
Abstract
Evaluate lisdexamfetamine dimesylate (LDX) augmentation of antidepressant monotherapy for executive dysfunction in partially or fully remitted major depressive disorder (MDD). This randomized, placebo-controlled study (NCT00985725) enrolled 143 adults (18–55 years) with mild MDD (Montgomery-Åsberg Depression Rating Scale (MADRS) score ⩽18) and executive dysfunction (Behavior Rating Inventory of Executive Function-Adult Version (BRIEF-A) Self-Report Global Executive Composite (GEC) T score ⩾60) on stable antidepressant monotherapy for ⩾8 weeks. After 2 weeks of screening, participants were randomized to 9 weeks of double-blind LDX (20–70 mg/day) or placebo augmentation, followed by 2 weeks of single-blind placebo. The primary end point was change from baseline to week 9/end of study (EOS) in BRIEF-A Self-Report GEC T score; secondary assessments included the BRIEF-A Informant Report, MADRS, and treatment-emergent adverse events (TEAEs). Of 143 randomized participants, 119 completed double-blind treatment (placebo, n=59; LDX, n=60). Mean±standard deviation (SD) BRIEF-A GEC T scores decreased from baseline (placebo, 74.2±8.88; LDX, 76.8±9.66) to week 9/EOS (placebo, 61.4±14.61; LDX, 55.2±16.15); the LS mean (95% CI) treatment difference significantly favored LDX (−8.0 (−12.7, −3.3); P=0.0009). The LS mean (95% CI) treatment difference for MADRS total score also significantly favored LDX (−1.9 (−3.7, 0.0); P=0.0465). TEAE rates were 73.6% with placebo and 78.9% with LDX; serious TEAE rates were 4.2 and 2.8%. In this trial, LDX augmentation significantly improved executive dysfunction and depressive symptoms in participants with mild MDD. The safety profile of LDX was consistent with prior studies in adults with attention-deficit/hyperactivity disorder.
Similar content being viewed by others

INTRODUCTION
Major depressive disorder (MDD) is often chronic and/or recurrent (Kupfer, 2005); the estimated lifetime prevalence is 16% (Kessler et al, 2003). Antidepressant pharmacotherapy does not always result in remission. The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial reported remission rates of 37, 31, 14, and 13% after successive treatments in individuals with non-psychotic MDD using the 16-item Quick Inventory of Depressive Symptomatology (QIDS)-Self-Report (Rush et al, 2006). Even when depressive symptoms abate, cognitive problems may persist (Preiss et al, 2009; Reppermund et al, 2009), compromising an individual’s coping ability (Gualtieri et al, 2006) and likelihood of successfully returning to work (Reppermund et al, 2009).
In the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition Text Revision, several of the criteria for MDD describe symptoms related to cognition, including changes in psychomotor activity, diminished ability to think or concentrate, and indecisiveness (American Psychiatric Association, 2000). Additionally, a recent meta-analysis noted that individuals with MDD exhibit non-specific cognitive deficits related to difficulty planning and slower processing speed (Snyder, 2013). Furthermore, several reports note that attention, memory, and executive function are commonly impaired in individuals with MDD (Gualtieri et al, 2006; Marazziti et al, 2010; Preiss et al, 2009; Reppermund et al, 2009), as is psychomotor function (Gualtieri et al, 2006; Preiss et al, 2009).
Of particular interest in MDD are executive function deficits (Reppermund et al, 2009), which comprise higher-order functions such as problem solving, goal setting, planning, and sequencing actions (Elliott, 2003), because it creates significant problems in coping with life events (Marazziti et al, 2010). Executive dysfunction is linked to disrupted prefrontal-subcortical circuitry (Rogers et al, 2004). Norepinephrine (NE) and dopamine (DA) are critical to maintain executive functions mediated by the prefrontal cortex (Arnsten and Li, 2005); thus, agents that modulate these systems may ameliorate executive dysfunction in MDD. Stimulants, such as those used to treat attention-deficit/hyperactivity disorder (ADHD), preferentially modulate central DA and, to a lesser extent, NE systems (Arnsten, 2006).
Lisdexamfetamine dimesylate (LDX), a pharmacologically inactive prodrug of D-amphetamine, is approved for treatment of ADHD in children, adolescents, and adults (Vyvanse (lisdexamfetamine dimesylate), 2012). In a small clinical study assessing LDX augmentation in individuals with residual depressive symptoms after escitalopram monotherapy (Harvey et al, 2012), LDX was superior to placebo in reducing depressive symptoms and self-reported executive dysfunction on the Behavior Rating Inventory of Executive Function-Adult Version (BRIEF-A), a predefined secondary end point.
The BRIEF-A is a psychometrically validated executive function assessment tool for adults (Roth et al, 2005), which demonstrates strong convergent validity with other executive function assessment instruments (ie, the Dysexecutive Questionnaire and the Frontal Systems Behavior Scale) (Roth et al, 2013) and modest correlations with standard neuropsychologic tests (Rabin et al, 2006). However, there has been debate over whether these two types of tools measure the same processes. It has been suggested that performance measures objectively assess cognitive skills and rating scales subjectively assess the application of those skills in everyday life function, the latter supported by evidence that executive function rating scales correlate with real-world measures of function such as academic performance (Isquith et al, 2013).
The current study included individuals reporting executive dysfunction on the BRIEF-A despite partially or fully remitted MDD (Montgomery-Åsberg Depression Rating Scale (MADRS) ⩽18) at screening and baseline. The primary objective was to evaluate the efficacy of LDX augmentation of selective serotonin reuptake inhibitor (SSRI) monotherapy for treating executive dysfunction. The primary end point was change in executive functioning on the BRIEF-A Self-Report form; secondary cognitive end points included executive functioning as observed by a knowledgeable informant (using the BRIEF-A Informant Report form), and objective cognitive functioning as assessed using the CNS Vital Signs computerized testing battery.
The decision to use the BRIEF-A Self-Report was partially driven by the recognition that demonstrations of improved cognitive function on laboratory-based measures have limited practical meaning if patients are unaware of improvement in their everyday lives. Thus, a self-report provides some degree of ecological validity (an approximation of the real-world experience) (Isquith et al, 2013), with objective end points (ie, CNS Vital Signs data) placing the subjective data in perspective. In this regard, it should be noted that the STAR*D study (Rush et al, 2006) used remission based on the QIDS Self-Report as a primary end point. Additionally, since BRIEF-A was included as a secondary end point in a separate LDX augmentation study of individuals with MDD who had not achieved remission on escitalopram monotherapy (Harvey et al, 2012), inclusion of the BRIEF-A in the current study provides data that will complement previously reported findings in a distinct population of individuals with MDD.
PATIENTS AND METHODS
Study Design
This randomized, double-blind, placebo-controlled, parallel-group, multicenter study was conducted from 29 October 2009 to 18 April 2011 at 27 US clinical research sites (clinicaltrials.gov registration: NCT00985725). The protocol was approved by institutional review boards and/or independent ethics committees at each clinic.
Participants
Eligible participants, individuals with self-reported executive dysfunction despite full or partial remission of depressive symptoms in MDD, were recruited across study sites via advertisements and from private practices of investigators. Participants included men and non-pregnant women (18–55 years) who, for ⩾2 years before screening, maintained a primary diagnosis of recurrent, non-psychotic MDD by Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition-Text Revision (DSM-IV-TR) criteria, confirmed by the Structured Clinical Interview for DSM-IV-TR disorders–Axis I. Eligible participants also had a BRIEF-A Self-Report GEC T score of ⩾60 (1 standard deviation (SD) above the normative mean) and MADRS total score ⩽18 at screening and baseline. All participants had maintained SSRI monotherapy (citalopram, escitalopram, fluoxetine, paroxetine, or sertraline) for ⩾8 weeks at a stabilized therapeutic dosage (ie, SSRI dosage did not vary by more than ±10% during a 4-week period before augmentation baseline); SSRI usage continued during the course of the study.
Key exclusion criteria included comorbid psychiatric disorders controlled with prohibited medications (stimulants or stimulant-like agents, cholinesterase inhibitors, N-methyl-D-aspartic acid receptor modulators, pramipexole, modafinil, bupropion, mirtazapine, monoamine oxidase inhibitors, serotonin-NE reuptake inhibitors, tricyclic antidepressants, mood stabilizers, antipsychotics, and narcotics) or uncontrolled and significantly symptomatic; history of psychotic symptoms; symptoms that contraindicated LDX treatment or could confound efficacy or safety assessments; previous ADHD diagnosis, treatment for ADHD, or fulfillment of DSM-IV-TR criteria before the age of 7 in a retrospective review based on participant interviews; first-degree relative with bipolar I disorder; suicidality (current risk or prior attempt within 3 years); history of non-response of depressive symptoms to a stimulant or hypersensitivity to stimulants; history of symptomatic cardiovascular disease or other serious cardiac problems; or a recent history of substance abuse (⩽6 months). All participants received a complete study description and provided written informed consent in accordance with Good Clinical Practice guidelines.
Treatment
After a 2-week screening period while participants remained on antidepressant medication, participants entered a double-blind phase and were randomized to placebo or LDX augmentation in a 1 : 1 ratio for 9 weeks. Randomization numbers and treatments were assigned using an interactive voice response system/interactive Web response system, which investigators accessed to obtain bottle numbers for double-blind treatment. Double-blind LDX treatment was initiated at 20 mg/day and, if tolerated, increased to 30 mg/day at the start of week 1. Participants continued to maintain or increase their dose in 10-mg increments at weekly intervals (maximum, 70 mg/day), depending on tolerability, until week 6 (the dose optimization period). During weeks 7–9, participants maintained their optimized dose (the dose maintenance period). All participants received single-blind placebo for 2 weeks upon completion of double-blind treatment, with participants remaining blinded to treatment, to determine whether cessation of LDX treatment was associated with safety or tolerability concerns. Compliance with treatment was assessed as (the total number of capsules taken × 100)/total number of dosing days; lost medication was recorded as taken.
Efficacy Assessments
The primary efficacy end point was change from baseline to week 9/EOS in BRIEF-A GEC Self-Report T score. The BRIEF-A (Roth et al, 2005) assesses aspects of executive function manifested in everyday life during the past month; it has been used in healthy and clinical populations (Garlinghouse et al, 2010; Rabin et al, 2011; Rabin et al, 2006). The BRIEF-A includes 75 items scored on 3-point Likert scales, with higher scores indicating worse executive function; both Self-Report and Informant Reports are available. The BRIEF-A provides an overall score (GEC) composed of the Behavioral Regulation Index (BRI) and Metacognition Index (MI). The BRI has four clinical scales (Inhibit, Shift, Emotional Control, and Self Monitor); the MI has five (Initiate, Working Memory, Plan/Organize, Task Monitor, and Organization of Materials). Executive dysfunction on the BRIEF-A is reported as a T score; T scores are linear transformations of the raw scores that normalize them with respect to a standardization sample, for which the mean T score is 50 (SD, 10) (Roth et al, 2005). Cronbach alpha coefficients for the clinical scales range from 0.73 to 0.90; 1-month test–retest reliabilities range from 0.82 to 0.93 (Roth et al, 2005). The current report focuses on changes in Self-Report GEC T scores. The mean percentage change from baseline to week 9/EOS in BRIEF-A Self-Report GEC T score was also calculated.
Secondary cognitive end points included changes from baseline to week 9/EOS in BRIEF-A Informant Report GEC T score, which complemented the Self-Report, and in the performance-based Neurocognitive Index (NCI) derived from the validated, computerized CNS Vital Signs testing battery. The CNS Vital Signs comprise seven neuropsychological tests (Gualtieri and Johnson, 2006). The NCI incorporates verbal, visual, working, and composite memory; sustained and complex attention; cognitive flexibility; executive function; processing speed; and reaction time (higher scores reflect better performance).
Raters received training in the use of all assessment instruments at an investigators meeting; follow-up training was provided as needed.
Secondary efficacy end points included MADRS total score changes and Clinical Global Impression-Severity (CGI-S) and -Improvement (CGI-I) response frequencies (categorical scores) from baseline to week 9/EOS. The MADRS is a validated 10-item questionnaire, with each item rated from 0 (best) to 6 (worst); overall score ranges from 0 (normal) to 60 (very severe depression) (Montgomery and Asberg, 1979). The CGI-S and CGI-I provide global evaluations of disease severity and improvement over time; both measures are rated from 1 (best) to 7 (worst) (Guy, 1976).
Changes in the 7-item Generalized Anxiety Disorder Scale (Spitzer et al, 2006), Short Form-12 Health Survey (Ware et al, 1996), Quality of Life Enjoyment and Satisfaction Questionnaire (Endicott et al, 1993), Endicott Work Productivity Scale (Endicott and Nee, 1997), and a more detailed presentation of CNS Vital Sign cognitive test battery changes will be described elsewhere.
Safety Assessments
Safety end points included treatment-emergent adverse events (TEAEs), which were recorded throughout the study and categorized by severity and relationship to treatment. TEAEs of special interest included psychiatric (psychosis/mania, suicide, aggression, or other) and non-psychiatric (weight, clinical laboratory measures, vital signs, and sexual dysfunction) events. Participants also received physical examinations (screening and week 9/EOS), vital sign assessments at each visit, clinical laboratory evaluations (screening, week 6, and week 9/EOS), and electrocardiograms (screening, baseline, and week 9/EOS).
Other safety evaluations assessed withdrawal, suicidality, and sexual functioning. The Amphetamine Cessation Symptom Assessment (ACSA; measured at baseline and weeks 10–11 (single-blind stage)) is a validated self-report with 16 items rated from 0 (not at all) to 4 (extremely) (McGregor et al, 2008). The Sheehan Suicidality Tracking Scale (STS; monitored at each visit) is a prospective scale that assesses suicidal ideation and behaviors, with 8 items rated from 0 (not at all) to 4 (extremely) (Coric et al, 2009). The Changes in Sexual Functioning Questionnaire Short-Form (CSFQ-14; measured at baseline, week 6, and end point) is a validated 14-item instrument rated on a 5-point scale, with higher scores indicating higher levels of sexual functioning (1 (never) to 5 (every day) for 12 items; 1 (every day) to 5 (never) for 2 items) (Keller et al, 2006).
Statistical Analysis
Statistical analysis was performed using SAS® Version 9.2 (SAS Institute, Cary, NC, USA). Efficacy analyses were based on the full analysis set (FAS: all randomized participants who took ⩾1 study drug dose and had ⩾1 post-baseline BRIEF-A assessment). The primary analysis used analysis of covariance (ANCOVA) assessing group differences in the LS mean change in BRIEF-A Self-Report GEC T score from baseline to week 9/EOS, with treatment as a factor and baseline score as a covariate. Last observation carried forward (LOCF) was used to account for missing data; statistical significance was set at two-sided P<0.05. Assuming an effect size of 0.56 for the LDX vs placebo comparison, a two-sided, two-sample t-test needed ⩾52 participants/group to provide 80% power. Secondary efficacy end points were assessed using ANCOVA (MADRS total score) or the Cochran–Mantel–Haenszel test stratified by baseline CGI-S score (CGI-I). The distribution of standardized CNS Vital Signs scores was assessed using Wilcoxon rank sum tests. Post hoc assessment of between-group differences in demographic and clinical characteristics at baseline used t-tests for continuous variables and Chi-square tests for categorical variables. Two post hoc regression models, based on ANCOVA with MADRS total score change as a dependent variable and treatment as an effect, assessed the independence of cognitive and depression symptom changes: one employed BRIEF-A GEC T score change as a covariate and the other employed CNS Vital Signs NCI score change as a covariate. No multiplicity adjustments were performed for efficacy end point analyses.
Safety end points were assessed using the safety analysis set (all randomized participants who took ⩾1 dose of study drug and who had ⩾1 follow-up safety assessment) and are presented descriptively.
RESULTS
Disposition, Demographics, and Drug Exposure
Participant disposition is summarized in Figure 1. All 143 enrolled participants were randomized (placebo, n=72; LDX, n=71) and included in the FAS and safety analysis sets; 119 (placebo, n=59; LDX, n=60) completed 9 weeks of double-blind treatment. Of the 24 participants (placebo, n=13; LDX, n=11) not completing the study, the reasons cited were participant withdrawal (placebo, n=4; LDX, n=4), protocol violation (placebo, n=3; LDX, n=1), other (placebo, n=3; LDX, n=1), adverse events (placebo, n=1; LDX, n=4), lost to follow-up (placebo, n=1; LDX, n=1), and lack of efficacy (placebo, n=1).

Participant disposition. LDX, lisdexamfetamine dimesylate.
With the exception of average citalopram dosage, which was significantly lower in participants randomized to LDX, demographic and primary clinical variables (BRIEF-A Self-Report GEC T score and MADRS total score) did not differ significantly between treatment groups at baseline (Table 1). Mean±SD age was 40.7±10.23 years; the majority were white and female. Executive dysfunction and depressive symptom levels, as measured by BRIEF-A Self-Report GEC T scores and MADRS scores, respectively, did not differ between groups. SSRI usage between treatment groups before double-blind augmentation is summarized in Table 1.
A majority of participants randomized to LDX were receiving doses between 50 and 70 mg/day at the end of dose optimization (week 6; 49/68) and at week 9 (45/63). Mean±SD optimized LDX doses were 53.2±17.42 mg/day and 53.2±16.76 mg/day at weeks 6 and 9, respectively. Compliance during double-blind treatment ranged from 80 to 120% for a majority of participants (placebo, 68/72 (94.4%); LDX, 69/71 (97.2%)).
Efficacy
Primary end point
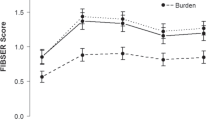
At baseline, mean±SD BRIEF-A Self-Report GEC T scores were 74.2±8.88 and 76.8±9.66 with placebo and LDX, respectively; at week 9/EOS, T scores decreased to 61.4±14.61 (mean±SD reduction: −12.8±13.89) and 55.2±16.15 (mean±SD reduction: −21.6±14.55), respectively (Figure 2, inset). LS mean (95% CI) reductions in BRIEF-A Self-Report GEC T score are depicted in Figure 2. The primary efficacy analysis showed LS mean (95% CI) GEC T score reductions of −13.2 (−16.5, −9.9) for placebo and −21.2 (−24.5, −17.9) for LDX at week 9/EOS; the LS mean (95% CI) treatment difference statistically favored LDX (−8.0 (−12.7, −3.3); |t140|=3.38, P=0.0009). The ANCOVA model-based effect size (95% CI) at week 9/EOS (LDX—placebo) was −0.6 (−0.9, −0.2).

LS mean (95% CI) change from baseline in (a) BRIEF-A Self-Report GEC T score and (b) Informant Report GEC T score; full analysis set, last observation carried forward. *P<0.05 vs placebo; †P<0.01 vs placebo; ‡P<0.001 vs placebo. INSETS: Mean (standard deviation) values at baseline and week 9/EOS; the horizontal line indicates a GEC T score of 60. BRIEF-A, Behavior Rating Inventory of Executive Function-Adult Version; CI, confidence interval; EOS, end of study; GEC, Global Executive Composite; LDX, lisdexamfetamine dimesylate; LS, least square.
Secondary end points
BRIEF-A Self-Report. Statistically significant LS mean treatment differences between placebo and LDX were observed at weeks 5–9 (all |t140|>1.9771=t0.025,140; all P<0.05, Figure 2). From baseline to week 9/EOS, the mean±SD percentage change in GEC T score was −17.0±18.09% with placebo and −28.1±18.07% with LDX. Using the linear regression model for the MADRS total score changes from baseline to week 9/EOS with treatment group and BRIEF-A GEC T score changes from baseline to week 9/EOS as a covariate revealed that the MADRS total score and BRIEF-A GEC T score changes from baseline to week 9/EOS were strongly correlated (r2=0.30). Additionally, the coefficient of change in BRIEF-A GEC T score was statistically significant (estimate, 0.21; t-statistic, 7.33; P<0.0001), but the analysis showed that there was no differential effect of treatment with LDX vs treatment with placebo (t-statistic, –0.41; P=0.6818).
BRIEF-A Informant Report. The mean±SD change in BRIEF-A Informant Report GEC T score from baseline to week 9/EOS was −3.0±9.29 with placebo and −9.6±11.94 with LDX. The LS mean (95% CI) treatment difference significantly favored LDX (−5.9 (−9.3, −2.6); |t128|=3.53, P=0.0006).
Other secondary end points. On the CNS Vital Signs-derived NCI, median (range) standardized scores increased from baseline (placebo, 99.5 (54, 117); LDX, 96.0 (40, 117)) to week 9/EOS (placebo, 100.0 (33, 118); LDX, 103.0 (73, 121)); there were no significant treatment differences in median NCI standardized scores at week 9/EOS (Wilcoxon rank sum test: Z=1.788, P=0.0738). Post hoc regression analysis revealed that NCI score and MADRS total score changes at week 9/EOS were not correlated (r2=0.06). There was a differential effect of treatment (t-statistic, −2.66; P=0.0090); the coefficient for change in CNS Vital Sign NCI score was not significant (estimate, 0.03; t-statistic, 0.69; P=0.4885).
MADRS total score reductions during double-blind treatment indicated depressive symptom improvement. Mean±SD MADRS total scores decreased from 11.8±3.77 with placebo and 12.7±3.23 with LDX at baseline to 8.9±5.67 and 7.6±6.28, respectively, at week 9/EOS, representing mean±SD changes of −2.9±5.44 with placebo and −5.1±5.94 with LDX. Changes in LS mean (95% CI) MADRS total scores from baseline to week 9/EOS were −3.1 (−4.4, −1.8) with placebo and −5.0 (−6.3, −3.6) with LDX; the LS mean (95% CI) treatment difference significantly favored LDX (−1.9 (−3.7, 0.0); |t140|=2.01, P=0.0465).
Based on CGI-S scores, a majority of participants in both treatment groups were borderline to moderately ill at baseline (Table 1). At week 9/EOS, the percentage of participants with improved symptoms (ie, CGI-I scores of 1 (very much improved) and 2 (much improved)) was 38.9% with placebo and 60.6% with LDX.
Safety and Tolerability
TEAE rates were 73.6% with placebo and 78.9% with LDX. Five serious TEAEs occurred during double-blind treatment (placebo, n=3 (viral gastroenteritis, Salmonellosis, rhabdomyolysis); LDX, n=2 (loss of consciousness, suicidal ideation)). Another serious AE occurred during single-blind treatment with placebo (pelvic inflammatory disease). With the exception of loss of consciousness with LDX, none of the serious TEAEs were deemed by investigators to be treatment related. Discontinuations due to AEs occurred in one placebo participant (1.4%; headache) and four LDX participants (5.6%; moderate rash on lower left arm, worsening of depression, loss of consciousness, and suicidal ideation). TEAEs reported by ⩾5% of participants in either treatment group are summarized in Table 2; those reported most frequently were headache and dry mouth with placebo and decreased appetite, headache, dry mouth, insomnia, and irritability with LDX.
Vital sign and physical examination changes were generally modest with both treatments (Table 3). There were no notable trends observed with body weight or body mass index. Changes in blood pressure were small and similar between treatment groups. Three participants (4.2%) in the placebo group and four (5.6%) in the LDX group were observed to have systolic blood pressure ⩾140 mm Hg and an increase from baseline of ⩾10 mm Hg on two consecutive visits; zero participants in the placebo group and one (1.4%) in the LDX group were observed to have systolic blood pressure of ⩾140 mm Hg and an increase from baseline of ⩾10 mm Hg on two consecutive visits when one of the visits was the last study visit. One participant (1.4%) in the placebo group and three (4.2%) in the LDX group were observed to have diastolic blood pressure of ⩾90 mm Hg and an increase from baseline of ⩾10 mm Hg on two consecutive visits; zero participants in the placebo group and one (1.4%) in the LDX group were observed to have diastolic blood pressure of ⩾90 mm Hg and an increase from baseline of ⩾10 mm Hg on two consecutive visits when one of the visits was the last study visit. Electrocardiogram results at week 9/EOS showed a small mean±SD decrease in heart rate with placebo (−0.9±7.16) and an increase with LDX (3.2±10.71). One participant (1.4%) in the placebo and three (4.2%) in the LDX group were observed to have pulse rates ⩾100 b.p.m. and increases of ⩾15 b.p.m. from baseline.
There were no apparent withdrawals or rebound effects based on the ACSA. Mean±SD aggregate ACSA scores decreased from baseline (placebo, 25.5±9.91; LDX, 26.4±11.04) to weeks 10 (placebo, 19.5±10.63; LDX, 17.1±11.85) and 11 (placebo, 19.7±10.26; LDX, 16.6±11.66). There were no results of concern on the STS; participants had low mean±SD scores at baseline (placebo, 0.0±0.12; LDX, 0.1±0.39) and showed virtually no change throughout double-blind and single-blind treatments. CSFQ-14 total scores increased slightly from baseline to week 9 (observed cases) for men (mean±SD change from baseline: placebo, 2.1±5.06; LDX, 2.4±4.72) and women (mean±SD change from baseline: placebo, 1.6±5.91; LDX, 2.6±8.65); total scores were similar between treatment groups at week 9.
DISCUSSION
Participants with partially to fully remitted MDD and executive dysfunction exhibited significantly greater improvements in executive function with LDX augmentation compared with placebo, as measured by BRIEF-A Self-Report GEC T scores. This patient-reported amelioration was supported by the significant improvement reported by knowledgeable informants; for the other secondary cognitive end point, a performance-based computerized test of neurocognitive function, the improvement seen with LDX was not statistically superior to placebo. Overall, these findings support previous evidence of the ability of dopaminergic agents to mitigate cognitive dysfunction (Killgore et al, 2008; Schachar et al, 2008).
However, because there are typically only modest correlations between BRIEF-A and performance-based measures of executive function (Isquith et al, 2013), this finding is not surprising. Furthermore, a population specifically selected for having high BRIEF-A GEC T scores may not have exhibited the same level of impairment on the CNS Vital Signs test battery. As such, the study may have a higher possibility to detect changes on the BRIEF-A than on the CNS Vital Signs. It is also possible that additional training on the CNS Vital Signs may have decreased variability in this end point, as is observed in many cognition studies (Brooks and Sherman, 2012; Iverson et al, 2009). To address these issues, future studies should consider stratifying participants according to both self-reported and performance-based cognitive measures. This would ensure that participants are impaired on both types of measures at baseline, thereby increasing the probability of detecting improved performance on both measures. Additionally, including assessments of a participant’s perception of their treatment regimen (ie, do they think they were administered active treatment or placebo) would help to determine whether treatment expectations differentially influence self-reported and performance-based end points. Finally, as studies are not typically powered to assess changes in secondary end points, the use of a larger study population would help reduce measurement variability and increase the probability of observing statistical differences vs placebo.
Executive dysfunction can impact daily functioning (Marazziti et al, 2010). Although the implications of these findings for clinical practice should be interpreted cautiously until the results are replicated, the clinical relevance of self-reported changes in executive function should be considered. In this study, executive dysfunction was operationally defined as a Self-Report GEC T score of ⩾60. By study end, mean Self-Report GEC T score with LDX augmentation was reduced to levels below the operationally defined inclusion criteria and shifts in BRIEF-A GEC T scores to within 1 SD of the normative sample mean were observed. This suggests that executive function improvements were clinically relevant. Overall, these findings indicate that some individuals with MDD report considerable executive dysfunction despite having only mild depressive symptoms; this self-reported dysfunction was ameliorated by LDX augmentation. However, it will be important for future studies to determine whether performance-based objective tests also demonstrate improved executive function with LDX augmentation.
The mechanism of action by which LDX augmentation may ameliorate executive dysfunction is uncertain. However, individuals with MDD exhibit hypoactivation in the prefrontal and anterior cingulate cortices (Rogers et al, 2004), so the ability of stimulants to alleviate executive dysfunction is likely related to their modulation of cortical dopaminergic and adrenergic systems (Arnsten and Li, 2005; Elliott, 2003).
In addition to improvements in cognitive function, residual depressive symptoms were also significantly improved by LDX augmentation, which is consistent with the results of a previous study of LDX augmentation for treatment of depressive symptoms in individuals who did not achieve remission after 8 weeks of open-label escitalopram (Trivedi et al, 2013). These results are important given previous data suggesting that residual depressive symptoms can have a clinically relevant impact on quality of life (ten Doesschate et al, 2010).
Previous studies of the pharmacotherapy for cognitive or executive dysfunction in MDD have reported somewhat inconsistent findings (DeBattista, 2005; Ferguson et al, 2003; Herrera-Guzman et al, 2010). These inconsistencies are likely related to the fact that cognitive function was usually a secondary assessment in individuals selected for having relatively severe depression with or without documented cognitive dysfunction at baseline in these previous studies (Herrera-Guzman et al, 2010; Raskin et al, 2007). In our study, the primary end point was assessed in individuals specifically selected for having self-reported executive dysfunction (ie, BRIEF-A Self-Report GEC T score ⩾60) and mild or no depression (MADRS score ⩽18). These inclusion criteria are important because only a subset of individuals with depression experience cognitive dysfunction (Grant et al, 2001; Iverson et al, 2011), so observing cognitive improvement in individuals with impairment may be difficult when they are grouped with individuals who are not impaired. Furthermore, cognitive problems may be exacerbated in those with more severe depression and improve when depressive symptoms abate. Thus, by including only those individuals with mild depressive symptoms, it is possible to assess changes in executive and cognitive function relatively independent of depressive symptoms. Post hoc analyses noted that changes in MADRS total score and BRIEF-A GEC T score were not independent; MADRS total score and CNS Vital Sign NCI change scores were independent, although relatively smaller sample size may have limited our ability to definitively assess this relationship.
Overall, there were no unexpected safety concerns. Consistent with previous findings with long-acting stimulant use (Medori et al, 2008; Reimherr et al, 2007), there was a small mean increase in heart rate with LDX administration, whereas changes in blood pressure, weight, and BMI were small and similar between groups. The most common adverse events with LDX, including decreased appetite, headache, dry mouth, and insomnia, were similar to those reported in another study of LDX augmentation to treat depressive symptoms (Trivedi et al, 2013). Results from the ACSA during single-blind placebo treatment showed no evidence of adverse withdrawal effects.
Several limitations should be noted. First, multiple factors that contribute to cognitive impairment (eg, duration of disease and age of onset) (Grant et al, 2001) were not analyzed separately. Second, although the executive function and neurocognitive measures suggested that function improved, the study duration may not have been long enough to show the full impact of these improvements on daily functioning over time. Third, although the blind was not broken, the perception of stimulant effects among study participants who received LDX augmentation may have contributed to the greater improvement observed on BRIEF-A Self-Report scores vs placebo. Improvement in a BRIEF-A informant rating scale may mitigate this concern, but it would be useful in future studies to ask participants which treatment they believe they received. Future studies could also incorporate features into their study designs that might mitigate or more clearly reveal the influence of a participant’s treatment expectation on cognitive end points. Such features could include using an independent rater who is blind to the treatment regimen, employing a randomized crossover design in which individual participants are rated while on active treatment and placebo, or employing a low-dose comparator arm which would have similar perceived effects as the active treatment but would be expected to be ineffective in treating cognitive dysfunction.
Fourth, response rates to placebo can be high in clinical trials of MDD (Brunoni et al, 2009; Walsh et al, 2002); however, the consistent observation of superiority of LDX over placebo across self-reports and informant reports provides additional support for our positive findings. In addition, post hoc analyses of all baseline demographic, patient clinical characteristics, and pivotal assessments reported only one significant difference, which was mean citalopram usage before augmentation. Although this baseline treatment difference was not observed in combination with baseline differences in depressive symptoms measured with MADRS total score or executive function measured with the BRIEF-A Self-Report GEC T score, it could potentially impact the benefit of LDX augmentation and should be further explored.
In summary, these findings suggest that LDX augmentation of SSRI monotherapy significantly improved self- and informant-reported executive function and reduced depressive symptoms in participants with partially or fully remitted MDD. In view of the limited attention often paid to patients’ perceptions of their own condition in drug efficacy trials, it is considered to be important that the improvement in executive function noted in this trial was demonstrated on both the self-report and informant-report versions of the BRIEF-A. The safety profile was similar to that observed in studies of LDX in adults with ADHD (Weisler et al, 2009; Wigal et al, 2010) and MDD (Trivedi et al, 2013) and was consistent with those of other long-acting stimulants (Medori et al, 2008; Reimherr et al, 2007).
FUNDING AND DISCLOSURE
Manisha Madhoo, James Wu, and Colleen S Anderson are employees of Shire Development LLC, and hold stock and/or stock options in Shire. Richard SE Keefe currently or in the past 3 years has received investigator-initiated research funding support from the Department of Veterans Affairs, GlaxoSmithKline, National Institute of Mental Health, Novartis, Psychogenics, Research Foundation for Mental Hygiene, and the Singapore National Medical Research Council. He currently or in the past 3 years has served as a consultant for Abbott, Amgen, Astellas, Asubio, BiolineRx, Bristol-Myers Squibb, Cypress Bioscience, Eli Lilly, EnVivo, Helicon, Lundbeck, Merck, Mitsubishi, Novartis, Pfizer, Roche, Shire, Sunovion, and Takeda. He also receives royalties from the Brief Assessment of Cognition in Schizophrenia (BACS) testing battery and the MATRICS Battery (BACS Symbol Coding) and is a shareholder in NeuroCog Trials (Durham, NC). Angelo Sambunaris has received research support from Alkermes, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, BrainCells, CeNeRx, Cephalon, Forest Pharmaceuticals, GlaxoSmithKline, Jazz, Johnson & Johnson Labopharm, Lilly, Lundbeck, MediciNova, Merck, Neurocrine, Novartis, Otsuka, Pfizer, Roche, Sanofi-Aventis, Sepracor/Sunovion, Shire, and Takeda. He has also served as a speaker for Forest Pharmaceuticals and as a consultant for Forest, Mylan, and Takeda. Madhukar H Trivedi is or has been an advisor/consultant for or on Speakers’ Bureaus for: Abbott Laboratories, Abdi Ibrahim, Akzo (Organon Pharmaceuticals), Alkermes, AstraZeneca, Axon Advisors, Bristol-Myers Squibb Company, Cephalon, Eli Lilly & Company, Evotec, Fabre Kramer Pharmaceuticals, Forest Pharmaceuticals, GlaxoSmithKline, Janssen Pharmaceutic a Products, LP, Johnson & Johnson PRD, Libby, Lundbeck, Meade Johnson, MedAvante, Medtronic, Merck, Naurex, Neuronetics, Otsuka Pharmaceuticals, Pamlab, Parke-Davis Pharmaceuticals, Pfizer, PgxHealth, Rexahn Pharmaceuticals, Roche Products, Sepracor, Shire Development, Sierra, SK Life and Science, Takeda, Transcept, VantagePoint, and Wyeth-Ayerst Laboratories. In addition, he has received research support from: Agency for Healthcare Research and Quality (AHRQ), Corcept Therapeutics, Cyberonics, National Alliance for Research in Schizophrenia and Depression, National Institute of Mental Health, National Institute on Drug Abuse, Novartis, Pharmacia & Upjohn, Predix Pharmaceuticals (Epix), and Solvay Pharmaceuticals. Robert M Roth is an author of the BRIEF-A and receives a royalty from the publisher and has served as a research consultant for Shire. R Lasser was formerly an employee of Shire Development LLC; he is currently an employee of F Hoffman—La Roche.
References
American Psychiatric Association (2000) Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition, Text Revision. American Psychiatric Association: Washington, DC.
Arnsten AF (2006). Stimulants: therapeutic actions in ADHD. Neuropsychopharmacology 31: 2376–2383.
Arnsten AF, Li BM (2005). Neurobiology of executive functions: catecholamine influences on prefrontal cortical functions. Biol Psychiatry 57: 1377–1384.
Brooks BL, Sherman EM (2012). Computerized neuropsychological testing to rapidly evaluate cognition in pediatric patients with neurologic disorders. J Child Neurol 27: 982–991.
Brunoni AR, Lopes M, Kaptchuk TJ, Fregni F (2009). Placebo response of non-pharmacological and pharmacological trials in major depression: a systematic review and meta-analysis. PLoS One 4: e4824.
Coric V, Stock EG, Pultz J, Marcus R, Sheehan DV (2009). Sheehan Suicidality Tracking Scale (Sheehan-STS): preliminary results from a multicenter clinical trial in generalized anxiety disorder. Psychiatry (Edgmont) 6: 26–31.
DeBattista C (2005). Executive dysfunction in major depressive disorder. Expert Rev Neurother 5: 79–83.
Elliott R (2003). Executive functions and their disorders. Br Med Bull 65: 49–59.
Endicott J, Nee J (1997). Endicott Work Productivity Scale (EWPS): a new measure to assess treatment effects. Psychopharmacol Bull 33: 13–16.
Endicott J, Nee J, Harrison W, Blumenthal R (1993). Quality of Life Enjoyment and Satisfaction Questionnaire: a new measure. Psychopharmacol Bull 29: 321–326.
Ferguson JM, Wesnes KA, Schwartz GE (2003). Reboxetine versus paroxetine versus placebo: effects on cognitive functioning in depressed patients. Int Clin Psychopharmacol 18: 9–14.
Garlinghouse MA, Roth RM, Isquith PK, Flashman LA, Saykin AJ (2010). Subjective rating of working memory is associated with frontal lobe volume in schizophrenia. Schizophr Res 120: 71–75.
Grant MM, Thase ME, Sweeney JA (2001). Cognitive disturbance in outpatient depressed younger adults: evidence of modest impairment. Biol Psychiatry 50: 35–43.
Gualtieri CT, Johnson LG (2006). Reliability and validity of a computerized neurocognitive test battery, CNS Vital Signs. Arch Clin Neuropsychol 21: 623–643.
Gualtieri CT, Johnson LG, Benedict KB (2006). Neurocognition in depression: patients on and off medication versus healthy comparison subjects. J Neuropsychiatry Clin Neurosci 18: 217–225.
Guy W (1976) Clinical Global Impressions. ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education, and Welfare; Public Health Service, Alcohol, Drug Abuse and Mental Health Administration; NIMH Psychopharmacology Research Branch: Rockville, MD. pp 218–222.
Harvey PD, Roth RM, Bilder RM, Richards C, Lasser R, Geibel B et al (2012) Assessment of executive dysfunction in adults with major depressive disorder receiving lisdexamfetamine dimesylate augmentation of escitalopram. 165th Annual Meeting of the American Psychiatric Association; May 5–9: Philadelphia, PA.
Herrera-Guzman I, Herrera-Abarca JE, Gudayol-Ferre E, Herrera-Guzman D, Gomez-Carbajal L, Pena-Olvira M et al (2010). Effects of selective serotonin reuptake and dual serotonergic-noradrenergic reuptake treatments on attention and executive functions in patients with major depressive disorder. Psychiatry Res 177: 323–329.
Isquith PK, Roth RM, Gioia G (2013). Contribution of rating scales to the assessment of executive functions. Appl Neuropsychol Child 2: 125–132.
Iverson GL, Brooks BL, Langenecker SA, Young AH (2011). Identifying a cognitive impairment subgroup in adults with mood disorders. J Affect Disord 132: 360–367.
Iverson GL, Brooks BL, Young AH (2009). Identifying neurocognitive impairment in depression using computerized testing. Appl Neuropsychol 16: 254–261.
Keller A, McGarvey EL, Clayton AH (2006). Reliability and construct validity of the Changes in Sexual Functioning Questionnaire short-form (CSFQ-14). J Sex Marital Ther 32: 43–52.
Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR et al (2003). The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 289: 3095–3105.
Killgore WD, Muckle AE, Grugle NL, Killgore DB, Balkin TJ (2008). Sex differences in cognitive estimation during sleep deprivation: effects of stimulant countermeasures. Int J Neurosci 118: 1547–1557.
Kupfer DJ (2005). The pharmacological management of depression. Dialogues Clin Neurosci 7: 191–205.
Marazziti D, Consoli G, Picchetti M, Carlini M, Faravelli L (2010). Cognitive impairment in major depression. Eur J Pharmacol 626: 83–86.
McGregor C, Srisurapanont M, Mitchell A, Longo MC, Cahill S, White JM (2008). Psychometric evaluation of the Amphetamine Cessation Symptom Assessment. J Subst Abuse Treat 34: 443–449.
Medori R, Ramos-Quiroga JA, Casas M, Kooij JJ, Niemelä A, Trott GE et al (2008). A randomized, placebo-controlled trial of three fixed dosages of prolonged-release OROS methylphenidate in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry 63: 981–989.
Montgomery SA, Asberg M (1979). A new depression scale designed to be sensitive to change. Br J Psychiatry 134: 382–389.
Preiss M, Kucerova H, Lukavsky J, Stepankova H, Sos P, Kawaciukova R (2009). Cognitive deficits in the euthymic phase of unipolar depression. Psychiatry Res 169: 235–239.
Rabin LA, Fogel J, Nutter-Upham KE (2011). Academic procrastination in college students: the role of self-reported executive function. J Clin Exp Neuropsychol 33: 344–357.
Rabin LA, Roth RM, Isquith PK, Wishart HA, Nutter-Upham KE, Pare N et al (2006). Self- and informant reports of executive function on the BRIEF-A in MCI and older adults with cognitive complaints. Arch Clin Neuropsychol 21: 721–732.
Raskin J, Wiltse CG, Siegal A, Sheikh J, Xu J, Dinkel JJ et al (2007). Efficacy of duloxetine on cognition, depression, and pain in elderly patients with major depressive disorder: an 8-week, double-blind, placebo-controlled trial. Am J Psychiatry 164: 900–909.
Reimherr FW, Williams ED, Strong RE, Mestas R, Soni P, Marchant BK (2007). A double-blind, placebo-controlled, crossover study of osmotic release oral system methylphenidate in adults with ADHD with assessment of oppositional and emotional dimensions of the disorder. J Clin Psychiatry 68: 93–101.
Reppermund S, Ising M, Lucae S, Zihl J (2009). Cognitive impairment in unipolar depression is persistent and non-specific: further evidence for the final common pathway disorder hypothesis. Psychol Med 39: 603–614.
Rogers MA, Kasai K, Koji M, Fukuda R, Iwanami A, Nakagome K et al (2004). Executive and prefrontal dysfunction in unipolar depression: a review of neuropsychological and imaging evidence. Neurosci Res 50: 1–11.
Roth RM, Isquith PK, Gioia G (2013). The Behavior Rating Inventory of Executive Function (BRIEF) family of measures. In: Goldstein S, Naglieri J (eds). Executive Functioning Handbook 2nd edn.
Roth RM, Isquith PK, Gioia GA (2005) Behavior Rating Inventory of Executive Function-Adult Version (BRIEF-A) Professional Manual. Psychological Assessment Resources: Lutz, FL.
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D et al (2006). Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 163: 1905–1917.
Schachar R, Ickowicz A, Crosbie J, Donnelly GA, Reiz JL, Miceli PC et al (2008). Cognitive and behavioral effects of multilayer-release methylphenidate in the treatment of children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol 18: 11–24.
Snyder HR (2013). Major depressive disorder is associated with broad impairments on neuropsychological measures of executive function: a meta-analysis and review. Psychol Bull 139: 81–132.
Spitzer RL, Kroenke K, Williams JB, Lowe B (2006). A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med 166: 1092–1097.
ten Doesschate MC, Koeter MW, Bockting CL, Schene AH (2010). Health related quality of life in recurrent depression: a comparison with a general population sample. J Affect Disord 120: 126–132.
Trivedi MH, Cutler A, Richards C, Lasser R, Geibel B, Gao J et al (2013). A randomized controlled trial of the efficacy and safety of lisdexamfetamine dimesylate as augmentation therapy in adults with residual symptoms of major depressive disorder after treatment with escitalopram. J Clin Psychiatry 74: 802–809.
Vyvanse (lisdexamfetamine dimesylate) (2012) Full Prescribing Information. Shire US Inc: Wayne, PA.
Walsh BT, Seidman SN, Sysko R, Gould M (2002). Placebo response in studies of major depression: variable, substantial, and growing. JAMA 287: 1840–1847.
Ware J Jr., Kosinski M, Keller SD (1996). A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care 34: 220–233.
Weisler R, Young J, Mattingly G, Gao J, Squires L, Adler L (2009). Long-term safety and effectiveness of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. CNS Spectr 14: 573–585.
Wigal T, Brams M, Gasior M, Gao J, Squires L, Giblin J (2010). Randomized, double-blind, placebo-controlled, crossover study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder: novel findings using a simulated adult workplace environment design. Behav Brain Funct 6: 34.
Acknowledgements
Under the direction of the authors, Elizabeth LaFlamme, PhD, employee of Complete Healthcare Communications (Chadds Ford, PA, USA), provided writing assistance for this publication. Editorial assistance in formatting, proofreading, copyediting, and fact checking was also provided by Craig Slawecki, PhD, from Complete Healthcare Communications. Brian Scheckner, PharmD, from Shire Development LLC also reviewed and edited the manuscript for scientific accuracy. Shire Development LLC also provided funding to Complete Healthcare Communications for support in writing and editing this manuscript. We wish to acknowledge all study investigators who worked at the sites at which the study was conducted. Clinical research was funded by Shire Development LLC (Wayne, PA, USA). Although the sponsor was involved in design, collection, analysis, interpretation, and fact checking of information, the content of this manuscript, the ultimate interpretation, and the decision to submit it for publication in Neuropsychopharmacology were made by the authors independently.
Author information
Authors and Affiliations
Corresponding author
Additional information
Portions of these data have been presented as a poster at the 50th American College of Neuropsychopharmacology (Waikoloa Beach, HI, 4–8 December 2011), the 15th College of Psychiatric and Neurologic Pharmacists (Tampa, FL, 29 April–2 May 2012), the 28th Collegium Internationale Neuro-Psychopharmacologicum (Stockholm, Sweden, 3–7 June 2012), and the 165th Annual Meeting of the American Psychiatric Association (Philadelphia, PA, 5–9 May 2012).
PowerPoint slides
Rights and permissions
About this article
Cite this article
Madhoo, M., Keefe, R., Roth, R. et al. Lisdexamfetamine Dimesylate Augmentation in Adults With Persistent Executive Dysfunction After Partial or Full Remission of Major Depressive Disorder. Neuropsychopharmacol 39, 1388–1398 (2014). https://doi.org/10.1038/npp.2013.334
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.334
Keywords
This article is cited by
-
Psychostimulant Augmentation of Antidepressant Therapy in Depression: a Systematic Review and Meta-Analysis
Current Treatment Options in Psychiatry (2023)
-
Alertness in patients with treatment-resistant depression: interface between sleep medicine and psychiatry—review article
Middle East Current Psychiatry (2021)
-
Altered GABA-mediated information processing and cognitive dysfunctions in depression and other brain disorders
Molecular Psychiatry (2021)
-
Adult ADHD and comorbid disorders: clinical implications of a dimensional approach
BMC Psychiatry (2017)
-
Cognitive remission: a novel objective for the treatment of major depression?
BMC Medicine (2016)
