs that were expressed in the NAcore. (a) Experimental timeline. B) A one-way ANOVA (F(2,30)=3.29, p=0.04) followed by a Scheffé post-hoc indicated that 3 mg/kg CNO decreased the EtOH breakpoint in rats expressing Gαq-DREADD compared to vehicle-treated rats expressing Gαq-DREADD or CNO-treated rats that were expressing GFP. (c) Cumulative active-lever responding during the test session. (d) A one-way repeated measures ANOVA revealed no effect on response rates across initial reinforcer deliveries (p=0.39). (e) Inactive-lever responding was not different (p=0.21). NS, nonsignificant. Data represent mean±SEM, *p<0.05.
DISCUSSION
We found that NAcore astrocytes can shape behavioral responding for rewarding stimuli as well as the motivation to self-administer EtOH after 3 weeks abstinence. In brief, GFAP+ astrocyte density increased in the NAcore after 3 weeks abstinence from EtOH, which positively correlated with breakpoint. In a separate cohort, the EtOH breakpoint increased following microinjection of gap-junction hemichannel blockers into the NAcore, but not NAshell. In contrast, NAcore astrocyte-specific Gαq-DREADD activation facilitated ICSS and decreased the EtOH breakpoint. Taken together, this work demonstrated the importance of astrocyte biology in modulating the behavioral responses to reinforcing stimuli and it is the first to use astrocyte-specific designer receptors to reduce ethanol self-administration.
Behavioral Relevance of Astrocyte Density
This study revealed that NAcore astrocyte density responded uniquely during abstinence from sucrose compared to EtOH. While not proof of causation, the positive correlation of NAcore astrocyte density with the EtOH breakpoint supported our hypothesis that NAcore astrocytes shape the motivation to self-administer EtOH after abstinence for at least three reasons. First, the NAcore is critically involved in circuitry guiding motivated behavior. Second, breakpoint is used to quantify motivation and reinforcing efficacy (Richardson and Roberts, 1996). Third, behaviorally relevant stimuli can impact astrocyte morphology that can, in turn, influence synaptic efficacy and neuronal responses to salient stimuli (Laming et al, 2000). Though the potential impact of EtOH-seeking behavior during testing cannot be dissociated from astrocyte density, a significant correlation was observed in both the operant and EtOH-IA cohorts that differed markedly in operant responding.
Given the increased density of GFAP+ astrocytes in the NAcore following EtOH abstinence, it was somewhat surprising that neither ALDH1L1+ cell counts nor ALDH1L1 protein expression changed. These data suggest that increased density only occurred in a subset of the NAcore astrocyte population, which is consistent with earlier reports showing that only a subset of ALDH1L1+ cells co-express GFAP (Cahoy et al, 2008). Additionally, a similar finding of altered GFAP+ astrocyte density, yet unchanged GFAP expression, was previously observed in EtOH-dependent human brain (Miguel-Hidalgo et al, 2002). While the specific ramifications of increased GFAP+ astrocyte density concerning motivated behavior remain unknown, it has been shown that increased astrocyte density can mitigate the impact of neurotoxic peroxide production that occurs during cytochrome P450-mediated metabolism of EtOH (Desagher et al, 1996). Additionally, it is important to note that our experiments expressed Gαq-DREADDs under the GFAP promoter.
Astrocyte Communication Impacts Neural Physiology and Behavior
Exactly how NAcore astrocytes contribute to the motivation to self-administer EtOH is not presently clear. However, a re-balancing of neuron-astrocyte bidirectional communication is likely to be an important aspect of this effect. This form of communication has been extensively reviewed (eg, Santello et al, 2012). In brief, the foundation of bidirectional communication is that astrocytes both respond to (Porter and McCarthy, 1996) and release (Parpura et al, 1994) transmitter, which ultimately impacts synaptic plasticity and behavior (Parpura et al, 2012). Additionally, bidirectional communication was also recently observed in human brain preparations (Navarrete et al, 2013). Increased [Ca++]i is both necessary and sufficient for most forms of astrocytic transmitter release (Parpura et al, 1994). EtOH can also increase astrocytic [Ca++]i and transmitter release (Allen et al, 2002). Thus, it is intriguing that Gαq-DREADD activation increased astrocytic [Ca++]i both ex vivo (Agulhon et al, 2013) and in vitro (results here), and also reduced the motivation for EtOH as shown by our data. While the precise mechanisms that lie downstream of Gαq-DREADD activation and that may underlie reduced breakpoint remain unknown, it has been shown that increased astrocytic [Ca++]i levels and subsequent glutamate release occur after activation of endogenous Gαq-coupled receptors (Woo et al, 2012). Further, the impact of astrocytic [Ca++]i on transmitter release, and subsequent neuronal responses have been reviewed (Santello et al, 2012). Nonetheless, future studies are needed to identify if these mechanisms and transmitter system are directly involved in the effects reported here.
Others have also shown that astrocytes can shape behavior. For example, cerebellar astrocyte optogenetic stimulation activated purkinje neuron glutamate receptors, induced synaptic plasticity, and modulated cerebellar-regulated behaviors (Sasaki et al, 2012). Additionally, methamphetamine conditioned-stimulus effects were increased by nucleus accumbens microinjection of astrocyte-conditioned medium (Narita et al, 2006). Given that astrocyte-conditioned medium can induce neural stem cell differentiation into astrocytes (Narita et al, 2006), our data correlating increased NAcore astrocyte density with increased motivation for EtOH breakpoint are especially intriguing.
Gap-Junction Hemichannel Pharmacology
A limitation of the present study is that gap-junction hemichannel blockers exhibit affinity for other structurally related channels, including the pannexin-1 containing pannexons and the P2X7 receptor pore as well as the connexin-containing gap channels and volume-regulated anion channels (VRACs; Ye et al, 2009). Thus, gap-junction hemichannel blockers are not astrocyte-specific. However, the channels exhibit unique biophysical properties that can influence pharmacological sensitivity under certain conditions. For example, gap-junction channels couple neighboring cells, but the other hemichannels can open to the extracellular space. Of those opening to the extracellular space, the connexin-containing hemichannels are usually closed under physiological conditions, as even brief opening can be cytotoxic (Halassa et al, 2007). Further, hemichannels are closed by EtOH (Adermark and Lovinger, 2006) or increased [Ca++]i (Scemes et al, 2007). In contrast, the pannexin-containing pannexons are open under physiological conditions (Dahl and Harris, 2008) including increased [Ca++]i (Scemes et al, 2007), and can exhibit approximately 5- to 10-fold more sensitivity to glycyrrhetinic acidic-related synthetic derivatives; eg, 18-α, than connexin-based channels (Bruzzone et al, 2005). EtOH can open both the pannexin-containing channels (Locovei et al, 2006) and VRAC (Allen et al, 2002). Thus, it is likely that the increased EtOH breakpoint may have emanated from pannexin and/or VRAC blockade. Future experiments can partially evaluate this hypothesis by using the VRAC-selective blocker DCPIB.
Despite this promiscuity, low mefloquine doses were previously shown to preferentially block neuronal channels (Cruikshank et al, 2004). Thus, while potential additive effects of simultaneous neuronal and astrocytic gap-junction hemichannel blockade cannot be cleanly discerned, it is clear that neither the sucrose nor EtOH breakpoints were altered by neuron-targeting mefloquine doses. Because 18-α is not cell-selective, it is important to note that both high-dose mefloquine and 18-α increased the EtOH breakpoint to the same extent (data here), given that microinjection of this same 18-α dose into the rat prefrontal cortex increased EtOH preference to the same extent as an astrocyte-specific toxin (Miguel-Hidalgo et al, 2008). Together, these data suggest that blockade of NAcore astrocytic gap-junction hemichannels increased the motivation to self-administer EtOH after 3 weeks abstinence.
Stimulating Astrocytes Decreased the Motivation for Ethanol
Due to its effect on the EtOH breakpoint, Gαq-DREADD activation can be conceptualized as the functional inverse of blocking gap-junction hemichannels. One could test this hypothesis by reversing the effect of Gαq-DREADD activation on breakpoint via microinjection of subthreshold doses of gap-junction hemichannel blockers. Nonetheless, given that astrocytic Gαq-DREADD activation facilitated ICSS, this postulate is partially supported by studies showing that NAcore gap-junction hemichannel blockers decreased ICSS (Kokarovtseva et al, 2009). Further, the facilitation of responding for ICSS observed following astrocytic Gαq-DREADD activation could, by extension, underlie the reduced EtOH breakpoint observed here because ICSS thresholds are elevated during EtOH abstinence (Schulteis et al, 1995). The Gαq-DREADD-mediated ICSS facilitation exhibited an inverted U-shaped dose–response and was small relative to cocaine, suggesting that future medications that stimulate astrocytes in this manner may not possess high abuse liability. Finally, the significance of being able to selectively manipulate astrocytic signaling can be illustrated by comparing our data to a recent report where EtOH consumption was not affected by stimulation of Gαq-DREADDs that were expressed in mouse NAcore neurons (Cassataro et al, 2014).
CONCLUSION
Astrocytes can actively shape neuronal plasticity and function. This is the first study revealing a dichotomous role of nucleus accumbens astrocytes in the motivation to self-administer EtOH and responding for rewarding brain stimulation. These data add to the rapidly growing appreciation that, in addition to critical homeostatic functions, discrete astrocyte populations can be manipulated to abate drug-seeking behavior.
FUNDING AND DISCLOSURE
Dr Bull’s postdoctoral position was funded by the Virginia Commonwealth University School of Medicine for the entire period of time during which this research was conducted. During the past 3 years, she has not received any compensation from private or publicly owned firms.
Freitas is a second year graduate student funded by the Virginia Commonwealth University School of Medicine for the entire period of time during which this research was conducted. During the past 3 years, she has not received any compensation from private or publicly owned firms.
Zou is a second year graduate student that has been funded through National Institutes of Health grants to Dr Knapp for the entire period of time during which this research was conducted. During the past 3 years, he has not received any compensation from private or publicly owned firms.
Sgt. Poland’s undergraduate research experiences were funded by a gift from the Lacey family to the Virginia Institute for Psychiatric and Behavioral Genetics located at the Virginia Commonwealth University School of Medicine. During the past 3 years, he has received compensation from Navistar for work unrelated to this project.
Syed’s undergraduate research experiences were funded by the Virginia Commonwealth University Honors College and Howard Hughes Medical Institute. During the past 3 years, he has not received any compensation from private or publicly owned firms.
Dr Urban’s graduate work was funded through National Institutes of Health grants to Dr Roth for the entire period of time during which this research was conducted. During the past 3 years, he has not received any compensation from private or publicly owned firms.
Minter’s research assistantship was by the Virginia Commonwealth University School of Medicine for the entire period of time during which this research was conducted. During the past 3 years, she has not received any compensation from private or publicly owned firms.
Dr Shelton, Knapp, and Hauser declare that their research has been funded by the National Institutes of Health. During the past 3 years, they have not received any compensation from private or publicly owned firms.
Dr Negus declares that his research has been funded by the National Institutes of Health. During the past 3 years, he has provided consulting to Alkermes Pharmaceutical Co for topics unrelated to this manuscript.
Dr Bowers declares that his research has been funded by the National Institutes of Health, the Alcohol Beverage Medical Research Foundation, and the Virginia Commonwealth University School of Medicine. During the past 3 years, he has not received any compensation from private or publicly owned firms.
Research reported in this publication was supported by ABMRF/The Foundation for Alcohol Research (MSB) and the Virginia Commonwealth University School of Medicine (MSB). A portion of this work was also supported by the National Institute on Drug Abuse (SSN). The content is solely the responsibility of the authors and does not necessarily represent the official views of these organizations.
References
Adermark L, Lovinger DM (2006). Ethanol effects on electrophysiological properties of astrocytes in striatal brain slices. Neuropharmacology 51: 1099–1108.
Agulhon C, Boyt KM, Xie AX, Friocourt F, Roth BL, McCarthy K (2013). Modulation of the autonomic nervous system by acute glial cell Gq-GPCR activation in vivo. J Physiol 591: 5599–5609.
Allen JW, Mutkus LA, Aschner M (2002). Chronic ethanol produces increased taurine transport and efflux in cultured astrocytes. Neurotoxicology 23: 693–700.
Anlauf E, Derouiche A (2013). Glutamine synthetase as an astrocytic marker: its cell type and vesicle localization. Front Endocrinol (Lausanne) 4: 144.
Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. PNAS 104: 5163–5168.
Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S et al (2003). Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6: 743–749.
Bauer CT, Banks ML, Blough BE, Negus SS (2013). Use of intracranial self-stimulation to evaluate abuse-related and abuse-limiting effects of monoamine releasers in rats. Br J Pharmacol 168: 850–862.
Beitner-Johnson D, Guitart X, Nestler EJ (1993). Glial fibrillary acidic protein and the mesolimbic dopamine system: regulation by chronic morphine and Lewis-Fischer strain differences in the rat ventral tegmental area. J Neurochem 61: 1766–1773.
Bowers MS, Hopf FW, Chou JK, Guillory AM, Chang SJ, Janak PH et al (2008). Nucleus accumbens AGS3 expression drives ethanol seeking through G betagamma. Proc Natl Acad Sci USA 105: 12533–12538.
Bowers MS, Kalivas PW (2003). Forebrain astroglial plasticity is induced following withdrawal from repeated cocaine administration. Eur J Neurosci 17: 1273–1278.
Bruzzone R, Barbe MT, Jakob NJ, Monyer H (2005). Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J Neurochem 92: 1033–1043.
Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22: 183–192.
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS et al (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28: 264–278.
Cassataro D, Bergfeldt D, Malekian C, Van Snellenberg JX, Thanos PK et al (2014). Reverse pharmacogenetic modulation of the nucleus accumbens reduces ethanol consumption in a limited access paradigm. Neuropsychopharmacology 39: 283–290.
Chaudhri N, Sahuque LL, Schairer WW, Janak PH (2010). Separable roles of the nucleus accumbens core and shell in context- and cue-induced alcohol-seeking. Neuropsychopharmacology 35: 783–791.
Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC, Srinivas M (2004). Potent block of Cx36 and Cx50 gap junction channels by mefloquine. PNAS 101: 12364–12369.
Dahl G, Harris A (2008). Pannexins or Connexins? In: Harris A, Locke D (eds) Connexins: a guide. Humana Press: New York. pp 287–301.
Davidson JS, Baumgarten IM (1988). Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap-junctional intercellular communication. Structure-activity relationships. J Pharmacol Exp Ther 246: 1104–1107.
Dermietzel R, Spray DC (1993). Gap junctions in the brain: where, what type, how many and why? Trends Neurosci 16: 186–192.
Desagher S, Glowinski J, Premont J (1996). Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci 16: 2553–2562.
Eng LF, Ghirnikar RS, Lee YL (2000). Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000). Neurochem Res 25: 1439–1451.
Epstein DH, Preston KL, Stewart J, Shaham Y (2006). Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology (Berl) 189: 1–16.
Feresten AH, Barakauskas V, Ypsilanti A, Barr AM, Beasley CL (2013). Increased expression of glial fibrillary acidic protein in prefrontal cortex in psychotic illness. Schizophr Res 150: 252–257.
Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y et al (2011). Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci 14: 22–24.
Guettier JM, Gautam D, Scarselli M, Ruiz de Azua I, Li JH, Rosemond E et al (2009). A chemical-genetic approach to study G protein regulation of beta cell function in vivo. PNAS 106: 19197–19202.
Gundersen HJ, Jensen EB (1987). The efficiency of systematic sampling in stereology and its prediction. J Microsc 147: 229–263.
Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG (2007). Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27: 6473–6477.
Hassinger TD, Guthrie PB, Atkinson PB, Bennett MV, Kater SB (1996). An extracellular signaling component in propagation of astrocytic calcium waves. Proc Natl Acad Sci USA 93: 13268–13273.
Heinz A, Beck A, Grusser SM, Grace AA, Wrase J (2009). Identifying the neural circuitry of alcohol craving and relapse vulnerability. Addict Biol 14: 108–118.
Hopf FW, Chang SJ, Sparta DR, Bowers MS, Bonci A (2010). Motivation for alcohol becomes resistant to quinine adulteration after 3 to 4 months of intermittent alcohol self-administration. Alcohol Clin Exp Res 34: 1565–1573.
John CS, Smith KL, Van't Veer A, Gompf HS, Carlezon WA Jr, Cohen BM et al (2012). Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology 37: 2467–2475.
Kokarovtseva L, Jaciw-Zurakiwsky T, Mendizabal Arbocco R, Frantseva MV, Perez Velazquez JL (2009). Excitability and gap junction-mediated mechanisms in nucleus accumbens regulate self-stimulation reward in rats. Neuroscience 159: 1257–1263.
Laming PR, Kimelberg H, Robinson S, Salm A, Hawrylak N, Müller C et al (2000). Neuronal-glial interactions and behaviour. Neurosci Biobehav Rev 24: 295–340.
Locovei S, Wang J, Dahl G (2006). Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett 580: 239–244.
Long JM, Kalehua AN, Muth NJ, Calhoun ME, Jucker M, Hengemihle JM et al (1998). Stereological analysis of astrocyte and microglia in aging mouse hippocampus. Neurobiol Aging 19: 497–503.
Ludwig AM, Wikler A, Stark LH (1974). The first drink: psychobiological aspects of craving. Arch Gen Psychiatry 30: 539–547.
Miguel-Hidalgo J, Shoyama Y, Wanzo V (2008). Infusion of gliotoxins or a gap junction blocker in the prelimbic cortex increases alcohol preference in Wistar rats. J Psychopharmacol 23: 550–557.
Miguel-Hidalgo JJ (2005). Lower packing density of glial fibrillary acidic protein-immunoreactive astrocytes in the prelimbic cortex of alcohol-naive and alcohol-drinking alcohol-preferring rats as compared with alcohol-nonpreferring and Wistar rats. Alcohol Clin Exp Res 29: 766–772.
Miguel-Hidalgo JJ (2006). Withdrawal from free-choice ethanol consumption results in increased packing density of glutamine synthetase-immunoreactive astrocytes in the prelimbic cortex of alcohol-preferring rats. Alcohol Alcohol 41: 379–385.
Miguel-Hidalgo JJ, Wei J, Andrew M, Overholser JC, Jurjus G, Stockmeier CA et al (2002). Glia pathology in the prefrontal cortex in alcohol dependence with and without depressive symptoms. Biol Psychiatry 52: 1121–1133.
Narita M, Miyatake M, Narita M, Shibasaki M, Shindo K, Nakamura A et al (2006). Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology 31: 2476–2488.
Navarrete M, Perea G, Maglio L, Pastor J, Garcia de Sola R, Araque A (2013). Astrocyte calcium signal and gliotransmission in human brain tissue. Cereb Cortex 23: 1240–1246.
Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG (1994). Glutamate-mediated astrocyte-neuron signalling. Nature 369: 744–747.
Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG et al (2012). Glial cells in (patho)physiology. J Neurochem 121: 4–27.
Pierce RC, Kumaresan V (2006). The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev 30: 215–238.
Porter JT, McCarthy KD (1996). Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci 16: 5073–5081.
Richardson NR, Roberts DC (1996). Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J Neurosci Methods 66: 1–11.
Santello M, Cali C, Bezzi P (2012). Gliotransmission and the tripartite synapse. Adv Exp Med Biol 970: 307–331.
Sasaki T, Beppu K, Tanaka KF, Fukazawa Y, Shigemoto R, Matsui K (2012). Application of an optogenetic byway for perturbing neuronal activity via glial photostimulation. Proc Natl Acad Sci USA 109: 20720–20725.
Scemes E, Suadicani SO, Dahl G, Spray DC (2007). Connexin and pannexin mediated cell-cell communication. Neuron Glia Biol 3: 199–208.
Schulteis G, Markou A, Cole M, Koob GF (1995). Decreased brain reward produced by ethanol withdrawal. PNAS 92: 5880–5884.
Sofroniew MV, Vinters HV (2010). Astrocytes: biology and pathology. Acta Neuropathol 119: 7–35.
Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W et al (2013). Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 339: 197–200.
Szatkowski M, Barbour B, Attwell D (1990). Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348: 443–446.
Toms NJ, Roberts PJ (1999). Group 1 mGlu receptors elevate [Ca2+]i in rat cultured cortical type 2 astrocytes: [Ca2+]i synergy with adenosine A1 receptors. Neuropharmacology 38: 1511–1517.
Turner JR, Ecke LE, Briand LA, Haydon PG, Blendy JA (2012). Cocaine-related behaviors in mice with deficient gliotransmission. Psychopharmacology (Berl) 226: 167–176.
Wissman AM, May RM, Woolley CS (2012). Ultrastructural analysis of sex differences in nucleus accumbens synaptic connectivity. Brain Struct Funct 217: 181–190.
Woo DH, Han KS, Shim JW, Yoon BE, Kim E, Bae JY et al (2012). TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 151: 25–40.
Ye ZC, Oberheim N, Kettenmann H, Ransom BR (2009). Pharmacological ‘cross-inhibition’ of connexin hemichannels and swelling activated anion channels. Glia 57: 258–269.
Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR (2003). Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci 23: 3588–3596.
Zahm DS (2000). An integrative neuroanatomical perspective on some subcortical substrates of adaptive responding with emphasis on the nucleus accumbens. Neurosci Biobehav Rev 24: 85–105.
Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF et al (2011). Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain 134: 3616–3631.
Acknowledgements
This study was supported by AMBRF (MSB), NIAAA/P30-AA019372 pilot (MSB), and NIDA/R01-DA026946 (SN). We thank Jürgen Wess, PhD (NIDDK), for CNO, Bryan Roth, MD, PhD (UNC), for the Gαq-DREADD virus, and the NIDA drug supply program for cocaine.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article

Cite this article
Bull, C., Freitas, K., Zou, S. et al. Rat Nucleus Accumbens Core Astrocytes Modulate Reward and the Motivation to Self-Administer Ethanol after Abstinence. Neuropsychopharmacol 39, 2835–2845 (2014). https://doi.org/10.1038/npp.2014.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.135
This article is cited by
-
Astrocyte regulation of synaptic signaling in psychiatric disorders
Neuropsychopharmacology (2023)
-
Astrocytic transcriptional and epigenetic mechanisms of drug addiction
Journal of Neural Transmission (2023)
-
Astrocyte-neuron signaling in the mesolimbic dopamine system: the hidden stars of dopamine signaling
Neuropsychopharmacology (2021)
-
Cortical astrocytes regulate ethanol consumption and intoxication in mice
Neuropsychopharmacology (2021)
-
Contribution of TSPO imaging in the understanding of the state of gliosis in substance use disorders
European Journal of Nuclear Medicine and Molecular Imaging (2021)
