
Abstract
Cocaine is thought to be addictive because it elevates dopamine levels in the striatum, reinforcing drug-seeking habits. Cocaine also elevates dopamine levels in the hippocampus, a structure involved in contextual conditioning as well as in reward function. Hippocampal dopamine promotes the late phase of consolidation of an aversive step-down avoidance memory. Here, we examined the role of hippocampal dopamine function in the persistence of the conditioned increase in preference for a cocaine-associated compartment. Blocking dorsal hippocampal D1-type receptors (D1Rs) but not D2-type receptors (D2Rs) 12 h after a single training trial extended persistence of the normally short-lived memory; conversely, a general and a specific phospholipase C-coupled D1R agonist (but not a D2R or adenylyl cyclase-coupled D1R agonist) decreased the persistence of the normally long-lived memory established by three-trial training. These effects of D1 agents were opposite to those previously established in a step-down avoidance task, and were here also found to be opposite to those in a lithium chloride-conditioned avoidance task. After returning to normal following cocaine injection, dopamine levels in the dorsal hippocampus were found elevated again at the time when dopamine antagonists and agonists were effective: between 13 and 17 h after cocaine injection. These findings confirm that, long after the making of a cocaine–place association, hippocampal activity modulates memory consolidation for that association via a dopamine-dependent mechanism. They suggest a dynamic role for dorsal hippocampal dopamine in this late-phase memory consolidation and, unexpectedly, differential roles for late consolidation of memories for places that induce approach or withdrawal because of a drug association.
Similar content being viewed by others


INTRODUCTION
The addicted brain differs from the non-addicted brain as a function of two forms of neuroplasticity that are important for addiction theory: neuroadaptations resulting from the pharmacological impact of the drug on the brain and synaptic changes that reflect the memory trace for the drug experience (Berke and Hyman, 2000; Hyman et al, 2006). Neuroadaptations that are caused by repeated passive exposure to the drug itself have been widely studied (Nestler, 1992; Lüscher and Malenka, 2011), but there are further changes in brain function that reflect the memory of drug experience. These further brain changes are seen in animals that self-administer the drug, but are absent in animals that have passively received ‘yoked’ injections of the same drug at the same dose and temporal pattern (Wang et al, 2005; You et al, 2007, 2008; Chen et al, 2008; Caillé et al, 2009). Here, we were concerned with brain changes involved in the consolidation of the memory for drug–environment association established by Pavlovian conditioning.
Much of the interest in the neuroplasticity associated with cocaine addiction has been focused on the ventral tegmental dopamine neurons (Bonci and Malenka, 1999; Saal et al, 2003) and their projection targets in the nucleus accumbens (Nestler, 1992; Hyman et al, 2006; Kalivas, 2009). Self-administered cocaine elevates dopamine levels there (Pettit et al, 1990; Wise et al, 1995), and the rewarding effects of self-administered cocaine are attenuated or blocked by lesions or infusion of dopamine antagonists into this area. Cocaine (Ikemoto, 2003) and dopamine agonists (Ikemoto and Goeders, 1998) are reinforcing when injected into the medial portions of the ventral striatum. However, while the striatum is thought to have the major role in the establishment of instrumental response habits (Everitt and Robbins, 2005), rats not only learn to self-administer addictive drugs but they also learn to approach and spend more time in the places where they have experienced addictive drugs in the past. Such learning is likely to implicate the hippocampal circuitry involved in spatial and contextual learning (O'Keefe and Nadel, 1978; Phillips and LeDoux, 1994). One such circuit may involve the functional loops linking the ventral tegmental area (VTA) and the hippocampus (Lisman and Grace, 2005; Luo et al, 2011). Recent findings point to the dorsal hippocampus, and particularly to its dopaminergic inputs, as important for the persistence of memory storage in behaving rats (Rossato et al, 2009). This modulation is mediated by dopamine (acting at D1-type receptors (D1Rs)) and brain-derived neurotrophic factor (BDNF) 12 h after training (but not 0, 9, or 24 h after training), and is selective for the maintenance, but not for the formation, of fear-associated long-term memories (LTMs) (Bekinschtein et al, 2007; Rossato et al, 2009). However, the mechanisms by which a drug-related memory becomes persistent over time remain to be elucidated. In the present study, we explore the role of dorsal hippocampal dopamine in modulating the persistence of memories for the association between rewarding cocaine or aversive lithium chloride (LiCl) and the experimental chamber in which the effects of the drugs were experienced.
MATERIALS AND METHODS
See Supplementary Materials and methods’ for further details.
Animals
A total of 949 male Sprague–Dawley rats (Facultad de Ciencias Veterinarias, Argentina; and Charles River, Raleigh, NC) were used. Groups of five rats each (weighting around 200 g upon arrival at the laboratory) were housed in a vivarium maintained on a reversed 12-h light–dark cycle (lights off at 0630 hours) at a constant temperature of 23 °C. Animal care was in strict accordance with institutional and international standards (National Research Council, 2011; NIDA Animal Care and Use Committee).
Drugs
Cocaine hydrochloride (Laboratorios Verardo y Cia., Buenos Aires, Argentina or NIDA intramural pharmacy, Baltimore, MD), SCH 23390 hydrochloride, quinpirole hydrochloride (Sigma-Aldrich, St Louis, MO), eticlopride hydrochloride (Tocris Bioscience, Bristol, UK), LiCl (Laboratorio Cicarelli, San Lorenzo, Argentina) were all dissolved in sterile 0.9% physiological saline. SKF 38393 hydrochloride, SKF 83959 hydrobromide (Sigma-Aldrich), and SKF 83822 hydrobromide (Tocris Bioscience) were dissolved in sterile 0.9% physiological saline supplemented with DMSO (10%, final concentration; Laboratorio Ernesto van Rossum y Cia., Buenos Aires, Argentina). All the antibodies, except for anti-phospho-ERK1/2 and anti-ERK1/2 (Cell Signaling Technology, Beverly, MA), were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Surgical and Intracerebral Infusion Procedures
Each rat was anesthetized with a mix of ketamine (85 mg/kg) and xylazine (10 mg/kg), and placed on a stereotaxic frame. The skull was exposed and leveled (flat skull, lambda, and bregma at the same elevation). Either 22-G guide cannulae for intracerebral infusions or CMA 11 microdialysis guide cannulae (CMA, Helsingborg, Sweden) were bilaterally implanted, aimed at the dorsal hippocampus (CA1 region). The stereotaxic coordinates used were as follows: AP: −3.9; ML: ±3.0; DV: −3.0 (Paxinos and Watson, 2004). Animals were allowed 5–7 days of recovery before any experimental manipulation.
For intracerebral infusions, 30-G needles connected to Hamilton syringes were used. The infusions were always bilateral, with volume infusions of 1 μl per side. The needle was left in place for an additional minute after infusion to allow diffusion and to prevent reflux. The vehicle used in each case corresponds to the solution in which the respective drug was dissolved (see Drugs section above). The doses used for each compound were as follows: 1 μg/μl for quinpirole and SKF 83822, 1.5 μg/μl for SCH 23390 and eticlopride, 10 μg/μl for SKF 83959, and 12.5 μg/μl for SKF 38393. At the end of each experiment, cannula placement was verified by infusion of 1 μl of methylene blue and histological analysis. Only the behavioral data of animals with a correct placement of cannulae were included in the study.
In Vivo Microdialysis
Animals were connected to the microdialysis systems the evening previous to the microdialysis experiment. The blocker of each guide cannula was removed and the microdialysis probe (CMA 11, 1 mm membrane length; CMA) was inserted and secured in place. Probes were connected to the microdialysis pump (CMA/100; CMA) with FEP tubing through a two-channel swivel and were perfused overnight with an artificial cerebrospinal fluid, at a flow rate of 0.3 μl/min. Next morning, the flow rate was increased to 0.6 μl/min, and after 1 h of equilibration, a series of four 30-min baseline samples were collected. Then, animals were intraperitoneally injected with either saline or cocaine (20 mg/kg) and confined to the corresponding side of a three-compartment apparatus (see Behavioral training and testing section below) during 30 min. They were returned to their home cage afterwards. Microdialysis samples were collected every hour during a 24-h period. Acetic acid (20 mM, 6 μl) was added to the vials where samples were collected to avoid dopamine degradation.
Biochemical Analysis of Microdialysis Samples
Dialysate dopamine concentrations were analyzed using high-performance liquid chromatography (HPLC) with electrochemical detection. Aliquots from dialysate samples (15 μl) were injected into an Eicom HTEC-500 HPLC system with an integrated amperometric detector, consisting a PP-ODS HPLC column (30 × 4.6 mm2, inner diameter, C-18, 2 μm particulate silica gel) and an HTEC-500 pump (Eicom, San Diego, CA). The mobile phase consisted of 100 mM NaH2PO4, 1.3 mM EDTA, 2.0 mM decane-1-sulfonate, and 1% MeOH, pH 6.0, resulting in a retention time of 2.2–2.5 min at a pump rate of 0.5 ml/min. Applied potential was set at +450 mV vs Ag/AgCl.
Western Blot Assays
Three different conditions were analyzed: (1) biochemical profiles of BDNF, p-TrkB and p-ERK1/2 12 h after cocaine (or saline in the case of controls) injection in one-trial training; (2) biochemical profiles of BDNF and p-TrkB 12 h after the last injection of cocaine (or saline in the case of controls) in three-trial training; and (3) biochemical profiles of BDNF and p-ERK1/2 after the infusion of the D1R antagonist SCH 23390 in the dorsal hippocampus. The dopaminergic antagonist was infused 11.5 h after the injection of cocaine in one-trial training. Thirty minutes later, animals were killed; the dorsal hippocampi were dissected and rapidly homogenized in ice-chilled buffer. Samples of homogenates (10 μg of protein) were subjected to SDS-PAGE (10 or 15% gels) under reducing conditions. Proteins were electrotransferred onto PVDF membranes for 2 h at 100 V at 4 °C for BDNF analysis. For ERK1/2 and p-TrkB analysis, proteins were transferred onto PVDF membranes overnight at 40 V at 4 °C. Immunoblots were performed by incubating membranes with antibodies anti-BDNF (1:1000, sc-546), anti-actin (1:10 000, sc-1615), anti-phospho-ERK1/2 (1:4000, no. 9101s), anti-ERK1/2 (1:3000, no. 9102), anti-phospho-TrkB (1:2000, sc-135645), and anti-TrkB (1:1000, sc-8316). The blots were scanned in a Storm 845 PhosphorImager (GE Healthcare Life Sciences, Little Chalfont, UK), and quantifications were carried out with ImageQuant software (GE Healthcare Life Sciences).
Behavioral Training and Testing
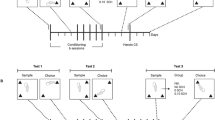
Conditioning
Place conditioning was carried out in three-compartment chambers; the center compartment was a short connecting passageway between the conditioning chambers in which the animals were placed after drug injections. One conditioning compartment had black walls (white squares pattern, grid floor) and the other had white walls (black lines pattern, perforated floor); the connecting passage was gray (no pattern, smooth floor). The experiments consisted of three phases: a pretest phase in which the animals were allowed to explore the entire apparatus for 15 min, a conditioning phase in which they were restricted to one side of the chamber after saline intraperitoneal injections and, on alternate days, the other side of the chamber after saline, cocaine, or LiCl injections, and a test phase in which they were allowed to explore the open apparatus again in a drug-free state. During the pretest phase, if an animal failed to spend at least 90 s in each compartment, it was excluded from the study. Five out of 954 animals were excluded for this reason.
In the one-trial version of the cocaine conditioning paradigm, the animals were given an intraperitoneal saline injection before placement in the initially preferred dark chamber for 30 min on the first conditioning day and were given an intraperitoneal cocaine injection before placement in the initially non-preferred white chamber for 30 min on the following day. Saline intraperitoneal injections were given in both compartments for control groups. The one-trial version was used to establish a weak memory for the cocaine-chamber association in experiments designed to assess the effects of treatments designed to increase memory persistence; the procedure was repeated two times more in a three-trial version to establish a stronger memory for the same association in experiments designed to assess treatments that might decrease memory persistence. Cocaine doses of 10, 15, or 20 mg/kg were used for the dose–response experiment, and 20 mg/kg was used for the remaining experiments. An increase in time spent in the cocaine-associated chamber after conditioning was taken as a reflection of the known rewarding properties of cocaine at this dose (Busse and Riley, 2004; dela Cruz et al, 2009; Crooks et al, 2010).
In the place conditioning studies with LiCl, the one-trial version was used. On the first day, the animals were given intraperitoneal saline and placed in the initially non-preferred white chamber for 60 min; on the next day, the animals were given intraperitoneal saline or LiCl (150 mg/kg) and placed in the initially preferred black compartment for 60 min. In each case, the animals were given open access to the entire apparatus on the day following the last conditioning day; this phase was identical to the pretest: animals in a drug-free state were left to explore the entire apparatus during 15 min and the time spent in each compartment was recorded. A decrease in time spent in the LiCl-associated chamber after conditioning was taken as a reflection of the known aversive properties of LiCl at this dose (Tenk et al, 2005).
Supplementary Table 1 shows the mean pre- and postconditioning times spent in the compartment associated with cocaine or LiCl for each of the main results in this study.
In experiments where intracerebral infusions were performed, the infusion time was always 12 h after conditioning. Animals were infused and returned immediately to their home cages. They were never exposed to the testing apparatus during or after the infusion procedure. When a three-trial training protocol was used, the dopaminergic drugs were infused 12 h after each conditioning session with either cocaine or LiCl.
Statistical Analyses
Data were presented as a score in s: time spent in the drug-associated compartment during the test minus time spent in the to-be drug-associated compartment during the pretest. Results are presented as mean±SEM. For behavioral and biochemical data, results were analyzed using a Student’s t-test in the case where two groups were to be compared and using a multifactorial analysis of variance (MANOVA) with cocaine/LiCl (saline vs cocaine/LiCl), dose (10, 15, or 20 mg/kg), place (home cage vs conditioning apparatus), time of testing (24 h, and 7 or 14 days), and challenge (vehicle vs dopaminergic agents) as between-subject factors, in the case where more than two groups were to be compared. For significant overall interactions, further analyses of partial interactions were carried out. Post hoc analyses were performed with Newman–Keuls test when the initial p-value was significant. A result was considered significant if p<0.05. All data were analyzed using Statistica software (Statsoft, Paris, France).
RESULTS
We first established conditioning parameters that would result in long-lasting (7 days or more) or short-lasting (24 h but not 7 days) increases in time spent in the cocaine-associated chamber using independent animal groups. Increased times in the cocaine-associated chamber were seen in rats trained with a single injection (20 mg/kg) and tested 1 day but not 7 or 14 days after the conditioning trial (Figure 1a, main effect of cocaine: F(1,58)=31.94, p<0.001; main effect of dose: F(2,58)=0.85, p=0.43, NS; cocaine × dose: F(2,58)=3.70, p<0.05; Figure 1b, main effect of cocaine: F(1,44)=2.05, p=0.16, NS; main × effect of time: F(2,44)=0.40, p=0.67, NS; cocaine × time: F(2,44)=4.24, p<0.05). The reversed order of injections of cocaine and saline gave similar results (Supplementary Figure S1a; p<0.05). Rats given two cocaine training days also showed an increase in time spent in the cocaine-associated chamber when tested (1 day but not 7 days) after conditioning (Supplementary Figure S1b; p=0.18). Independent groups of rats given three cocaine–chamber pairings showed increased time in the cocaine-associated chamber when tested 7, 14, or 21 days after training (Figure 1c, main effect of cocaine: F(1,62)=27.74, p<0.001; main effect of time: F(3,62)=0.83, p=0.48, NS; cocaine × time: F(3,62)=4.43, p<0.05). We subsequently used the single pairing condition to establish weakly persistent memory and the three-pairing condition to establish stronger, more persistent memory for the cocaine–chamber association.

Temporal duration and dopaminergic modulation of the persistence of a short-lasting cocaine-associated memory. (a) Three doses of cocaine were tested. The dose of 20 mg/kg induced a significant retention of the memory for cocaine–place association when tested 24 h after a one-trial training (n=5–21 per group). Therefore, this dose was used in all the remaining approach experiments. (b) A weak (one-trial training) memory for the cocaine–place association was retained for 24 h but not for 7 or 14 days (n=7–11 per group). (c) A strong (three-trial training) memory for the cocaine–place association was retained for at least 21 days (n=8–10 per group). Asterisks indicate significant differences between cocaine-injected animals and its corresponding saline controls (Newman–Keuls, *p<0.05; **p<0.01). (d–g) Effects of D1-type dopamine agonist (SKF 38393) or antagonist (SCH 23390) injections—given 12 h after the training trial—on the persistence of the memory for cocaine–place associations established by one-trial training. (d) The D1-type agonist had no effect on the 24 h memory for cocaine–place associations established by one-trial training (n=8–10 per group). (e) The D1-type agonist had no effect on the persistence of the memory for cocaine–place associations when tested at 7 days after one-trial training (n=10–12 per group). (f) The D1-type antagonist had no effect on the 24 h memory for cocaine–place associations established by one-trial training (n=16–17 per group). (g) The D1-type antagonist prolonged the duration of the memory for cocaine–place associations after one-trial training (n=11–14 per group). Post hoc analyses demonstrated that the group conditioned with cocaine, infused with SCH 23390 12 h later, and tested at 7 days showed a higher score when compared with the other groups (Newman–Keuls, **p<0.01). Asterisks upon bracket indicate a significant difference between saline- and cocaine-injected animals, collapsed across intracerebral infusion treatments (Newman–Keuls, *p<0.05; **p<0.01). Coc, cocaine; Sal, saline; Veh, vehicle.
To determine the effects of dorsal hippocampal dopamine receptor activation on the late post-training consolidation of the memory for the cocaine–chamber association, we microinjected dopamine receptor agonists or antagonists into the dorsal hippocampus 12 h after one- or three-trial training and tested for retention 24 h or 7 days later (see Supplementary Figure S2 to check for the location and extension of the infusions). Rats tested 24 h after one-trial training showed normal retention following bilateral hippocampal microinjections of either the D1R agonist SKF 38393 or the D1R antagonist SCH 23390 (Figure 1d, main effect of cocaine: F(1,31)=16.54, p<0.001; main effect of challenge: F(1,31)=2.12, p=0.16, NS; cocaine × challenge: F(1,31)=0.04, p=0.84, NS; Figure 1f, main effect of cocaine: F(1,62)=23.87, p<0.001; main effect of challenge: F(1,62)=3.69, p=0.06, NS; cocaine × challenge: F(1,62)=0.04, p=0.84, NS). The dopamine D1R agonist failed to modify the retention of the cocaine–chamber association when tested 7 days after training (Figure 1e, main effect of cocaine: F(1,40)=0.34, p=0.57, NS; main effect of challenge: F(1,40)=0.009, p=0.93, NS; cocaine × challenge: F(1,40)=0.05, p=0.82, NS). However, treatment with the D1R antagonist unexpectedly extended retention of the association to 7 days after one-trial training (Figure 1g, main effect of cocaine: F(1,47)=4.64, p<0.05; main effect of challenge: F(1,47)=4.33, p<0.05; cocaine × challenge: F(1,47)=7.60, p<0.01).
We next determined the effects of the same treatments on the persistence of the stronger memory established with the three-trial training paradigm. As in the case of the short-lasting cocaine-associated memory, neither the D1R antagonist nor the D1R agonist, given 12 h after training, affected retention as tested 24 h after training (Figure 2a, main effect of cocaine: F(1,35)=28.69, p<0.001; main effect of challenge: F(1,35)=0.24, p=0.63, NS; cocaine × challenge: F(1,35)=0.73, p=0.40, NS; Figure 2c, main effect of cocaine: F(1,31)=28.18, p<0.001; main effect of challenge: F(1,31)=0.11, p=0.74, NS; cocaine × challenge: F(1,31)=0.48, p=0.49, NS). The D1R antagonist also failed to affect retention as tested at 7 days (Figure 2d, main effect of cocaine: F(1,53)=19.96, p<0.001; main effect of challenge: F(1,53)=0.03, p=0.85, NS; cocaine × challenge: F(1,53)=0.04, p=0.85, NS). Conversely, when the D1R agonist was administered during the three training trials, the persistence of a normally long-lasting cocaine-associated memory was abolished (Figure 2b, main effect of cocaine: F(1,31)=7.44, p<0.05; main effect of challenge: F(1,31)=5.65, p<0.05; cocaine × challenge: F(1,31)=4.25, p<0.05). Interestingly, the D1R agonist was ineffective in three cases where the cannula placement was asymmetrical and injections did not reach the dorsal hippocampus or reached it on one side only (Supplementary Figure S3).

Dopaminergic modulation of the persistence of a long-lasting cocaine-associated memory. Effects of D1-type dopamine agonist (SKF 38393) or antagonist (SCH 23390) injections—given 12 h after the training trial—on the persistence of the memory for cocaine–place associations established by 3-trial training. (a) The D1-type agonist (SKF 38393) had no effect on the 24 h memory for cocaine–place associations established by three-trial training (n=9–10 per group). (b) Treatment with the D1-type agonist (SKF 38393) attenuated the 7-day persistence of the memory for cocaine–place associations established by three-trial training (n=7–10 per group). (c) The D1-type antagonist (SCH 23390) had no effect on the 24 h memory for cocaine–place associations established by three-trial training (n=8–10 per group). (d) The D1-type antagonist (SCH 23390) had no effect on the persistence of the memory for cocaine–place associations when tested at 7 days after three-trial training (n=13–15 per group). Post hoc analyses demonstrated that the group conditioned with cocaine, infused with SKF 38393 12 h later, and tested at 7 days showed a higher score when compared with the other groups (Newman–Keuls, **p<0.01). Asterisks upon bracket indicate a significant difference between saline and cocaine-injected animals, collapsed across intracerebral infusion treatments (Newman–Keuls, **p<0.01; ***p<0.001). Coc, cocaine; Sal, saline; Veh, vehicle.
Neither the D2-type dopamine receptor agonist quinpirole (1 μg per side; main effect of cocaine: F(1,41)=2.23, p=0.14, NS; main effect of challenge: F(1,41)=0.0004, p=0.98, NS; cocaine × challenge: F(1,41)=0.06, p=0.80, NS) nor the D2-type dopamine receptor antagonist eticlopride (1.5 μg per side; main effect of cocaine: F(1,31)=1.03, p=0.32, NS; main effect of challenge: F(1,31)=0.04, p=0.83, NS; cocaine × challenge: F(1,31)=0.13, p=0.72, NS) affected the persistence of the weak cocaine-associated memory (Supplementary Figure S4).
To gain better insight on the molecular mechanisms underlying the persistence of cocaine-associated memories, we next used agonists for D1Rs that are selectively coupled to different intracellular messenger cascades (Undieh, 2010). Whereas the adenylyl cyclase (AC)-selective agonist SKF 83822 given 12 h after three-trial training had no effect on retention tested 7 days after training (Figure 3a, main effect of cocaine: F(1,35)=12.16, p<0.01; main effect of challenge: F(1,35)=1.31, p=0.26, NS; cocaine × challenge: F(1,35)=0.07, p=0.79, NS), the phospholipase C (PLC)-coupled agonist SKF 83959 significantly decreased retention at the same time-point (Figure 3b, main effect of cocaine: F(1,26)=12.40, p<0.01; main effect of challenge: F(1,26)=8.33, p<0.01; cocaine × challenge: F(1,26)=5.72, p<0.05).

Persistence of cocaine-related memories is regulated by dopaminergic receptors coupled to phospholipase C (PLC) cascade and is not mediated by brain-derived neurotrophic factor (BDNF). (a) Delayed (12 h after conditioning trial) infusion of the adenylyl cyclase (AC)-selective D1-type agonist SKF 83822 had no effect on the 7-day retention of the memory for cocaine–place associations established by three-trial training (n=9–10 per group). (b) Delayed (12 h after conditioning trial) infusion of the PLC-selective D1-type agonist SKF 83959 significantly attenuated 7-day retention of the memory for cocaine–place associations established by three-trial training (n=7–9 per group). Post hoc analyses demonstrated that the group conditioned with cocaine, infused with SKF 83959 12 h later, and tested at 7 days showed a higher score when compared with the other groups (Newman–Keuls, *p<0.05). Asterisks upon bracket indicate a significant difference between saline- and cocaine-injected animals, collapsed across intracerebral infusion treatments (Newman–Keuls, **p<0.01). (c–e) Dorsal hippocampal levels of BDNF of animals administered either saline or 20 mg/kg cocaine. Animals were injected and returned to their home cages (HC groups) or were conditioned as described (C groups). In all the figures, control group is represented by animals injected with saline and left undisturbed in their home cages (Sal/HC groups). (c) Levels of dorsal hippocampal BDNF measured at 12 h after one-trial training. BDNF levels did not vary across treatments (n=5 per group). (d) Levels of dorsal hippocampal BDNF measured at 12 h after three-trial training. BDNF levels did not vary across treatments (n=3–4 per group). (e) SCH 23390 administration did not modify the dorsal hippocampal levels of BDNF measured at 12 h after one-trial training. Animals were conditioned with either saline or cocaine; 11.5 h later they were infused with SCH 23390 or its vehicle and were killed 30 min later (n=15 per group). Coc, cocaine; Sal, saline; Veh, vehicle.
To further investigate the hypothesis that persistent cocaine-associated memories depend on D1Rs not coupled to AC, we measured the expression of BDNF, a downstream component of D1Rs/AC/PKA/BDNF/ERK pathway known to be elevated in the dorsal hippocampus 12 h after training in long-lasting fear memories (Bekinschtein et al, 2007; Rossato et al, 2009). When BDNF levels were measured 12 h after training, no differences were observed either in animals subjected to the one-trial (Figure 3c, main effect of cocaine: F(1,16)=0.88, p=0.36, NS; main effect of place: F(1,16)=0.24, p=0.63, NS; cocaine × place: F(1,16)=0.26, p=0.61, NS) or to the three-trial (Figure 3d, main effect of cocaine: F(1,10)=0.0003, p=0.99, NS; main effect of place: F(1,10)=2.15, p=0.17, NS; cocaine × place: F(1,10)=0.89, p=0.37, NS) training. Furthermore, BDNF levels were not affected by dorsal hippocampal injections of the D1R antagonist SCH 23390, a treatment that improved the persistence of a weak cocaine-related memory (Figure 3e, main effect of group: F(2,42)=0.99, p=0.38, NS). In addition, no alterations were observed in the phosphorylation state of TrkB, or in the activation of ERK1/2, which are downstream steps of BDNF-induced memory persistence in the case of fear memories (Supplementary Figure S5).
It was not expected that dopamine agonists would inhibit the late-phase of cocaine-associated memory consolidation because dopamine is known to be necessary for promoting the late-phase of aversive memory consolidation in a step-down avoidance task (Rossato et al, 2009). To determine the role of dorsal hippocampal dopamine in an avoidance context, we substituted the rewarding agent cocaine by an aversive agent. Here we used LiCl, an aversive agent known to be capable of establishing taste and place avoidance (Nachman, 1963; Tenk et al, 2005) as our unconditioned stimulus. In our place-conditioning paradigm, it had the opposite effect of cocaine; where rats increased the time they spent in the cocaine-associated chamber, other independent group of rats decreased the time spent in the LiCl-associated chamber. This effect was seen 24 h but not 7 days after training (Supplementary Figure S6; p<0.05). Bilateral dorsal hippocampal infusions of the D1R agonist SKF 38393 given 12 h after training facilitated the persistence of the LiCl-associated memory, which remained strong 7 days after training (Figure 4, main effect of LiCl: F(1,30)=5.98, p<0.05; main effect of challenge: F(1,30)=13.81, p<0.001; LiCl × challenge: F(1,30)=4.55, p<0.05). Thus, late post-training activation of hippocampal D1Rs had opposite effects on the persistence of the memory for learned cocaine–context and LiCl–context associations, increasing the persistence of the memory for the association between LiCl and the place where it was given and decreasing the persistence of the memory for the association between cocaine and the place where it was given.

Effects of D1-type dopamine agonist (SKF 38393) on the persistence of the memory for one-trial LiCl–place associations. Infusion of the D1-type agonist (SKF 38393) enhanced the retention of the 7-day memory for the place associations induced by LiCl (n=7–9). Asterisk indicates significant post hoc comparisons (Newman–Keuls, *p<0.05). LiCl, lithium chloride; Sal, saline; Veh, vehicle.
The dopamine-dependent potentiation of late-phase memory consolidation through PLC-coupled D1Rs might require only normal baseline levels of hippocampal dopamine. To determine if there are normal or fluctuating levels of dopamine at the time when dopamine inhibits late-phase memory consolidation for cocaine–place associations, we used microdialysis to monitor dorsal hippocampal dopamine levels over the 24 h after cocaine treatment. Here, we used the one-trial paradigm. After a large but variable (and thus statistically unreliable) surge following the training injection of cocaine, dopamine levels returned to a stable baseline within about 5 h, but increased again for a period of 4 h starting 13 h after the training session (Figure 5). The increase began 3 h after the onset of the light phase of the light–dark cycle, at a time when striatal dopamine levels normally become depressed (Castañeda et al, 2004); thus, it was not obviously time-locked to the light change. The cocaine-injected animals showed a reliable increase in approach to the cocaine-associated chamber when tested 4 h after the completion of the microdialysis experiments (Supplementary Figure S7; p<0.05).

Dopamine (DA) fluctuations in microdialysis samples from the dorsal hippocampus of animals following one-trial training for cocaine–place associations. Cocaine or saline was given at the arrow after the baseline period (B) and hourly samples were taken for the next 24 h. The inset shows the results of four baseline samples pooled together. There were no significant differences in baseline between the two groups (n=7 per group; Student’s t-test, t(12)=−0.31; p=0.76). There were no significant differences in DA measurements between groups during the first 12-h period of the experiment (main effect of cocaine: F(1,12)=1.72; p=0.21, NS; main effect of time: F(11,132)=1.30; p=0.23, NS; cocaine × time interaction: F(11,132)=0.69; p=0.74, NS). However, during the second 12-h period, there was a significant main effect of time, F(11,132)=2.04; p=0.03 and a significant cocaine × time interaction, F(11,132)=4.17; p=0.00003. Asterisks indicate significant post hoc comparisons (Newman–Keuls, **p<0.01; ***p<0.001). The horizontal bar in the bottom of the figure indicates the dark–light cycle (black: lights out).
DISCUSSION
The present findings have two important implications. First, we report for the first time a role for dopamine in the dorsal hippocampus on the long-term storage for a cocaine-associated memory, an influence on memory storage that develops long after the elevation in dopamine levels caused by cocaine itself. Second, we report opposite effects of hippocampal dopamine actions on the memory for the association of neutral environmental place cues with the rewarding drug cocaine on the one hand or the memory for the association of the environmental place cues with the aversive drug LiCl on the other; stimulating dorsal hippocampal D1-type dopamine receptors decreased the persistence of the memory for a cocaine–place association, but increased the persistence of LiCl–place memory.
The persistence of a weak cocaine-conditioned memory was enhanced by blocking dorsal hippocampus D1Rs 12 h after conditioning; it was attenuated by stimulating these receptors. The effects were not due to enhancing or blocking the impact of the acute extracellular dopamine elevation resulting from cocaine’s blockade of the dopamine transporter, as dopamine levels had returned to normal within 5–6 h of cocaine injection. However, there was a 4-h surge in extracellular dopamine concentration that began about 13 h after the cocaine injection, and there was neither a pharmacological treatment nor a change in environmental stimulus conditions at that time. This surge in dopamine levels was presumably caused by an increase in dopamine cells firing in the VTA, and the cause of such an increase is a matter for future investigation. That microinjection of D1R agonists attenuated retention of the cocaine-associated memory suggests that the increase in dopamine 13 h after cocaine place conditioning is itself a limiting factor in the persistence of cocaine-associated memories.
This observation is surprising in that it is inconsistent with what is known of the role of dorsal hippocampal dopamine in late-phase consolidation of instrumental avoidance memories (Rossato et al, 2009). Indeed, the effects of dopamine receptor manipulations in our task were opposite to those previously reported for a step-down avoidance task; in the avoidance task, dopamine agonists administered to the dorsal hippocampus 12 h (but not 0 or 9 h) after conditioning promoted late-stage memory consolidation, whereas dopamine antagonists restricted it (Rossato et al, 2009). To be sure that our finding was not unique to our place-conditioning paradigm, we confirmed that a D1R agonist facilitates late-phase avoidance memory persistence in a paradigm similar to ours, but using an aversive rather than a rewarding stimulus. Our LiCl conditioning task involved the same equipment and protocol as was used for cocaine conditioning, but using the aversive agent LiCl rather than the appetitive agent cocaine, as the unconditioned stimulus. Our results thus suggest opposite roles for dorsal hippocampal dopamine in the late phase of LTM storage of memories associated with rewarding and aversive stimuli.
The obtained results suggest that the dopaminergic agents used involved actions at a given subtype of the D1-like dopamine receptors family. D1-like receptors are subdivided into D1 and D5 subtypes (Monsma et al, 1990; Sunahara et al, 1991), each of which is expressed in hippocampal neurons. The D5 subtype, presumably the one subtype coupled to PLC (Sahu et al, 2009; Undieh, 2010), is densely expressed in the hippocampus (Laurier et al, 1994) and has a 10-fold higher affinity for DA than does the D1 subtype (Sunahara et al, 1991). We found that SKF 83959 (thought to act specifically at the D5 receptor because it induces the selective activation of PLC intracellular cascade; Undie et al, 1994) had the inhibitory effect we saw with the more generic agonist, whereas SKF 83822, which induces activation of the AC intracellular cascade (O’Sullivan et al, 2004), failed to do so. These data suggest that the D5 subtype is important for modulating the persistence of contextual memories associated with the rewarding drug cocaine, whereas the D1 subtype is important for modulating the persistence of contextual memories associated with the aversive agent LiCl (present study), along with those associated with the aversive effects of painful footshock (Rossato et al, 2009). Consistent with this suggestion, the levels of BDNF and the activation of TrkB and ERK1/2, which are downstream effectors of D1Rs coupled to AC in controlling the persistence of an aversive memory (Rossato et al, 2009), were unchanged 12 h after cocaine-context conditioning.
These and earlier studies involving step-down avoidance (Rossato et al, 2009) make it clear that dopamine-dependent events taking place in the dorsal hippocampus around 12 h after a learning experience can enhance or limit the persistence of the memory for that experience (Supplementary Figure S8). One possibility is that dopamine is enhancing or inhibiting the continuing process of long-term storage of the original memory for the learning event. Another possibility to consider is whether dopamine is enhancing or inhibiting the reconsolidation of a reactivated trace. Consistent with this latter view is that the dopamine system, at least, is clearly reactivated at about this time. Against that view, however, are several facts. First, there is no obvious source of reactivation of the initial memory; the animals were not exposed to the contextual stimuli associated with the learning experience until they were tested 24 h or more hours later. Second, the effects of dopamine manipulations were time-locked to the original training; when given in the same way with the same attendant handling and lab activity at earlier times, the dopamine manipulations are ineffective (Rossato et al, 2009). Also seemingly inconsistent with any hypothesis involving spontaneous reactivation and reconsolidation during subsequent sleep is the fact that D1R activation had opposite effects on the duration of memories for place associations with rewarding and aversive stimuli.
The fact that the memory for a cocaine–place association is still labile 12 h after the event has potential implications for addiction therapy. Addicts who continue taking drug even after becoming tolerant to them often report that they are ‘chasing the remembered high’; thus, the memory for an early drug experience can have a significant role in subsequent drug-seeking. The fact that cocaine-associated memories are still labile and that their persistence can still be affected several hours after the experience should encourage recent suggestions (Mihindou et al, 2011; Xue et al, 2012; Wang et al, 2013) that the memory traces of drug experience itself may offer useful new targets for addiction treatment.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua RL, Izquierdo I, Medina JH (2007). Persistence of long-term memory storage requires a late protein synthesis- and BDNF-dependent phase in the hippocampus. Neuron 53: 261–277.
Berke JD, Hyman SE (2000). Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25: 515–532.
Bonci A, Malenka RC (1999). Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci 19: 3723–3730.
Busse GD, Riley AL (2004). Cocaine, but not alcohol, reinstates cocaine-induced place preferences. Pharmacol Biochem Behav 78: 827–833.
Caillé S, Guillem K, Cador M, Manzoni O, Georges F (2009). Voluntary nicotine consumption triggers in vivo potentiation of cortical excitatory drives to midbrain dopaminergic neurons. J Neurosci 29: 10410–10415.
Castañeda TR, de Prado BM, Prieto D, Mora F (2004). Circadian rhythms of dopamine, glutamate and GABA in the striatum and nucleus accumbens of the awake rat: modulation by light. J Pineal Res 36: 177–185.
Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM et al (2008). Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59: 288–897.
Crooks KR, Kleven DT, Rodriguiz RM, Wetsel WC, McNamara JO (2010). TrkB signaling is requiered for behavioral sensitization and conditioned place preference induced by a single injection of cocaine. Neuropharmacology 58: 1067–1077.
dela Cruz AM, Herin DV, Grady JJ, Cunningham KA (2009). Novel approach to data analysis in cocaine conditioned place preference. Behav Pharmacol 20: 720–730.
Everitt BJ, Robbins TW (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8: 1481–1489.
Hyman SE, Malenka RC, Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598.
Ikemoto S (2003). Involvement of the olfactory tubercle in cocaine reward: intracranial self-administration studies. J Neurosci 23: 9305–9511.
Ikemoto S, Goeders NE (1998). Microinjections of dopamine agonists and cocaine elevate plasma corticosterone: dissociation effects among the ventral and dorsal striatum and medial prefrontal cortex. Brain Res 81: 171–178.
Kalivas PW (2009). The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10: 561–572.
Laurier LG, O’Dowd BF, George SR (1994). Heterogeneous tissue-specific transcription of dopamine receptor subtype messenger RNA in rat brain. Brain Res Mol Brain Res 25: 344–350.
Lisman JE, Grace AA (2005). The hippocampal–VTA loop: controlling the entry of information into long-term memory. Neuron 46: 703–713.
Luo AH, Tahsili-Fahadan P, Wise RA, Lupica CR, Aston-Jones G (2011). Linking context with reward: a functional circuit from hippocampal CA3 to ventral tegmental area. Science 333: 353–357.
Lüscher C, Malenka RC (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69: 650–663.
Mihindou C, Vouillac C, Koob GF, Ahmed SH (2011). Preclinical validation of a novel cocaine exposure therapy for relapse prevention. Biol Psychiatry 70: 593–598.
Monsma FR Jr, Mahan LC, McVittie LD, Gerfen CR, Sibley DR (1990). Molecular cloning and expression of a D1 dopamine receptor linked to adenylyl cyclase activation. Proc Natl Acad Sci USA 87: 6723–6727.
Nachman M (1963). Learned aversion to the taste of lithium chloride and generalization to other salts. J Comp Physiol Psychol 56: 343–349.
National Research Council (2011) Guide for the Care and Use of Laboratory Animals 8th edn The National Academies Press: Washington, DC.
Nestler EJ (1992). Molecular mechanisms of drug addiction. J Neurosci 12: 2439–2450.
O'Keefe J, Nadel L (1978) The Hippocampus as a Cognitive Map. Oxford University Press: Oxford, UK.
O’Sullivan GJ, Roth BL, Kinsella A, Waddington JL (2004). SK&F 83822 distinguishes adenylyl cyclase from phospholipase C-coupled dopamine D1-like receptors: behavioural topography. Eur J Pharmacol 486: 273–280.
Paxinos G, Watson C (2004) The Rat Brain in Stereotaxic Coordinates 5th edn Elsevier Academic Press: San Diego, CA.
Pettit HO, Pan HT, Parsons LH, Justice JB Jr (1990). Extracellular concentrations of cocaine and dopamine are enhanced during chronic cocaine administration. J Neurochem 55: 798–804.
Phillips RG, LeDoux JE (1994). Lesions of the dorsal hippocampal formation interfere with background but not foreground contextual fear conditioning. Learn Mem 1: 34–44.
Rossato JI, Bevilaqua LR, Izquierdo I, Medina JH, Cammarota M (2009). Dopamine controls persistence of long-term memory storage. Science 325: 1017–1020.
Saal D, Dong Y, Bonci A, Malenka RC (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582.
Sahu A, Tyeryar KR, Vongtau HO, Sibley DR, Undie AS (2009). D5 dopamine receptors are required for dopaminergic activation of phospholipase C. Mol Pharmacol 75: 447–453.
Sunahara RK, Guan HC, O’Dowd BF, Seeman P, Laurier LG, Ng G et al (1991). Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 350: 614–619.
Tenk CM, Kavaliers M, Ossenkopp KP (2005). Dose response effects of lithium chloride on conditioned place aversions and locomotor activity in rats. Eur J Pharmacol 515: 117–127.
Undie AS, Weinstock J, Sarau HM, Friedman E (1994). Evidence for a distinct D1-like dopamine receptor that couples to activation of phosphoinositide metabolism in brain. J Neurochem 62: 2045–2048.
Undieh AS (2010). Pharmacology of signaling induced by dopamine D1-like receptor activation. Pharmacol Ther 128: 37–60.
Wang B, Shaham Y, Zitzman D, Azari S, Wise RA, You ZB (2005). Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci 25: 5389–5396.
Wang B, You ZB, Oleson EB, Cheer JF, Myal S, Wise RA (2013). Conditioned contribution of peripheral cocaine actions to cocaine reward and cocaine-seeking. Neuropsychopharmacology 38: 1763–1769.
Wise RA, Newton P, Leeb K, Burnette B, Pocock D, Justice JB Jr (1995). Fluctuations in nucleus accumbens dopamine concentration during intravenous cocaine self-administration in rats. Psychopharmacology 120: 10–20.
Xue YX, Luo YX, Wu P, Shi HS, Xue LF, Chen C et al (2012). A memory retrieval–extinction procedure to prevent drug craving and relapse. Science 336: 241–245.
You ZB, Wang B, Zitzman D, Azari S, Wise RA (2007). A role for conditioned ventral tegmental glutamate release in cocaine seeking. J Neurosci 27: 10546–10555.
You ZB, Wang B, Zitzman D, Wise RA (2008). Acetylcholine release in the mesocorticolimbic dopamine system during cocaine seeking: conditioned and unconditioned contributions to reward and motivation. J Neurosci 28: 9021–9029.
Acknowledgements
We gratefully acknowledge Dr Gustavo Murer and Verardo y Cía. laboratory for their kind donation of quinpirole and cocaine hydrochloride, respectively. We thank Catherine Schweppe, Fernando Garagoli, Eduardo Gigante, and Carolina González for excellent technical help. CP Kramar was supported by a doctoral fellowship from CONICET, Argentina. This work was supported by CONICET, UBA, Foncyt (Grants PICT 2010-1169 to Dr Medina and PICT 2011-1941 to Dr Barbano) and by the Intramural Research Program of the National Institute on Drug Abuse (NIDA/NIH).
Authors contribution
CPK and MFB carried out the behavioral experiments. MFB conducted the microdialysis experiments. VIC performed the biochemical analyses of the microdialysis samples. CPK carried out the western blot assays. JHM and MFB designed the study with CPK and RAW. MFB, RAW, and JHM wrote the manuscript with the help of CPK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article
Cite this article
Kramar, C., Chefer, V., Wise, R. et al. Dopamine in the Dorsal Hippocampus Impairs the Late Consolidation of Cocaine-Associated Memory. Neuropsychopharmacol 39, 1645–1653 (2014). https://doi.org/10.1038/npp.2014.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.11
Keywords
This article is cited by
-
Time-dependent inhibition of Rac1 in the VTA enhances long-term aversive memory: implications in active forgetting mechanisms
Scientific Reports (2023)
-
Memory enhancing effects of nicotine, cocaine, and their conditioned stimuli; effects of beta-adrenergic and dopamine D2 receptor antagonists
Psychopharmacology (2021)
-
Involvement of Hippocampal D1-Like Dopamine Receptors in the Inhibitory Effect of Cannabidiol on Acquisition and Expression of Methamphetamine-Induced Conditioned Place Preference
Neurochemical Research (2021)
-
Molecular and circuit mechanisms regulating cocaine memory
Cellular and Molecular Life Sciences (2020)
-
Dopamine Neurotransmission in the Ventral Tegmental Area Promotes Active Forgetting of Cocaine-Associated Memory
Molecular Neurobiology (2019)
