
Abstract
We previously reported linkage of schizophrenia and schizoaffective disorder to 13q32–34 in the European descent Afrikaner population from South Africa. The nature of genetic variation underlying linkage peaks in psychiatric disorders remains largely unknown and both rare and common variants may be contributing. Here, we examine the contribution of common variants located under the 13q32–34 linkage region. We used densely spaced SNPs to fine map the linkage peak region using both a discovery sample of 415 families and a meta-analysis incorporating two additional replication family samples. In a second phase of the study, we use one family-based data set with 237 families and independent case–control data sets for fine mapping of the common variant association signal using HapMap SNPs. We report a significant association with a genetic variant (rs9583277) within the gene encoding for the myosin heavy-chain Myr 8 (MYO16), which has been implicated in neuronal phosphoinositide 3-kinase signaling. Follow-up analysis of HapMap variation within MYO16 in a second set of Afrikaner families and additional case–control data sets of European descent highlighted a region across introns 2–6 as the most likely region to harbor common MYO16 risk variants. Expression analysis revealed a significant increase in the level of MYO16 expression in the brains of schizophrenia patients. Our results suggest that common variation within MYO16 may contribute to the genetic liability to schizophrenia.
Similar content being viewed by others

INTRODUCTION
Susceptibility to schizophrenia is determined by multiple genetic and possibly environmental factors. Recent studies addressing the role of high-penetrant rare variants (Walsh et al, 2008; Xu et al, 2008, 2009, 2011, 2012) or common genetic variants with low effect (ISC, 2008; Lee et al, 2012; O’Donovan et al, 2008; Ripke et al, 2011; Shi et al, 2009, 2011; Stefansson et al, 2009) suggest that patient genomes contain risk alleles at a wide range of frequencies, some driving and some merely modifying the disease risk and expression, which in concert may affect the structure and function of neural circuits (ISC, 2008; O'Donovan et al, 2008; Rodriguez-Murillo et al, 2012; Shi et al, 2009; Stefansson et al, 2009; Xu et al, 2008, 2009, 2011).
In complex diseases, the genetic structure of linkage signals most likely involves one or several rare alleles with strong effect on disease risk, or a combination of rare and common alleles, in the same or different genes (Bowden et al, 2010). Also, linkage analyses of inbred mice have shown that more than one gene can contribute to the same linkage signal for a given QTL trait (Karst et al, 2011). Along the same lines, association studies coupled with targeted resequencing have suggested that the same genes carrying common risk variants can also show an excess of rare risk variants implicated in the disease (Cirulli and Goldstein, 2010; Di Rienzo, 2006; Manolio et al, 2009; Trynka et al, 2011).
A 9-cM genome-wide linkage scan on families from the European descent Afrikaner population from South Africa identified three linkage signals on chromosomes 1, 9, and 13 (Abecasis et al, 2004). Subsequently, we increased the genomic coverage to better define the linkage regions, and performed a 2-cM genome-wide linkage scan on an extended set of Afrikaner families. The results from this genome scan identified chromosome 13q32–34 as the most robustly linked locus in this population. We also addressed the contribution of rare CNVs to schizophrenia in this cohort and found that, at the level of resolution of the linkage scan, none of the linkage signals observed in these families may be caused by the presence of CNVs within these genomic intervals (Xu et al, 2009).
Here, we present the results of our ongoing systematic effort to elucidate the genetic structure of our 13q32–34 linkage peak obtained in our 2-cM genome scan, by analyzing the contribution of local common variants via a multistage association study. In addition to genuine contributions to the risk associated with a given linkage signal, even in cases where the linkage signal is accounted for only by rare variants, common variants may in some cases help pinpoint with more accuracy the location of rare risk variants (Dickson et al, 2010; Lin et al, 2004; Sanna et al, 2011). With this aim, first, we genotyped 1223 individuals from 415 Afrikaner families for 723 SNPs localized within 13q32–34. Subsequently, the most significant SNPs were followed up in two independent family-based replication samples of European origin. One SNP showed replicated association in one of the two independent samples and remained significant after meta-analysis and correction for multiple testing. This SNP is located within the MYO16 (myosin XVI) gene (Patel et al, 2001; Yokoyama et al, 2011). Second, we performed a comprehensive fine-scale mapping of the genetic contribution of this gene with respect to common variation, by genotyping an independent set of families from the Afrikaner population for 102 SNPs within MYO16 and by imputing the rest of the HapMap SNPs within the gene boundaries. These analyses identified a preponderance of common variants implicated in schizophrenia within introns 2–6 of the gene MYO16. Furthermore, expression analysis of the MYO16 gene in brain samples from patients and controls identified a significantly elevated level of expression in patients with schizophrenia.
MATERIALS AND METHODS
We used a family-based approach studying families with at least one affected individual per family. Data sets are presented in Supplementary Table 1.
Afrikaner Cohorts
Affected families were recruited and diagnosed as part of our ongoing, large-scale genetic study of schizophrenia in the European descent Afrikaner population from South Africa, as described previously (Abecasis et al, 2004; Karayiorgou et al, 2004; Xu et al, 2008, 2009). Affected subjects were classified as either narrowly or broadly affected. The narrow diagnosis includes subjects with schizophrenia or schizoaffective disorder-depressive type, as described previously (Abecasis et al, 2004; Xu et al, 2009). The broad diagnosis includes all individuals classified under the narrow definition, as well as individuals with schizoaffective disorder-bipolar type (Xu et al, 2009).
Afrikaner Set 1
This data set includes the 143 families used for the linkage scan, plus an additional 272 families. The entire set comprises 474 affected individuals who meet the narrow diagnostic criteria or 741 who meet the broad diagnostic criteria.
Afrikaner Set 2
This data set includes 237 families, 85 of whom have family history of schizophrenia in the previous two generations. Two hundred and thirty-two individuals in these families meet the narrow diagnostic criteria, whereas 266 individuals meet the broad diagnostic criteria.
Rutgers Families
From the entire set of families collected under the NIMH Schizophrenia Genetics Initiative, maintained by the Rutgers University Cell and DNA Repository, we selected a subset of 301 families matched according to ethnicity. Our selected Caucasian, European ancestry, subset includes a total of 1241 individuals (631 affected with schizophrenia).
US Families
Two hundred and ten trios (consisting of one affected individual and both biological unaffected parents for a total of 630 individuals) were included in this sample of Caucasian, European descent families recruited from the United States. All probands met full diagnostic criteria for schizophrenia or schizoaffective disorder. Description of this data set and the methods of subject selection and clinical evaluation have been described previously by Sobin et al (2001, 2003).
GAIN Data Set
This study is part of the Genetic Association Information Network (GAIN) (ID phs000021.v.2.p1). Details on inclusion criteria and participants are available at dbGap (database of Genotype and Phenotype) (see URLs) (Suarez et al, 2006). In total, 1314 cases and 1368 controls of European descent were included in the final set.
MGS_nonGAIN Data Set
This study is part of the Molecular Genetics of Schizophrenia (MGS) genome-wide association study (ID phs000167). Details on inclusion criteria and participants are available at dbGap (see URLs). In all, 1405 cases and 1347 controls of European descent were included in the final set.
PGC Data Set
This data set is part of the Schizophrenia Psychiatric Genome-wide association study consortium (Ripke et al, 2011). We included the results from stage 1 mega-analysis published by Ripke et al (2011) that correspond to the MYO16 gene region overlapping SNPs genotyped or imputed in our Afrikaner Set 2 (SAF2) data set. This data set included 9394 schizophrenia cases.
Genotyping, Quality Control, and Imputation
SAF1
Family members were genotyped for 723 SNPs covering 14.65 Mb under the 13q32–34 linkage peak and within candidate genes (ZIC2, ZIC5, NALCN, FGF14, G72, and EFNB2) in the immediate vicinity of the linkage peak (Supplementary Table 2), on the Illumina GoldenGate platform at the Center for Inherited Disease Research (CIDR).
Rutgers and US samples
Family members were genotyped for 22 SNPs on a Taq Man Open Array Genotyping Platform (Applied Biosystems). These 22 SNPs were chosen among the top-associated SNPs resulting from the association analysis in stage 1 (Afrikaner Set 1 (SAF1)) or surrogates of those (ie, in strong LD with at least one of the top-associated SNPs).
SAF2
This set of families was genotyped as part of a wider genotyping project on a Human Genome-Wide SNP Array 5.0 (Affymetrix), which contains 500 568 SNPs (manuscript in preparation). Samples were processed as described previously (Xu et al, 2008). Average call rate on arrays used in this study was 99.43%. All microarray experiments were performed in the Vanderbilt Microarray Shared Resource.
GAIN and MGS
Individual genotypes as well as phenotypic information were available to download from the dbGap website. Only individuals of European descent were included in the analysis.
For all data sets, quality control procedures per family, individual, and marker were performed with PLINK (Purcell et al, 2007) and PedStats (see URLs). All data sets went through quality control and we only selected samples with a call rate >95%. We eliminated from the analysis duplicated SNPs, monomorphic SNPs, and SNPs with Hardy–Weinberg exact test P<10−6. Only SNPs with a minor allele frequency over 0.01 were included in the downstream analyses. We also checked for Mendelian inheritance errors among families, and removed SNPs with more than four Mendelian errors in the total sample. For the case–control data sets, we corrected for population stratification with the program EIGENSTRAT, eliminating outliers from the downstream analyses.
Imputation of non-genotyped HapMap SNPs for SAF2, GAIN and MGS data sets was performed with MACH (see URLs) using 100 Markov iterations with the two-step procedure recommended in the manual. HapMap Phased Haplotypes (release 22) on CEU subjects were used in the imputation. After imputation, only SNPs with a MACH R2 over 0.3 were further considered. This estimates the correlation between imputed and true genotypes; a value <0.3 flags poorly imputed SNPs (Li et al, 2010). In addition, Mendelian checks (for the family-based samples) and Hardy–Weinberg equilibrium tests were performed to eliminate unreliable imputation calls to include imputed genotypes in downstream analyses. Imputed SNPs were then analyzed as the genotyped SNPs.
Statistical Analyses
Family-based association testing for single SNPs was performed using LAMP (see URLs) (Li et al, 2005, 2006). We adopted a free model for the analysis that does not constrain the penetrances for the three genotypes. Haplotype-based associations were assessed by means of the transmission disequilibrium test (TDT) for haplotypes implemented in PLINK. For the case–control data sets, a trend test was performed to evaluate the SNP association. We applied Bonferroni correction in all tests to obtain an α-corrected threshold. We calculated the number of independent tests in each case based on LD patterns between SNP pairs. These procedures were performed in PLINK (see URLs).
Meta-analysis of the results for the independent samples was performed with Metal (see URLs). The algorithm checks for heterogeneity and performs meta-analysis under a fixed-effects model. All base pair positions are based on the current Human genome assembly (hg19) (see URLs).
To identify duplicated individuals and family relationships between individuals across data sets, we performed identity-by-descent analysis of GAIN, MGS_nonGAIN, and Rutgers samples merged together using PLINK. Duplicated and related individuals across data sets were removed from all but one of the data sets to avoid bias in the analysis. Specifically, GAIN and MGS_nonGAIN included 10 duplicated individuals that were removed from the larger MGS_nonGAIN data set.
Expression Analysis
Total RNA from frontal cortex was obtained from the Stanley Medical Research Institute (SMRI) (Bethesda, MD) (see URLs). The SMRI Array Collection includes 35 individual subjects in each of three groups: control, schizophrenia, and bipolar disorder subjects (Torrey et al, 2000). qRT-PCR was performed with pre-designed TaqMan Gene Expression assay by ABI (Applied Biosystems; ABI assay number Hs01031284_m1) on a 7900HT Fast Real-Time PCR system (Applied Biosystems). Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous control. Relative quantitation of expression comparing the three groups (schizophrenia, bipolar disorder, and controls) was tested with generalized linear models (GLM) and incorporating covariates into the model. Descriptive statistics, means comparison, and GLM were analyzed with R statistics software.
RESULTS
SNP Association in a Discovery set of Afrikaner Families, Replication, and Meta-Analysis
To follow-up the 13q32–34 linkage signal obtained through our 2-cM coverage linkage scan, we genotyped 723 SNPs from chromosome 13, on 1223 individuals from the 143 Afrikaner families included in the 2-cM linkage scan plus 272 additional families from the same homogeneous population (SAF1). We performed family-based association tests on these 415 families using LAMP (Li et al, 2005, 2006). One hundred and fifteen SNPs reached nominal significance at this stage (Figure 1 and Supplementary Table 2). None of these SNPs were within candidate genes (ZIC2, ZIC5, NALCN, FGF14, G72, and EFNB2) abutting the linkage peak. Twenty-two associated SNPs were followed-up in two independent samples of European descent (Rutgers and US samples, see Materials and methods). These 22 SNPs were selected from the top-associated SNPs, or surrogates of these, chosen based on the LD structure, each one representing one independent LD block, and the availability of a genotyping assay in a TaqMan Open Array Genotyping platform (Applied Biosystems). Family members from the replication samples (Rutgers and US) were genotyped for these 22 SNPs (Table 1). Following quality control procedures, seven SNPs were removed from the analysis, two because of bad calls, four because of Mendelian errors (these were not concentrated in specific families), and one because of deviations from Hardy–Weinberg equilibrium. The remaining 15 SNPs were tested for association in the replication samples by using LAMP. Table 1 shows the p-values and odd ratios for the association in the discovery sample (SAF1) and in the replication samples (Rutgers and US). Subsequently, meta-analysis was performed combining p-values obtained from the SAF1 and both replication samples. Meta-analysis identified one SNP with combined P-values that survive Bonferroni correction for multiple testing (Table 1) (αcorrected=0.0023). The top-associated SNP, rs9583277, has a meta-analysis p-value of 1.86 × 10−4 and 2.25 × 10−4 for the combined sample SAF1–Rutgers–US families, for both narrow and broad definition of schizophrenia, respectively. It is worth noting that the Bonferroni correction we used to declare significance reflects the number of independent tests (n) we performed (n=22, αcorrected=0.05/22=0.0023) and therefore are not as stringent as thresholds used in genome-wide association study (GWAS) that reflect corrections for ∼1 million tests performed. The identified variant (rs9583277) maps to 109 333 749 bp on chromosome 13q33.3, within the second intron of the MYO16 gene. Our previous linkage analysis indicated dominant inheritance for the risk locus at 13q32–34 (Xu et al, 2009). Consistent with this finding, we did not detect any excess of homozygosity (an indication of recessive mode of inheritance) at rs9583277, either when the entire SAF1 data set was considered or upon stratified analysis including only families linked to 13q32–34 (data not shown). Overall, our analysis, using densely spaced SNPs to fine map the prior 13q32–34 linkage peak region on a discovery and two replication family samples (a total of 923 families), highlighted a potential contribution of the MYO16 gene locus.

Genome-wide linkage and fine mapping. Bd, broad; LOD, logarithm of the odds; Na, narrow; SNP, single-nucleotide polymorphism.
Fine Mapping of the Common Variant Association Signal Using MYO16 HapMap SNPs
Having identified a significant association with a genetic variant (rs9583277) within the MYO16 gene, we then performed a comprehensive fine-scale mapping of the common variant association signal in an independent set of families and cases. To this end, we genotyped, or imputed when necessary, all HapMap SNPs within the MYO16 gene boundaries, according to UCSC genome browser genome positions (hg19). First, we examined a sample of 228 Afrikaner families with an average of 3.2 individuals per family (SAF2). Following quality control procedures (see Materials and methods), 102 genotyped and 470 imputed HapMap SNPs were available for analysis of the MYO16 gene locus with respect to underlying common risk variants. It should be noted that there is no overlap with MYO16 SNPs genotyped in SAF1 and that the SNP previously found associated in the SAF1 sample (rs9583277) was not genotyped in the SAF2 sample as it is not a HapMap SNP nor is it present in the common genotyping platforms. Therefore, this stage is not intended to be a replication of the previous findings, but a deeper characterization of the common variation within MYO16 in the context of schizophrenia. Even though several SNPs genotyped at this stage are located in the general vicinity of rs9583277 within the MYO16 gene, rs9583277 is in a region of low LD.
Figure 2 shows LAMP p-values for the association of MYO16 SNPs for both narrow and broad definitions of schizophrenia, along with the recombination frequency across the region. Table 2 shows the top-associated SNPs within this data set. Notably, four SNPs showed significant association with narrow definition schizophrenia after correction for multiple testing (αcorrected=2.17 × 10−4, based on a Bonferroni correction after estimating the number of independent tests to 230, taking into account the LD pattern among SNP pairs). Three of these four SNPs also showed association with the disease under its broader definition. All these four SNPs were located within intron 3 of the MYO16 gene, within an LD block that expands from intron 2 to 6.

Plot depicts the negative logarithm of the SNP association p values for the Afrikaner Set 2 data set with narrow (SAF2(NARROW)) and broad (SAF2(BROAD)) schizophrenia diagnosis, as well as the meta-analysis p-values for the combined sample SAF2(BROAD)/GAIN/MGS data sets (META(BROAD)). The background graph represents the recombination rate throughout the region. GAIN, Genetic Association Information Network; MGS, Molecular Genetics of Schizophrenia.
We also investigated if there was any specific configuration of alleles or haplotypes conferring susceptibility to schizophrenia for either narrow or broad definition. Only directly genotyped SNPs were used to test association on haplotypes. Haplotype-based association in Afrikaner families was assayed with the TDT. First, we estimated haplotype blocks based on the LD structure by means of the default procedure implemented in Haploview. Subsequently, each haplotype within each block was tested for association with the hap-tdt option implemented in PLINK. In this manner, we tested 74 haplotypes, each comprising 2–10 SNPs. Table 3 shows the top-associated haplotypes for either schizophrenia definition. Two distinct two-SNP haplotypes show undertransmission and significant association with schizophrenia (αcorrected=6.76 × 10−4, 74 independent tests). It is worth noting that, of the two haplotypes with significant P-values, the CG haplotype including SNPs rs558322 and rs4976845 is associated with the narrow definition, and the GA haplotype including rs4578513 and rs10492418 is associated with the broad definition. Notably, these two haplotypes reside within distinct haplotype blocks, suggesting that the observed association signals are independent of each other. Of note, there are other examples where independent haplotypes are associated with distinct forms of a disease (Cruz et al, 2008). In our study, the two independent haplotypes might be acting as modifiers of the clinical presentation or reflect two distinct patient sub-populations.
We extended our follow-up studies to two additional, independent, case–control data sets, which are part of genome-wide genotyping projects (GAIN and MGS). To this end, we extracted SNP genotypes located within the MYO16 gene boundaries and also imputed non-genotyped HapMap SNPs from this region to facilitate comparison with the SNPs in our SAF2 data set. The SNPs extracted from GAIN and MGS data sets matched the SNPs in SAF2, and so the LD patterns were equivalent. Therefore, we used the same significance threshold for the SAF2 as well as the GAIN and MGS data sets. Following quality control procedures, 572 single SNP P-values from the three data sets, SAF2, GAIN, and MGS (a total of 3307 cases), were meta-analyzed. The lowest combined P-value after meta-analysis was 1.1 × 10−3 for the combined sample SAF2 (broad status of SCZ)-GAIN-MGS for SNP rs4772996 (Table 4). Although this SNP does not survive the correction for multiple testing when considering 230 independent tests (corrected α level, 0.05/230=2.17 × 10−4), it is important to note that the direction of association is consistent across all three data sets for the top-associated SNPs. Moreover, all top-associated SNPs following meta-analysis are located within intron 4 of the gene, in complete LD with the top-associated SNPs in the SAF2 data set (D′=1), strongly suggesting that the association signal obtained upon meta-analysis points to the same associated region within the MYO16 gene. The fact that these SNPs do not reach significance after correction for multiple testing likely reflects the presence of heterogeneity across data sets (Table 4). Sample heterogeneity also likely explains change of ranking among top SNPs. Specifically, although the top-associated SNPs in the SAF2 data set continue to show nominally significant association in the meta-analysis, they are not present among the top-ranking SNPs (Table 4). However, top-ranking SNPs from either data set are in high LD with each other and likely represent the same association signal.
We further compared the results obtained in our SAF2 data set with recently available results from the Schizophrenia Psychiatric GWAS Consortium (Ripke et al, 2011) The PGC study is a meta-analysis that combines various data sets, including GAIN and MGS. Therefore, meta-analysis of our data set and the PGC data set is not intended as a replication, but as a test of our hypothesis using a more extensive set of data. We extracted results for SNPs mapping within MYO16 and performed a meta-analysis following the same procedure as with the SAF2-GAIN-MGS data sets. We meta-analyzed 248 SNPs overlapping across data sets, for a total of 22 640 individuals. The top-associated SNP following meta-analysis is rs9284246 (Supplementary Table 3) located within intron 2 of the MYO16 gene (109 327 788 bp). This finding further points to the region across introns 2–6 as the most likely region to harbor common variants implicated in schizophrenia.
Expression Analysis
In seeking convergent supporting evidence, we also tested the expression levels of the MYO16 gene in brains of patients with schizophrenia. Our analysis of the SMRI Array Collection using qRT-PCR showed that mean levels of expression of MYO16 were significantly higher in the frontal cortex of schizophrenia patients as compared with controls (F(1, 66)=4.2; p=0.044). The significance holds when we incorporate either sex or age at death (F(3,64)=3.008; p=0.037) or brain pH and post-mortem interval (F(3,64)=2.778; p=0.048) as covariates in our analysis. The comparison of the bipolar group to controls did not result in a significant difference, although mean levels of expression were slightly higher in the bipolar group (see Supplementary Figure 1 for a scatter plot of expression levels). Furthermore, 6 out of 11 expression studies that have profiled the SMRI Array Collection samples using array technology reported increased levels of MYO16 expression in schizophrenia patients vs controls.
We also tested 28 SNPs genotyped in the Stanley Array Collection, located at both ends of our significant SNPs, but none of these SNPs showed association with MYO16 expression levels (p>0.05). It should be noted that SNP rs9583277 as well as most of the significant SNPs in SAF2 were not included in this set as they had not been genotyped in the Stanley Array Collection.
DISCUSSION
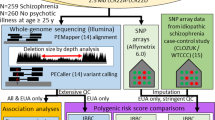
This study used seven patient cohorts and a dense array of SNPs to fine map the prior linkage region at the 13q32–34 locus. We provide evidence suggesting that variants within MYO16 contribute to the genetic liability to schizophrenia conferred by the 13q32–34 locus. The MYO16 gene stretches along 611 856 bp on chromosome 13q33. It consists of 35 exons and has several isoforms. All associated SNPs from SAF1, SAF2, and meta-analysis, and one haplotype from SAF2 are located within introns 2–6 of the gene. Considering that there was no significant excess of total genotyped SNPs in this region, this finding indicates that the signal related to common risk variation from this gene is likely localized in this region of the gene. It should be noted that incorporating the initial findings in the meta-analyses is necessary because of the small effect sizes of common variants and the need to increase the power of our association study, albeit the potential for introducing biases (Zeggini and Ioannidis, 2009). The effect of the associated SNPs on the function of the MYO16 gene remains unknown. It should be noted, however, that one of the most significant SNPs in the SAF2 data set (rs9301323, p-value=1.7 × 10−3), which is in strong LD with the top-associated SNP in the same data set (rs9520990), is located within a splice site region in intron 6 (see URLs), and could affect the pattern of splicing of the MYO16 gene. This position is conserved in the mouse. The Human Splicing Finder program (Desmet et al, 2009) indicates that the minor allele (G) of rs9301323 disrupts a predicted branch point sequence in intron 6.
Myosin XVI appeared very recently during the evolution of mammals and is unique in both its structure and function (Thompson and Langford, 2002). Earlier evidence suggested that MYO16 is important for neuronal migration and brain development (Patel et al, 2001). More recently, MYO16 has been implicated in neuronal phosphoinositide 3-kinase (PI3K) signaling (Yokoyama et al, 2011), an extensively studied pathway involved in neuronal function and morphogenesis, as well as in a number of neurological and psychiatric disorders, including schizophrenia and autism (Waite and Eickholt, 2010). MYO16 is a member of the neuronal tyrosine-phosphorylated adaptor for the PI3K (NYAP) family of phosphoproteins, which is comprised of NYAP1, NYAP2, and Myo16/NYAP3. The NYAPs are expressed predominantly in developing neurons and upon stimulation with Contactin5, they are tyrosine phosphorylated by Fyn. Phosphorylated NYAPs interact with PI3K p85 and activate PI3K, Akt, and Rac1. In addition, NYAPs interact with the WAVE1 complex, thus serving as a bridge for a PI3K–WAVE1 interaction, which mediates PI3K-dependent remodeling of the actin cytoskeleton. Importantly, disruption of the NYAP genes in mice affects brain size and neurite elongation (Yokoyama et al, 2011). Notably, meta-analysis of the SAF2, GAIN, and MGS data sets (a total of 2956 cases) showed a gene-wise significant association (p-value of 1.8 × 10−5) with a SNP located within the third intron of the NYAP2 gene (rs1897227), suggesting that variation within this gene family may be modulating the risk of schizophrenia (data not shown).
Additional supporting evidence was provided by expression analysis in brain samples (frontal cortex), which revealed a significant increase in the levels of MYO16 expression in schizophrenia patients compared with controls. Finally, convergent supporting evidence could be found in the existing literature. First, according to the SCAN database (see URLs), the top-associated MYO16 SNP rs9583277 is a potential trans-acting eQTL (expression quantitative trait locus) for MAP3K13 (mitogen-activated protein kinase 13) gene on chromosome 3q27 (p=8 × 10−5). Given a potential convergence of MAP3K13 and PI3K pathways (Ambacher et al, 2012), regulation in trans of MAP3K13 may be mediated by altered MYO16 activity. Interestingly, MAP3K13 can phosphorylate MAP2K7 (mitogen-activated kinase protein 7), which has been recently implicated in schizophrenia (Winchester et al, 2012). In addition, the seven top-associated SNPs identified by our meta-analysis of the SAF2-GAIN-MGS data sets (Table 4) are reported by the SCAN database to have a trans-acting effect on the expression of PAG1 (phosphoprotein associated with glycosphingolipid microdomains 1) on chromosome 8q21.23 (p=2 × 10−6), a gene implicated in brain maturation (Lindquist et al, 2011). Notably, we have previously reported a non-synonymous de novo mutation within PAG1 in a schizophrenia proband (Xu et al, 2011, 2012). Finally, a recent study (Nakayama et al, 2002) reported a physical interaction between the gene products of MYO16 and NRXN1, a synaptic neuronal adhesion molecule that connects presynaptic and postsynaptic neurons and has an important role in cognitive process (Sudhof, 2008). Rare and recurrent deletions disrupting NRXN1 have been reported in patients with schizophrenia and neurodevelopmental disorders. Furthermore, MYO16 has been identified as a candidate risk gene in a genome-wide association study of autism where suggestive association signals were reported in two independent discovery cohorts (Wang et al, 2009) as well as in GWAS of alcohol response (Joslyn et al, 2010) and smoking cessation (Rose et al, 2010).
Although our results suggest that common variation within MYO16 may contribute to the genetic liability to schizophrenia, we cannot exclude the possibility that common variants within MYO16 act in combination with or as surrogates of rare alleles with strong effect in the same or different genes to generate the observed linkage signal in the 13q32–34 locus. We started addressing this question using inherited exonic variant data extracted from our recent whole-exome sequencing study in 146 Afrikaner and 85 US parent–proband trios afflicted with schizophrenia or schizoaffective disorder (Xu et al, 2012). Trios used in the present study and the study by Xu et al (2012) overlap by ∼50% (∼72% if we considered just the South African sample). None of the MYO16 variants located in exons 2–6 (Supplementary Table 4) are in LD with associated SNPs, show differential enrichment in cases vs controls, or show strong allele transmission distortion in affected families. Also, no homozygous or compound heterozygous carriers were identified. Although further analysis in expanded samples and in linked families is required, these results suggest that the association observed with common variants of the MYO16 gene is unlikely to be due to rare exonic variants.
Our results establish MYO16 as a novel candidate gene for schizophrenia. Interpretation of our findings awaits replication in independent data sets.
FUNDING AND DISCLOSURE
This work was supported in part by National Institute of Mental Health (NIMH) Grant MH061399 (to MK) and the Lieber Center for Schizophrenia Research at Columbia University. LRM was partially supported by a Gray Matters Fellowship and BX was partially supported by an NARSAD Young Investigator Award. The authors declare no conflict of interest.
References
Abecasis GR, Burt RA, Hall D, Bochum S, Doheny KF, Lundy SL et al (2004). Genomewide scan in families with schizophrenia from the founder population of Afrikaners reveals evidence for linkage and uniparental disomy on chromosome 1. Am J Hum Genet 74: 403–417.
Ambacher KK, Pitzul KB, Karajgikar M, Hamilton A, Ferguson SS, Cregan SP (2012). The JNK- and AKT/GSK3beta-signaling pathways converge to regulate Puma induction and neuronal apoptosis induced by trophic factor deprivation. PLoS One 7: e46885.
Bowden DW, An SS, Palmer ND, Brown WM, Norris JM, Haffner SM et al (2010). Molecular basis of a linkage peak: exome sequencing and family-based analysis identify a rare genetic variant in the ADIPOQ gene in the IRAS Family Study. Hum Mol Genet 19: 4112–4120.
Cirulli ET, Goldstein DB (2010). Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet 11: 415–425.
Cruz E, Whittington C, Krikler SH, Mascarenhas C, Lacerda R, Vieira J et al (2008). A new 500 kb haplotype associated with high CD8+ T-lymphocyte numbers predicts a less severe expression of hereditary hemochromatosis. BMC Med Genet 9: 97.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37: e67.
Di Rienzo A (2006). Population genetics models of common diseases. Curr Opin Genet Dev 16: 630–636.
Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB (2010). Rare variants create synthetic genome-wide associations. PLoS Biol 8: e1000294.
ISC (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455: 237–241.
Joslyn G, Ravindranathan A, Brush G, Schuckit M, White RL (2010). Human variation in alcohol response is influenced by variation in neuronal signaling genes. Alcohol Clin Exp Res 34: 800–812.
Karayiorgou M, Torrington M, Abecasis GR, Pretorius H, Robertson B, Kaliski S et al (2004). Phenotypic characterization and genealogical tracing in an Afrikaner schizophrenia database. Am J Med Genet B 124B: 20–28.
Karst S, Cheng R, Schmitt AO, Yang H, de Villena FP, Palmer AA et al (2011). Genetic determinants for intramuscular fat content and water-holding capacity in mice selected for high muscle mass. Mamm Genome 22: 530–543.
Lee SH, DeCandia TR, Ripke S, Yang J, Sullivan PF, Goddard ME et al (2012). Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet 44: 247–250.
Li M, Boehnke M, Abecasis GR (2005). Joint modeling of linkage and association: identifying SNPs responsible for a linkage signal. Am J Hum Genet 76: 934–949.
Li M, Boehnke M, Abecasis GR (2006). Efficient study designs for test of genetic association using sibship data and unrelated cases and controls. Am J Hum Genet 78: 778–792.
Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR (2010). MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34: 816–834.
Lin S, Chakravarti A, Cutler DJ (2004). Exhaustive allelic transmission disequilibrium tests as a new approach to genome-wide association studies. Nat Genet 36: 1181–1188.
Lindquist S, Karitkina D, Langnaese K, Posevitz-Fejfar A, Schraven B, Xavier R et al (2011). Phosphoprotein associated with glycosphingolipid-enriched microdomains differentially modulates SRC kinase activity in brain maturation. PLoS One 6: e23978.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ et al (2009). Finding the missing heritability of complex diseases. Nature 461: 747–753.
Nakayama M, Kikuno R, Ohara O (2002). Protein-protein interactions between large proteins: two-hybrid screening using a functionally classified library composed of long cDNAs. Genome Res 12: 1773–1784.
O'Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V et al (2008). Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet 40: 1053–1055.
Patel KG, Liu C, Cameron PL, Cameron RS (2001). Myr 8, a novel unconventional myosin expressed during brain development associates with the protein phosphatase catalytic subunits 1alpha and 1gamma1. J Neurosci 21: 7954–7968.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575.
Ripke S, O'Dushlaine C, Chambert K, Moran J, Kähler A, Akterin S et al (2011). Genome-wide association study identifies five new schizophrenia loci. Nat Genet 43: 969–976.
Rodriguez-Murillo L, Gogos JA, Karayiorgou M (2012). The genetic architecture of schizophrenia: new mutations and emerging paradigms. Annu Rev Med 63: 63–80.
Rose JE, Behm FM, Drgon T, Johnson C, Uhl GR (2010). Personalized smoking cessation: interactions between nicotine dose, dependence and quit-success genotype score. Mol Med 16: 247–253.
Sanna S, Li B, Mulas A, Sidore C, Kang HM, Jackson AU et al (2011). Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet 7: e1002198.
Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe'er I et al (2009). Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460: 753–757.
Shi Y, Li Z, Xu Q, Wang T, Li T, Shen J et al (2011). Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat Genet 43: 1224–1227.
Sobin C, Blundell ML, Conry A, Weiller F, Gavigan C, Haiman C et al (2001). Early, non-psychotic deviant behavior in schizophrenia: a possible endophenotypic marker for genetic studies. Psychiatry Res 101: 101–113.
Sobin C, Roos JL, Pretorius H, Lundy LS, Karayiorgou M (2003). A comparison study of early non-psychotic deviant behavior in Afrikaner and US patients with schizophrenia or schizoaffective disorder. Psychiatry Res 117: 113–125.
Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D et al (2009). Common variants conferring risk of schizophrenia. Nature 460: 744–747.
Suarez BK, Duan J, Sanders AR, Hinrichs AL, Jin CH, Hou C et al (2006). Genomewide linkage scan of 409 European-ancestry and African American families with schizophrenia: suggestive evidence of linkage at 8p23.3-p21.2 and 11p13.1-q14.1 in the combined sample. Am J Hum Genet 78: 315–333.
Sudhof TC (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455: 903–911.
Thompson RF, Langford GM (2002). Myosin superfamily evolutionary history. Anat Rec 268: 276–289.
Torrey EF, Webster M, Knable M, Johnston N, Yolken RH (2000). The stanley foundation brain collection and neuropathology consortium. Schizophr Res 44: 151–155.
Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A et al (2011). Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet 43: 1193–1201.
Waite K, Eickholt BJ (2010). The neurodevelopmental implications of PI3K signaling. Curr Top Microbiol Immunol 346: 245–265.
Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM et al (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320: 539–543.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS et al (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459: 528–533.
Winchester CL, Ohzeki H, Vouyiouklis DA, Thompson R, Penninger JM, Yamagami K et al (2012). Converging evidence that sequence variations in the novel candidate gene MAP2K7 (MKK7) are functionally associated with schizophrenia. Hum Mol Genet 21: 4910–4921.
Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y et al (2012). De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet 44: 1365–1369.
Xu B, Roos JL, Dexheimer P, Boone B, Plummer B, Levy S et al (2011). Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet 43: 864–868.
Xu B, Roos JL, Levy S, van Rensburg EJ, Gogos JA, Karayiorgou M (2008). Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet 40: 880–885.
Xu B, Woodroffe A, Rodriguez-Murillo L, Roos JL, van Rensburg EJ, Abecasis GR et al (2009). Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proc Natl Acad Sci USA 106: 16746–16751.
Yokoyama K, Tezuka T, Kotani M, Nakazawa T, Hoshina N, Shimoda Y et al (2011). NYAP: a phosphoprotein family that links PI3K to WAVE1 signalling in neurons. EMBO J 30: 4739–4754.
Zeggini E, Ioannidis JP (2009). Meta-analysis in genome-wide association studies. Pharmacogenomics 10: 191–201.
Acknowledgements
We thank all the families who participated in this research. For our Afrikaner cohorts, we thank H Pretorius and nursing sisters R van Wyk, C Botha, and H van den Berg for their assistance with subject recruitment, family history assessments, and diagnostic evaluations. We thank Yan Sun for technical assistance with all DNA sample preparations. We also thank the Center for Inherited Disease Research (CIDR), and in particular J Roberts and KF Doheny, for custom genotyping services under NIH Contract Number N01-HG-65403. We thank the Rutgers University Cell and DNA Repository for samples and also the Stanley Medical Research Institute for brain samples. The Rutgers samples were collected in three projects that participated in the NIMH Schizophrenia Genetics Initiative. From 1991 to 1997, the Principal Investigators and Co-Investigators were as follows—(i) Harvard University: MT Tsuang, S Faraone, and J Pepple; (ii) Washington University: CR Cloninger, T Reich, and D Svrakic; and (iii) Columbia University: C Kaufmann, D Malaspina, and JH Friedman.
For the GAIN and MGS_nonGAIN data sets, funding was provided through U01s MH79469 and MH79470. Assistance with data cleaning was provided by the National Center for Biotechnology Information. The GAIN and MGS data sets used for the analyses included in this manuscript were obtained from the database of dbGaP found at dbGaP (see URLs) through dbGaP accession numbers phs000021.v.2.p1 (GAIN) and phs000167.v.1.p1 (nonGAIN). Samples and associated phenotype data for the MGS GWAS study were collected under the following grants: NIMH Schizophrenia Genetics Initiative U01s: MH46276 (CR Cloninger), MH46289 (C Kaufmann), and MH46318 (MT Tsuang); and MGS Part 1 (MGS1) and Part 2 (MGS2) R01s: MH67257 (NG Buccola), MH59588 (BJ Mowry), MH59571 (PV Gejman), MH59565 (Robert Freedman), MH59587 (F Amin), MH60870 (WF Byerley), MH59566 (DW Black), MH59586 (JM Silverman), MH61675 (DF Levinson), and MH60879 (CR Cloninger). Further details of collection sites, individuals, and institutions may be found in Supplementary Table 1 of Sanders et al (2008; PMID: 18198266) and at the study dbGaP pages.
URLs:
dbGaP: http://www.ncbi.nlm.nih.gov/gap/
Plink: http://pngu.mgh.harvard.edu/~purcell/plink/
PedStats: http://www.sph.umich.edu/csg/abecasis/PedStats/
MACH: http://www.sph.umich.edu/csg/abecasis/MACH
LAMP: http://www.sph.umich.edu/csg/abecasis/LAMP
Metal: http://www.sph.umich.edu/csg/abecasis/Metal
UCSC genome browser: http://genome.ucsc.edu/
Stanley Medical Research Institute: http://stanleyresearch.org
Scan database: http://scandb.org
1000 genomes project: www.1000genomes.org, accession no.: ENST00000357550
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
PowerPoint slides
Rights and permissions
About this article
Cite this article
Rodriguez-Murillo, L., Xu, B., Roos, J. et al. Fine Mapping on Chromosome 13q32–34 and Brain Expression Analysis Implicates MYO16 in Schizophrenia. Neuropsychopharmacol 39, 934–943 (2014). https://doi.org/10.1038/npp.2013.293
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.293
Keywords
This article is cited by
-
Altered gene expression in antipsychotic-induced weight gain
npj Schizophrenia (2019)
-
Gene-body 5-hydroxymethylation is associated with gene expression changes in the prefrontal cortex of depressed individuals
Translational Psychiatry (2017)

{kind=link}