
Abstract
What is the defining property of addiction? We dust off a several-decades-long debate about the relative importance of two forms of reinforcement—positive reinforcement, subjectively linked to drug-induced euphoria, and negative reinforcement, subjectively linked to the alleviation of pain—both of which figure importantly in addiction theory; each of these forms has dominated addiction theory in its time. We agree that addiction begins with the formation of habits through positive reinforcement and that drug-opposite physiological responses often establish the conditions for negative reinforcement to come into play at a time when tolerance, in the form of increasing reward thresholds, appears to develop into positive reinforcement. Wise’s work has tended to focus on positive-reinforcement mechanisms that are important for establishing drug-seeking habits and reinstating them quickly after periods of abstinence, whereas Koob’s work has tended to focus on the negative-reinforcement mechanisms that become most obvious in the late stages of sustained addiction. While we tend to agree with each other about the early and late stages of addiction, we hold different views as to (i) the point between early and late at which the diagnosis of ‘addiction’ should be invoked, (ii) the relative importance of positive and negative reinforcement leading up to this transition, and (iii) the degree to which the specifics of negative reinforcement can be generalized across the range of addictive agents.
Similar content being viewed by others
POSITIVE REINFORCEMENT IN ADDICTION: ISSUES OF CAUSE AND CONSEQUENCE (ROY A WISE)
All things have a root and a top
All events an end and a beginning
Whoever understands correctly
What comes first and what follows
Draws nearer to Tao
What is the defining property of addiction; what comes first and what follows? Is positive reinforcement sufficient to establish an addiction? Or must negative reinforcement come into play before an individual is past the point of improbable return? Here we have little agreement. The continuing debate over whether cannabis is addictive; the historical debate over whether nicotine is addictive; and the emerging debates over whether compulsive gambling, video gaming, and sexual behavior are addictive all result from the fact that there is no widely accepted scientific definition that allows us to identify the point at which addiction emerges from a casual or ‘recreational’ habit. In the first place, the committees of clinical experts that set forth diagnostic criteria for such groups as the American Psychiatric Association (APA) and the World Health Organization do not offer a scientific definition (Schuckit, 2012); in the second, there is little agreement between (Edwards, 2012), or even within, these scholarly groups (O’Brien et al, 2006). Instead of ‘addiction,’ the Diagnostic and Statistical Manual of Mental Disorders (DSM) (like Koob in his first quotation below) stresses the term ‘dependence’ (also a fuzzy term, one that sometimes refers to ‘physical’ dependence and sometimes to questionably defined ‘psychic’ dependence). Thus, not surprisingly, the reliability of diagnosis using the DSM—the authority on which new animal models have recently been based (Deroche-Gamonet et al, 2004; Vanderschuren and Everitt, 2004)—has been poor and is getting worse (Hasin et al, 2013). In truth, in our use of the word ‘addiction,’ we share the sorry condition articulated by Humpty Dumpty: ‘When I use a word it means just what I choose it to mean—neither more nor less’ (Norman White used this quotation earlier in a related context; White, 1989).
There are good reasons that the word ‘addiction’ does not appear as a diagnostic category in the DSM. It simply means different things to different people—even to different experts (including Koob and me). When some people think of addiction, they think of the condition of addiction, the condition of someone at the stage of asking for treatment or of causing problems for others: someone who has progressed to the later stages of this progressive process. This is a useful clinical perspective; it focuses on the variety of symptoms that require attention and for which medications might be developed. Others, however, are thinking of the process of addiction, looking for the necessary and sufficient conditions for the development of addiction, the transition from volitional to compulsive drug use. This is a scientific perspective, probing the differences between cause (what comes first) and consequence (what follows). The first use of the word differentiates the extremes of addiction: it differentiates the end points from the drug-naive starting point. The second seeks an intermediate point, a point of no return, a point partway down the slippery slope of increasingly compulsive intake. It is problematic for those interested in process that addiction does not suddenly appear like a fever or a boil at some relatively well-defined and finite point along the way. Rather, it begins to grow with the first reinforced response and strengthens incrementally thereafter, even in the case of a classic addiction such as opiate addiction (McAuliffe and Gordon, 1980).
Opiate addiction offers a good case in point. Opiate addiction is the addiction against which all others are traditionally compared (Tatum and Seevers, 1931). Repeated use of opiates usually, but not always (Zinberg and Jacobson, 1976), leads to tolerance and dependence (Tatum and Seevers, 1931), conditions in which progressively higher doses are required to maintain initial drug effectiveness and in which abrupt drug abstinence results in a largely autonomic withdrawal syndrome involving thermoregulatory (chills and sweats) and gastrointestinal (cramps, diarrhea) disturbances. The chills, sweats, cramps, and diarrhea associated with opiate withdrawal have traditionally been considered to be what maintains opiate addiction; the idea was that opiate use becomes compulsive when continued intake becomes required to avoid these and other unpleasant withdrawal symptoms. Much of classic addiction theory has dealt with the mechanisms of the tolerance and withdrawal distress on this assumption that they motivate increasing intake and increasing compulsion to continue opiate use (Collier, 1980; Goldstein and Goldstein, 1961; Himmelsbach, 1943; Jaffe and Sharpless, 1968; Koob et al, 1989a; Lindesmith, 1947). However, while the desire to relieve withdrawal distress can add to the motivation to continue self-administration of opiates, it has long been recognized that compulsive drug intake can develop before the development of physiological dependence: ‘Dependence and tolerance are neither necessary nor sufficient conditions for the compulsive self-administration of a drug, a behavior that common parlance has defined very well as ‘addiction’’(Goldstein, 1983; see also Deneau et al, 1969; Seevers and Schuster, 1967; and Woods and Schuster, 1971). Indeed, rats will repeatedly self-administer morphine locally into the ventral tegmental area of the brain without signs of tolerance or dependence, whereas they will not self-administer the drug into the periaqueductal gray, where it does cause classic dependence signs (Bozarth and Wise, 1984).
What comes after cannot explain what comes first. That is, withdrawal distress cannot explain the habit that initially established physical dependence, nor can it explain the rapidity of relapse in addicts that have been detoxified (Stewart and Wise, 1992). Consider the cases of cocaine and alcohol self-administration. The ultimate end points of intravenous cocaine addiction in laboratory animals (Bozarth and Wise, 1985; Deneau et al, 1969; Johanson et al, 1976) and alcoholism in humans (McKeon et al, 2008) are convulsions and death, but we do not consider convulsions and impending death to be causes of addiction—they are consequences that need treatment and that are worth modeling, but they are not consequences that help us understand the nature of the compulsion that led to their development. They are part of the disease state caused by addiction; they are not part of the cause of addiction.
While dependence theory is intuitively satisfying and was central to early theories of addiction (Collier, 1965; Goldstein and Goldstein, 1961; Himmelsbach, 1943; Lindesmith, 1947; Tatum and Seevers, 1931), it did not explain the rapid acquisition of intravenous drug taking in laboratory animals (Deneau et al, 1969; Schuster and Thompson, 1969) and it did not appear to explain the self-administration of psychomotor stimulants such as cocaine and amphetamine (Pickens and Harris, 1968; Wise and Bozarth, 1987).
Although the simple laws of instrumental habit reinforcement (Johanson, 1978; Katz, 1989; Wise, 1987) dominated the addiction literature in the 1970s and 1980s, in recent years it has been pointedly argued that mere drug self-administration—the dominant animal model of addiction over the past half-century—does not, by itself, constitute addiction. This is, of course, true and is seemingly confirmed by self-report in a widely cited comorbidity study (Anthony et al, 1994). However, it is dangerous to generalize across drug classes, particularly from survey or introspectional data. Such statements as ‘This pathological behavior appears only in a small proportion (15–17%) of those using drugs’ (Deroche-Gamonet et al, 2004) generalizes across such different drugs as cannabis, on which less than 10% of users reported dependence, and tobacco, on which more than 30% reported dependence. It also generalizes from a single culture; surely the numbers worldwide depend on conditions of drug availability, social disapproval, and other environmental factors (compare the rates of alcoholism between the Czech Republic and Saudi Arabia). Finally, it lumps together self-reported intranasal use of cocaine by some and smoking or intravenous use of the drug by others; the statement that not all individuals who use drugs become addicted to them should be qualified by some kind of objective and quantitative statement as to the degree of drug use that is insufficient to establish addiction. In the Anthony survey (Anthony et al, 1994), there were only minimal guidelines as to what constituted significant ‘using’ of drugs, and there was no differentiation between routes of administration. Trying cocaine by the intranasal route and getting away with it should not encourage us to try it by smoking it or taking it intravenously. Moreover, the question of how many who use a drug become addicted to it depends on what level of drug use or drug seeking we define as the first stage of addiction.
While it is obviously true that simply trying drugs is not a sufficient condition for becoming addicted to them, I would argue that this is not equally true for humans and for laboratory animals. It is clearly true in the case of alcohol; like most humans, most laboratory rats will not drink alcohol to the point of physical dependence (Wise, 1975), let alone to the point of convulsions and death. Alcohol is simply not a very powerful reinforcer for laboratory rats; this is not surprising, as it causes only a modest 100% increase in extracellular levels of the reward transmitter dopamine (Di Chiara and Imperato, 1988). But what about intravenous cocaine or amphetamine, which drive extracellular dopamine levels many times higher (Pettit and Justice, 1989; Ranaldi et al, 1999; Wise et al, 1995b)? In the case of intravenous cocaine, I believe that quite limited initial self-administration of the drug is a sufficient condition for addiction, at least in laboratory rats.
If given unlimited access to intravenous cocaine, rats or monkeys that learn to respond regularly for the drug will, unless their veins or catheters fail, almost invariably take cocaine to the point of convulsions and death (Bozarth and Wise, 1985; Deneau et al, 1969; Johanson et al, 1976). Even if given access to intravenous cocaine for as little as 2 h per day, most animals take the drug steadily, and, if testing continues for weeks or perhaps months, many reach the point of being willing to take the drug despite painful footshock (Deroche-Gamonet et al, 2004) or merely the threat of painful footshock (Vanderschuren and Everitt, 2004). In my lab in Montreal, where we gave animals 4 h of daily access to intravenous cocaine at 1 mg/kg/injection, 89% learned within 1–5 days to take cocaine steadily and without stopping until the drug was withdrawn. While the time between injections was initially quite variable, the variability narrowed to levels that meet my operational definition of compulsive drug taking (Figure 1) (Wise, 1996). We had similar results (88 and 91%) with amphetamine (0.25 mg/kg/injection) and heroin (0.1 mg/kg/injection). We did not food-restrict, prime, or shape these animals; we simply placed rats with jugular catheters for 4 h a day in chambers where accidental or intentional lever pressing resulted in intravenous injections. For the most part, the 10% of our animals that did not learn to take the drug regularly within 5 days could be attributed to catheter or vein problems rather than to ambivalence for the drug. Animals trained in the same way and given unlimited access to intravenous cocaine invariably continued to take the drug, with little variation in rate (except for periods of collapse and sleep), to the point of convulsions and death (Bozarth and Wise, 1985).

From the Wise perspective. Interresponse interval histograms from a rat lever-pressing for intravenous cocaine in 4-h daily sessions at a dose per injection of 1 mg/kg. Of interest is the decrease in variability across days of testing. The narrowing of the distribution of interresponse times over days offers an objective measure of the steady progress to compulsive cocaine taking that develops even with limited daily access to the drug. Wise offers the ratio of mean to standard deviation (M/SD) interresponse time as an objective measure of the subjective label ‘compulsive.’
Thus, in the case of intravenous cocaine, amphetamine, or heroin, my view is that simple unfettered exposure to the drug is a sufficient condition for addiction in rats—rats sampling these drugs at above-threshold doses and by this route of administration learn to take it compulsively and would continue to do so without remission if we did not limit their access to the drug. I would not generalize this to other drugs, to other routes of administration, or even to marginal doses of these drugs (an ED50 means, by definition, a dose that is ineffective in half the population), nor would I generalize it to the specific conditions of limited access used in some animal studies (Ahmed and Koob, 1998; Deroche-Gamonet et al, 2004; Vanderschuren and Everitt, 2004) or to the addiction-resistant Fischer rat, which, although equally sensitive to cocaine reward, is reluctant to lever-press the first time, let alone enough times to learn about the drug’s rewarding consequences (Ranaldi et al, 2001). Most of all, I would not generalize it to humans. Humans are informed about health risks that are not understood by laboratory animals, and it is anticipated rather than immediate consequences that are arguably the strongest deterrent to human addiction, particularly in the case of tobacco smoking. No such communicated deterrent exists for laboratory animals, and in my experience laboratory animals that start taking intravenous cocaine, amphetamine, or heroin never stop taking them voluntarily except when they collapse from exhaustion or convulsions (Bozarth and Wise, 1985; Deneau et al, 1969; Johanson et al, 1976; Pickens and Harris, 1968). I would argue that animals are addicted when they take these drugs as compulsively as do our animals, even when we limit their collateral health problems by restricting their drug access to a few hours a day.
I do not question that addiction can often develop through the three stages outlined by Koob (Koob and Le Moal, 1997b), nor do I question the argument that avoidance of aversive consequences of drug withdrawal can contribute importantly to compulsive drug seeking in established addicts. I do, however, argue that the animals can become addicted long before these stages have progressed significantly. How do we make objective the criteria of compulsive drug intake? To my mind the most important criteria are not whether the animal develops allostatic neuronal or hormonal adaptations and whether the animal persists in drug taking despite footshock. To my mind the rat is addicted when its drug taking becomes regular, predictable, and uninterrupted except by exhaustion or restricted drug intake. Figure 1 illustrates the regularity of intake that I offer as evidence of compulsive cocaine intake in the rat.
My definition of addiction is behavioral. I see addiction as the result of self-administration (Wise, 1988) of drugs that more strongly elevate extracellular dopamine levels (Ranaldi et al, 1999; Wise et al, 1995a; Wise et al, 1995b) than do such natural rewards as food (Hernandez and Hoebel, 1988) or sex (Pfaus et al, 1990), and from the stamping in (Wise, 2004) of the memory traces associated with that self-administration. The alternatives proposed by Koob and others (Kalivas, 2009; Nestler, 1992; Robinson and Berridge, 1993) involve the neuropharmacological consequences of—that is, the neuroadaptations caused by—the drug itself, and if that were a sufficient condition for addiction, then simply making animals dependent on alcohol should give us rodents that drink compulsively to maintain their dependent state by self-medicating withdrawal symptoms. To the surprise of many, this approach has failed multiple times to work in rodents (Lester, 1966; Wise, 1975), and, indeed, even human alcoholics often fail to drink to alleviate withdrawal distress but nonetheless begin drinking again after the severe distress signs have abated (Mello and Mendelson, 1972).
Addiction changes the brain in two ways (Berke and Hyman, 2000). First, the brain is changed by repeated drug exposure, whether self-administered or not (Lüscher and Malenka, 2011; Nestler, 1992); this is the drug-generated sensitization of anti-reward in Koob’s formulation. Second, the brain is changed by the development of memory traces for the drug experience. I would argue that it is this second form of brain change—the emerging memory for the drug experience—that is the more critical difference between the addicted and the nonaddicted brain. Just as the memory of one’s first ejaculation can cause sexual cravings and compulsive masturbation, so can the memory of an initial euphoric drug experience cause drug cravings and compulsive drug seeking (Bijerot, 1980; McAuliffe and Gordon, 1980). It is becoming increasingly clear that self-administration of addictive drugs causes brain and peripheral changes above and beyond those caused by passive receipt of the drug (Caillé et al, 2009; Chen et al, 2008; Dumont et al, 2005; Krawczyk et al, 2013; Wang et al, 2005; Wang et al, 2013; Wise et al, 2008; You et al, 2007). These changes determine, in part, when drug availability is expected (Wang et al, 2013; Wise et al, 2008; You et al, 2007). In the case of smokers, at least, it is when the next cigarette will become available, rather than how long the smoker has been without one, that is the primary stimulus for craving (Dar et al, 2010; Dar et al, 2005). Thus, my feeling is that we should use the term ‘addiction’ in relation to early signs of compulsive drug taking and the cravings associated with it rather than in relation to the subsequent bodily consequences that often—as with nicotine and alcohol, for example—develop only after a long history of drug self-administration.
NEGATIVE REINFORCEMENT IN DRUG ADDICTION: THE DEVIL IN THE DYSPHORIA (GEORGE F KOOB)
The notion of dependence on a drug, object, role, activity or any other stimulus-source requires the crucial feature of negative affect experienced in its absence. The degree of dependence can be equated with the amount of this negative affect, which may range from mild discomfort to extreme distress, or it may be equated with the amount of difficulty or effort required to do without the drug, object, etc. (Russell, 1976)
My view is that drug addiction, in contrast to recreational use of drugs, involves not recruitment or sensitization of reward but rather a drug-generated sensitization of anti-reward: the ‘devil in the dysphoria.’ We have conceptualized addiction as a three-stage cycle: binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation. These stages worsen over time and ultimately involve elements of impulsivity and compulsivity and both positive and negative (Shen and Kalivas, 2013) reinforcement within these frameworks (Koob et al, 1997a). Our argument has been that the stages of the addiction cycle feed into each other, become more intense, and ultimately lead to the pathological state known as addiction, in which negative reinforcement ultimately predominates.
In this context, my argument is that, in addition to the positive reinforcement of the early stages of the addiction process, an additional source of motivation is recruited: negative reinforcement. Negative reinforcement is defined as the process by which removal of an aversive stimulus (or aversive state, in the case of addiction) increases the probability of a response. The aversive, negative emotional state that drives the negative reinforcement of addiction is produced by opponent processes. As such, the negative emotional state is hypothesized to be mediated not only by deficits in the brain systems that mediate positive reinforcement but also by recruitment of brain stress/dysphoria systems that mediate negative reinforcement. We have argued that drug addiction progresses from a source of positive reinforcement that may indeed involve more elements of impulsivity (defined behaviorally as ‘actions which are poorly conceived, prematurely expressed, unduly risky, or inappropriate to the situation and that often result in undesirable consequences’ (Daruna and Barnes, 1993) to sensitization of the brain stress and anti-reward systems that may involve more elements of compulsivity (defined as actions inappropriate to the situation that persist, have no obvious relationship to the overall goal, and often result in undesirable consequences). Note that each drug of abuse with addiction potential may enter the cycle at different points in the cycle, with emphasis on different elements constituting each stage. For example, the binge-intoxication stage is a prominent part of the transition to psychostimulant addiction but plays a much more minor role in tobacco addiction, in which dependence and withdrawal and the ‘craving’ of the preoccupation-anticipation stage predominate. Also, the prominent classic opioid-withdrawal syndrome driven by a significant painful dysphoria is a prominent part of the transition to opioid addiction, in contrast to psychostimulant addiction, which has few ‘physical’ symptoms of withdrawal.
So, what are the historical and current disagreements of Koob with Wise’s positions? First, historically, I was never convinced that dopamine was the reward system or the reward neurotransmitter. For me, dopamine was ‘oil in the machine,’ to quote H Chris Fibiger (sometime in the 1980s). I was both wrong and right about that issue, as it is clear that phasic release of dopamine does drive incentive salience (see definition above and in Figure 2), but, as much research has confirmed, tonic dopamine does have a general facilitory effect on basal ganglia motor function. However, I still insist that reward can take place in parallel or independent of even the dopamine phasic activation. Second, I discovered some 25 years ago that opioid antagonists had the most profound motivational effects in opioid-dependent subjects when injected into reward- and stress-associated brain regions (Koob et al, 1989b) leading me to hypothesize that it was not the physical signs of withdrawal that drove addiction but rather the motivational signs of withdrawal. This led to the reward-deficit/stress-surfeit hypothesis that guides my current thinking.

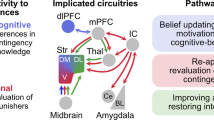
From the Koob perspective. Diagram showing the neurocircuitry of addiction divided heuristically into the three stages of the addiction cycle: binge/intoxication, blue; withdrawal/negative affect, red; and preoccupation/anticipation, green. The neurocircuits involved are also color-coded, with the basal ganglia, including the nucleus accumbens (NAc), dorsal striatum (DS), globus pallidum (GP) and thalamus (Thal) as key elements of the binge/intoxication stage; the extended amygdala, including the central nucleus of the amygdala (AMG), bed nucleus of the stria terminals (BNST), and a transition area in the shell of the nucleus accumbens (NAc) as key elements of the withdrawal/negative affect stage; and the frontal cortex and allocortex, including the prefrontal cortex (PFC), orbitofrontal cortex (OFC), hippocampus (Hippo), and insula (Insula) as key elements of the preoccupation/anticipation stage. Molecular, synaptic, and neurocircuitry neuroadaptations combine to render the four key elements of the transition to addiction: increased incentive salience (Koob’s shortcut translation of Wise’s ‘dominance for the cues that guide and motivate drug seeking over the cues that guide and motivate the seeking of the more natural pleasures of life’), decreased reward, increased stress, and decreased executive function.
My thesis is that addiction involves long-term, persistent dysregulation of the activity of neural circuits that mediate these two sources of reinforcement, deriving from two mechanisms: decreased function of the brain reward systems that normally mediate natural rewards and recruitment of brain stress/anti-reward systems that drive aversive states. Anti-reward, a concept developed by Koob and Le Moal, 2008, is based on an opponent-process framework that forms a general feature of biological systems with the hypothesis that brain systems are in place to limit reward (see footnote in Koob and Le Moal, 1997b). My concept of an anti-reward system is derived from the hypothesis of both within- and between-system neuroadaptations to excessive activation of the reward system at the neurocircuitry level (Koob and Bloom, 1988). Within-system neuroadaptations are defined as the process by which the primary cellular response element to the drug (circuit A) itself adapts to neutralize a drug’s effects. Persistence of the opposing effects after the drug disappears produces adaptation. Between-system neuroadaptations are defined as circuitry changes—circuit B (ie, the anti-reward circuit) is activated, opposing the action of circuit A, potentially at multiple levels (ie, the reward circuit).
As noted by Wise, important neuroanatomical circuits for positive reinforcement include the mesocorticolimbic dopamine system that originates in the ventral tegmental area and projects to the nucleus accumbens and opioid peptides that interface with not only the nucleus accumbens but also the ventral tegmental area (to drive dopamine) and the extended amygdala. However, I argue that what really drives addiction is excessive activation of this reward and incentive salience circuitry that in turn triggers key within-system and between-system neuroadaptive changes that produce the withdrawal/negative affect stage of the addiction cycle.
For the present discussion, the withdrawal/negative affect stage can be defined as the presence of the negative emotional signs of withdrawal in humans, including malaise, dysphoria, alexithymia, chronic irritability, physical pain, emotional pain (ie, hyperkatifeia; Shurman et al, 2010), sleep disturbances, and loss of motivation for natural rewards. It can be characterized in animals by increases in anxiety-like behavior, increases in dysphoric-like responses (conditioned place aversion), decreases in pain thresholds, and increases in reward thresholds during withdrawal from all major drugs of abuse. Particularly compelling for my position, in animal models of extended access, where one observes compulsive-like responding for intravenous self-administration of drugs of abuse, increases in brain reward thresholds occur that temporally precede and are highly correlated with escalation in drug intake (Ahmed et al, 2002; Jang et al, 2013; Kenny et al, 2006).
Such acute withdrawal is associated with notable molecular neuroadaptive changes within the nucleus accumbens and decreased activity of the mesocorticolimbic dopamine system. Molecular neuroadaptive changes during acute withdrawal include changes within medium spiny neurons of the nucleus accumbens such as decreased long-term potentiation (Shen and Kalivas, 2013; Thomas et al, 2001), increased trafficking of AMPA receptors to the surface of the neurons (Wolf and Tseng, 2012), increased adenylate cyclase activity, and phosphorylation (activation) of cyclic adenosine monophosphate response element binding protein (Carlezon et al, 1998). Decreased activity of the mesocorticolimbic dopamine system is reflected by decreased firing in electrophysiological recordings (Diana et al, 1993, 1995) and decreased in vivo release during withdrawal (Weiss et al, 1996). Human imaging studies of individuals with addiction during withdrawal or protracted abstinence have generated results that are consistent with such animal studies. There are decreases in dopamine D2 receptors (presumably reflecting hypodopaminergic functioning) and hyporesponsiveness to dopamine challenge (Volkow et al, 2003). These are hypothesized to be within-system neuroadaptations that may reflect presynaptic release or postsynaptic receptor plasticity.
Perhaps less obvious and less accepted by the field, as dependence and withdrawal develop with compulsive drug use, brain stress/anti-reward systems such as corticotropin-releasing factor (CRF), norepinephrine, and dynorphin are recruited in the extended amygdala, ventral striatum, ventral tegmental area, and prefrontal cortex. The extended amygdala, which is composed of the central nucleus of the amygdala, the bed nucleus of the stria terminalis, and a transition area in the medial (shell) part of the nucleus accumbens, is a system with a massive projection to the lateral hypothalamus, among other areas. For example, extracellular CRF in the extended amygdala is increased during acute withdrawal from all drugs of abuse. Critically, CRF receptor antagonists injected into the extended amygdala block the anxiety-like effect of withdrawal from all drugs of abuse, block the elevation in reward thresholds measured by intracranial self-stimulation associated with acute withdrawal, and blunt excessive drug taking during escalated drug taking with extended access to all drugs of abuse (Koob, 2003, 2009). We have hypothesized that the brain stress neurotransmitter CRF that is known to be activated during the development of excessive drug taking comprises a between-system opponent process, and this activation is manifest when the drug in removed, producing anxiety, hyperkatifeia, and irritability symptoms associated with acute and protracted abstinence.
The aversive stimulus effects of drug withdrawal and stress are also mediated by activation of the dynorphin-κ opioid system (Chartoff et al, 2012; Knoll et al, 2007; Land et al, 2008, 2009; Mague et al, 2003; McLaughlin et al, 2003, 2006; Redila and Chavkin, 2008; Schindler et al, 2010). Even more compelling is that excessive drug self-administration can be blocked by κ antagonists (Walker et al, 2011; Wee et al, 2009) and may be mediated by the shell of the nucleus accumbens (Nealey et al, 2011) and the extended amygdala. These results suggest a between-system neuroadaptation that was originally hypothesized by Carlezon and Nestler (Carlezon et al, 1998), in which activation of cyclic adenosine monophosphate response element binding protein by excessive dopamine and opioid peptide receptor activation in the nucleus accumbens (within-system neuroadaptation) triggers the induction of dynorphin (between-system neuroadaptation) to feed back to suppress dopamine release and glutamate release. Thus, our hypothesis is that anti-reward circuits are recruited as between-system neuroadaptations (Koob and Bloom, 1988) during the development of addiction, producing aversive, dysphoric-like, or stress-like states (Aston-Jones et al, 1999; Koob, 2003; Nestler, 2001) via two mechanisms: direct activation of stress-like, fear-like states in the extended amygdala (CRF-norepinephrine) and activation of dysphoric-like states in the ventral striatum and extended amygdala (dynorphin).
Perhaps a more radical view that differs sharply from Wise’s position at an interpretational level, but perhaps not at a phenotypic level, is that such ‘opponent-like’ processes begin with a single injection of drug but take an allostasis-like hold only if that injection pattern leaves no room for reestablishment of hedonic homeostasis. Indeed, I would argue not only that the Wise ‘habitual process’ begins with the first injection but that the ‘dark side’ also begins with the first injection of the drug and for some individuals there is no return (what determines why some individuals do not return to homeostasis and others do is a current compelling issue for the field). However, that said, the allostasis-like changes in the reward system that I consider compulsive-like are generally evident only in animal models with the extended-access paradigms associated with escalation in drug intake, not with the short-access paradigms illustrated by Wise in Figure 1 (see Kenny et al, 2013, for an explicit example of allostatic changes in reward processes with short- and long-access cocaine self-administration; see also the Supplementary Material online for an experiment to test this hypothesis explicitly).
In summary, my argument is that the combination of decreases in reward neurotransmitter function and recruitment of anti-reward systems provides a powerful source of negative reinforcement that defines compulsive drug-seeking behavior and addiction. The development of the aversive emotional state that drives the negative reinforcement of addiction derives from the excessive engagement of the constructs we term reward, incentive salience, and motivation. We have speculated that the brain motivational systems are a limited resource, and the proper reward balance that we call hedonic homeostasis requires a hedonic Calvinist approach (Koob and Le Moal, 1997b). Drug addiction thus becomes a compelling example of a failure to return to homeostasis, driven by an allostatic negative-reinforcement process.
HOW TO MOVE THE FIELD FORWARD (ROY A WISE AND GEORGE F KOOB)
Each of us has described a personal view of addiction. The question of our editor’s interest is: how are our views similar and how do they differ? They are identical in their starting point: positive reinforcement leads to the initial repetition of drug taking that becomes habitual and eventually compulsive. Wise does not question that positive reinforcement is necessarily limited by opponent processes—positive reinforcement involves a positive feedback system, and positive feedback mechanisms, if not limited by some form of opponent process, necessarily self-destruct—nor does he question that negative reinforcement comes to contribute very significantly to the compulsive chronic use of addictive drugs. Where the two disagree are largely in semantic areas. First, Wise does not accept ‘dependent’ as a synonym for ‘addicted.’ That said, where along the continuum between volitional and compulsive drug seeking—between initial positive reinforcement and progressively developing positive+negative reinforcement—is it most appropriate to draw a line and consider an individual or a laboratory animal ‘addicted’?
From a conceptual framework, even if one accepts Koob’s ‘dark side’ hypothesis, an argument can be made that, instead of anti-reward processes increasing, perhaps reward selection narrows (rats are no longer interested in alternative rewards, including intracranial self-stimulation, because the other rewards are not cocaine). In other words, the value of the drug increases at the expense of other natural (or unnatural) rewards, and instead of reflecting a general negative affective state it in fact reflects a very specific selection by the animal to focus its resources on obtaining and consuming only its drug of choice and discounting any other source. However, such a formulation does not account for the observations in animal models that pronounced stress-like responses and dysphoric-like responses dominate during drug abstinence and persist into protracted abstinence. In addition, CRF antagonists can reverse the elevation in reward thresholds associated with withdrawal from opioids, nicotine, and alcohol, and both CRF and κ-antagonists can reverse compulsive drug seeking and anxiety-like and dysphoric-like responses. It is true that individuals with substance use disorders prefer their drug of choice, but Koob would attribute that more to the specific pattern or quality of the ‘malaise dysphoria’ induced by chronic excessive use of the drug. Thus, for example, he would speculate that κ-antagonists may be more effective in reversing compulsive psychostimulant seeking and CRF antagonists may be more effective in reversing compulsive alcohol seeking, although such data have yet to be obtained.
We also differ as to the kinds of the animal models we study. One of us works largely with the early stages of addiction, for which a minimalist model is appropriate; the other works extensively on addiction stages where a longer-term, more inclusive model is appropriate. Wise is more interested in the second of the two kinds of difference between the addicted and the nonaddicted brain that are outlined by Berke and Hyman: the mechanisms of ‘the association of drug-related stimuli with specific learned behaviors’ (Berke and Hyman, 2000). Koob’s interests extend more into the ‘neuronal (and hormonal) adaptations, which are mostly homeostatic responses to excessive stimulation’ (Berke and Hyman, 2000). Each of us appreciates the value of the other’s models; neither type of model should be seen as being in conflict with the other. Models are not the real thing, and we discovered while writing this paper that we each have favorite quotes to underscore this point. Wise suggests a favorite of Yavin Shaham: ‘The best material model for a cat is another, or preferably the same, cat’ (Rosenblueth and Wiener, 1945). And, as Koob says about the obsession with face validity in animal models, ‘What you are looking for is a rat standing on the street corner smoking a cigarette with a bottle of liquor in a brown paper bag, and one will never attain such face validity in an animal model.’ The fact that our models and our emphases differ is primarily a reflection of our individual interests in different parts of the proverbial elephant. The field will continue to move forward as animal models are used to probe more deeply the variety of dimensions of the complex condition we variously call addiction.
FUNDING AND DISCLOSURE
Preparation of this essay was supported by the Intramural Research Program (RW) and extramural grant AA006420 (GFK) from the National Inistitutes of Health. Dr Wise and Dr Koob declare no potential conflict of interest.
BLOG COMMENTS
For discussion of this Circumspective, go to http://www.acnp.org/resources/acnpblog.aspx
References
Ahmed SH, Kenny PJ, Koob GF, Markou A (2002). Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat Neurosci 5: 625–626.
Ahmed SH, Koob GF (1998). Transition from moderate to excessive drug intake: change in hedonic set point. Science 282: 298–300.
Anthony JC, Warner LA, Kessler RC (1994). Comparative epidemiology of dependence on tobacco, alcohol, controlled substances, and inhalants: Basic findings from the national ocmorbiditiy survey. Exper Clin Psychopharmacol 2: 244–268.
Aston-Jones G, Delfs JM, Druhan J, Zhu Y (1999). The bed nucleus of the stria terminalis. A target site for noradrenergic actions in opiate withdrawal. Ann NY Acad Sci 877: 486–498.
Berke JD, Hyman SE (2000). Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25: 515–532.
Bijerot N (1980). Addiction to pleasure: A biological and social-psychological theory of addiction. In: Lettieri DJ, Sayers M, Pearson HW (eds). Theories on Drug Abuse: Selected Contemporary Perspectives. National Institute on Drug Abuse: Rockville, MD. pp 246–255.
Bozarth MA, Wise RA (1984). Anatomically distinct opiate receptor fields mediate reward and physical dependence. Science 224: 516–517.
Bozarth MA, Wise RA (1985). Toxicity associated with long-term intravenous heroin and cocaine self- administration in the rat. JAMA 254: 81–83.
Caillé S, Guillem K, Cador M, Manzoni O, Georges F (2009). Voluntary nicotine consumption triggers in vivo potentiation of cortical excitatory drives to midbrain dopaminergic neurons. J Neurosci 29: 10410–10415.
Carlezon WAJ, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N et al (1998). Regulation of cocaine reward by CREB. Science 282: 2272–2275.
Chartoff E, Sawyer A, Rachlin A, Potter D, Pliakas A, Carlezon WA (2012). Blockade of kappa opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology 62: 167–176.
Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM et al (2008). Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59: 288–897.
Collier HO (1965). A general theory of the genesis of drug dependence by induction of receptors. Nature 205: 181–182.
Collier HOJ (1980). Cellular site of opiate dependence. Nature 283: 625–629.
Dar R, Rosen-Korakin N, Shapira O, Gottlieb Y, Frenk H (2010). The craving to smoke in flight attendants: relations with smoking deprivation, anticipation of smoking, and actual smoking. J Abnorm Psychol 119: 248–253.
Dar R, Stronguin F, Marouani R, Krupsky M, Frenk H (2005). Craving to smoke in orthodox Jewish smokers who abstain on the Sabbath: a comparison to a baseline and a forced abstinence workday. Psychopharmacology 183: 294–299.
Daruna JH, Barnes PA (1993). A neurodevelopmental view of impulsivity. In: McCown WG, Johnson JL, Shure MB (eds). The Impulsive Client: Theory, Research, and Treatment. American Psychological Association: Washington, DC. pp 23–37.
Deneau G, Yanagita T, Seevers MH (1969). Self-administration of psychoactive substances by the monkey. Psychopharmacologia 16: 30–48.
Deroche-Gamonet V, Belin D, Piazza PV (2004). Evidence for addiction-like behavior in the rat. Science 305: 1014–1017.
Di Chiara G, Imperato A (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85: 5274–5278.
Diana M, Pistis M, Carboni S, Gessa GL, Rossetti ZL (1993). Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: electrophysiological and biochemical evidence. Proc Natl Acad Sci USA 90: 7966–7979.
Diana M, Pistis M, Muntoni A, Gessa G (1995). Profound decrease of mesolimbic dopaminergic neuronal activity in morphine withdrawn rats. J Pharmacol Exp Ther 272: 781–785.
Dumont E, Mark GP, Mader S, Williams JT (2005). Self-administration enhances excitatory synaptic transmission in the bed nucleus of the stria terminalis. Nat Neurosci 8: 413–414.
Edwards G (2012). ‘The evil genius of the habit’: DSM-5 seen in historical context. J Stud Alcohol Drugs 73 (4): 699–701.
Goldstein A (1983). Some thoughts about endogenous opioids and addiction. Drug Alcohol Depend 11: 11–14.
Goldstein DB, Goldstein A (1961). Possible role of enzyme inhibition and repression in drug tolerance and addiction. Biochem Pharmacol 8: 48.
Hasin DS, Auriacombe M, Borges G, Bucholz K, Budney A, Crowley T et al (2013). The DSM-5 field trials and reliability of alcohol use disorder. Am J Psychiatry 170: 442–443.
Hernandez L, Hoebel BG (1988). Food reward and cocaine increase extracellular dopamine in the nucleus accumbens as measured by microdialysis. Life Sci 42: 1705–1712.
Himmelsbach CK (1943). Morphine, with reference to physical dependence. Federat Proc 2: 201–203.
Hughart B (1984) Bridge of Birds. Ballantine: New York.
Jaffe JH, Sharpless SK (1968). Pharmacological denervation supersensitivity in the central nervous system: A theory of physical dependence. In: Wikler AH (ed). The Addictive States. Williams and Wilkins: Baltimore, MD. pp 226–246.
Jang CG, Whitfield T, Schulteis G, Koob GF, Wee S (2013). A dysphoric-like state during early withdrawal from extended access to methamphetamine self-administration in rats. Psychopharmacology 225: 753–763.
Johanson CE (1978). Drugs as reinforcers. In: Blackman DE, Sanger DJ (eds) Contemporary Research in Behavioral Pharmacology. Plenum Press: New York. pp 325–390.
Johanson CE, Balster RL, Bonese K (1976). Self-administration of psychomotor stimulant drugs: the effects of unlimited access. Pharmacol Biochem Behav 4: 45–51.
Kalivas PW (2009). The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10: 561–572.
Katz JL (1989). Drugs as reinforcers:Pharmacological and behavioural factors. In: Liebman JM, Cooper SJ (eds). The neuropharmacological basis of reward. Oxford University Press: New York. pp 164–213.
Kenny PJ, Chen SA, Kitamura O, Markou A, Koob GF (2006). Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J Neurosci 26: 5894–5900.
Kenny PJ, Voren G, Johnson PM (2013). Dopamine D2 receptors and striatopallidal transmission in addiction and obesity. Curr Opin Neurobiol 23: 535–538.
Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA Jr. (2007). Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. J Pharmacol Exp Ther 323: 838–845.
Koob GF (2003). Alcoholism: allostasis and beyond. Alcohol Clin Exper Res 27: 232–243.
Koob GF (2009). Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology 56 (Suppl 1): 18–31.
Koob GF, Bloom FE (1988). Cellular and molecular mechanisms of drug dependence. Science 242: 715–723.
Koob GF, Caine SB, Parsons L, Markou A, Weiss F (1997a). Opponent process model and psychostimulant addiction. Pharmacol Biochem Behav 57: 513–521.
Koob GF, Le Moal M (1997b). Drug abuse: hedonic homeostatic dysregulation. Science 278: 52–58.
Koob GF, Le Moal M (2008). Addiction and the brain antireward system. Ann Rev Psychol 59: 29–53.
Koob GF, Stinus L, Le Moal M, Bloom FE (1989a). Opponent process theory of motivation: Neurobiological evidence from studies of opiate dependence. Neurosci Biobehav Rev 13: 135–140.
Koob GF, Wall TL, Bloom FE (1989b). Nucleus accumbens as a substrate for the aversive stimulus effects of opiate withdrawal. Psychopharmacology 98: 530–534.
Krawczyk M, Mason X, Debacker J, Sharma R, Normandeau CP, Hawken ER et al (2013). D1 dopamine receptor-mediated LTP at GABA synapses encodes motivation to self-administer cocaine in rats. J Neurosci 33: 11960–11971.
Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C (2008). The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci 28: 407–414.
Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D et al (2009). Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proce Nat Acad Sci USA 106: 19168–19173.
Lester D (1966). Self-selection of alcohol by animals, human variation and the etiology of alcoholism: A critical review. Quart J Stud Alcohol 27: 395–438.
Lindesmith AR (1947) Opiate Addiction. Principia Press: Bloomington, IN.
Lüscher C, Malenka R.C. (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69: 650–663.
Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC Jr. et al (2003). Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther 305: 323–330.
McAuliffe WE, Gordon RA (1980). Reinforcement and the combination of effects: summary of a theory of opiate addiction. NIDA Res Monogr 30: 137–141.
McKeon A, Frye MA, Delanty N (2008). The alcohol withdrawal syndrome. J Neurol Neurosurg Psychiatry 79: 854–862.
McLaughlin JP, Li S, Valdez J, Chavkin TA, Chavkin C (2006). Social defeat stress-induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacology 31: 1241–1248.
McLaughlin JP, Marton-Popovici M, Chavkin C (2003). Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci 23: 5674–5683.
Mello NK, Mendelson JH (1972). Drinking patterns during work-contingent and noncontingent alcohol acquisition. Psychosom Med 34: 139–164.
Nealey KA, Smith AW, Davis SM, Smith DG, Walker BM (2011). kappa-opioid receptors are implicated in the increased potency of intra-accumbens nalmefene in ethanol-dependent rats. Neuropharmacology 61: 35–42.
Nestler EJ (1992). Molecular mechanisms of drug addiction. J Neurosci 12: 2439–2450.
Nestler EJ (2001). Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119–128.
O’Brien CP, Volkow N, Li TK (2006). What’s in a word? Addiction versus dependence in DSM-V. Amer J Psychiatry 163: 764–765.
Pettit HO, Justice JB (1989). Dopamine in the nucleus accumbens during cocaine self-administration as studied by in vivo microdialysis. Pharmacol Biochem Behav 34: 899–904.
Pfaus JG, Damsma G, Nomikos GG, Wenkstern DG, Blaha CD, Phillips AG et al (1990). Sexual behavior enhances central dopamine transmission in the male rat. Brain Res 530: 345–348.
Pickens R, Harris WC (1968). Self-administration of d-amphetamine by rats. Psychopharmacologia 12: 158–163.
Ranaldi R, Bauco P, McCormick S, Cools AR, Wise RA (2001). Equal sensitivity to cocaine reward in addiction-prone and addiction-resistant rat genotypes. Behav Pharmacol 12: 527–534.
Ranaldi R, Pocock D, Zereik R, Wise RA (1999). Dopamine fluctuations in the nucleus accumbens during maintenance, extinction, and reinstatement of intravenous D-amphetamine self- administration. J Neurosci 19: 4102–4109.
Redila VA, Chavkin C (2008). Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology 200: 59–70.
Robinson TE, Berridge KC (1993). The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev 18: 247–291.
Rosenblueth A, Wiener N (1945). The role of models in science. Philos Sci 12: 316–321.
Russell MAH (1976). What is dependence? In: Edwards G (ed). Drugs and Drug Dependence. Lexington Books: Lexington, MA. pp 182–187.
Schindler AG, Li S, Chavkin C (2010). Behavioral stress may increase the rewarding valence of cocaine-associated cues through a dynorphin/kappa-opioid receptor-mediated mechanism without affecting associative learning or memory retrieval mechanisms. Neuropsychopharmacology 35: 1932–1942.
Schuckit MA (2012). Editor’s corner: editorial in reply to the comments of Griffith Edwards. J Stud Alcohol Drug 73: 521–522.
Schuster CR, Thompson T (1969). Self administration of and behavioral dependence on drugs. Ann Rev Pharmacol 9: 483–502.
Seevers MH, Schuster CR (1967). Self-administration of psychoactive drugs by the monkey: a measure of psychological dependence. Science 158: 535.
Shen H, Kalivas PW (2013). Reduced LTP and LTD in prefrontal cortex synapses in the nucleus accumbens after heroin self-administration. Int J Neuropsychopharmacol 16: 1165–1167.
Shurman J, Koob GF, Gutstein HB (2010). Opioids, pain, the brain, and hyperkatifeia: a framework for the rational use of opioids for pain. Pain Med 11: 1092–1098.
Stewart J, Wise RA (1992). Reinstatement of heroin self-administration habits: morphine prompts and naltrexone discourages renewed responding after extinction. Psychopharmacology 108: 79–84.
Tatum AL, Seevers MH (1931). Theories of drug addiction. Physiological Review 11: 107–120.
Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4: 1217–1223.
Vanderschuren LJ, Everitt BJ (2004). Drug seeking becomes compulsive after prolonged cocaine self-administration. Science 305: 1017–1019.
Volkow ND, Fowler JS, Wang GJ (2003). The addicted human brain: insights from imaging studies. J Clin Invest 111: 1444–1451.
Walker BM, Zorrilla EP, Koob GF (2011). Systemic kappa-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addict Biol 16: 116–119.
Wang B, Shaham Y, Zitzman D, Azari S, Wise RA, You ZB (2005). Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci 25: 5389–5396.
Wang B, You ZB, Oleson EB, Cheer JF, Myal S, Wise RA (2013). Conditioned contribution of peripheral cocaine actions to cocaine reward and cocaine-seeking. Neuropsychopharmacol 38: 1763–1769.
Wee S, Orio L, Ghirmai S, Cashman JR, Koob GF (2009). Inhibition of kappa opioid receptors attenuated increased cocaine intake in rats with extended access to cocaine. Psychopharmacology 205: 565–575.
Weiss F, Parsons LH, Schulteis G, Hyytia P, Lorang MT, Bloom FE et al (1996). Ethanol self-administration restores withdrawal-associated deficiencies in accumbal dopamine and 5-hydroxytryptamine release in dependent rats. J Neurosci 16: 3474–3485.
White NM (1989). Reward or reinforcement: what’s the difference? Neurosci Biobehav Rev 13: 181–186.
Wise RA (1975). Maximization of ethanol intake in the rat. Adv Exp Med Biol 59: 279–294.
Wise RA (1987). Intravenous drug self-administration: A special case of positive reinforcement. In: Bozarth MA (ed). Methods of Assessing the Reinforcing Properties of Abused Drugs. Springer: New York. pp 117–141.
Wise RA (1988). The neurobiology of craving: implications for the understanding and treatment of addiction. J Abnorm Psychol 97: 118–132.
Wise RA (1996). Neurobiology of addiction. Curr Opin Neurobiol 6: 243–251.
Wise RA (2004). Dopamine, learning and motivation. Nat Rev Neurosci 5: 483–494.
Wise RA, Bozarth MA (1987). A psychomotor stimulant theory of addiction. Psychol Rev 94: 469–492.
Wise RA, Leone P, Rivest R, Leeb K (1995a). Elevations of nucleus accumbens dopamine and DOPAC levels during intravenous heroin self-administration. Synapse 21: 140–148.
Wise RA, Newton P, Leeb K, Burnette B, Pocock P, Justice JB (1995b). Fluctuations in nucleus accumbens dopamine concentration during intravenous cocaine self-administration in rats. Psychopharmacology 120: 10–20.
Wise RA, Wang B, You ZB (2008). Cocaine serves as a peripheral interoceptive conditioned stimulus for central glutamate and dopamine release. PLoS One 3: e2846.
Wolf ME, Tseng KY (2012). Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front Molec Neurosci 5: 72.
Woods JH, Schuster CR (1971). Opiates as reinforcing stimuli. In: Thompson T, Pickens R (eds). Stimulus Properties of Drugs. Appleton, Century, Crofts: New York. pp 163–175.
You ZB, Wang B, Zitzman D, Azari S, Wise RA (2007). A role for conditioned ventral tegmental glutamate release in cocaine seeking. J Neurosci 27: 10546–10555.
Zinberg NE, Jacobson RC (1976). The natural history of ‘chipping’. Am J Psychiat 133: 37–40.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article
Cite this article
Wise, R., Koob, G. The Development and Maintenance of Drug Addiction. Neuropsychopharmacol 39, 254–262 (2014). https://doi.org/10.1038/npp.2013.261
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.261
Keywords
This article is cited by
-
The genetic susceptibility analysis of TAAR1 rs8192620 to methamphetamine and heroin abuse and its role in impulsivity
European Archives of Psychiatry and Clinical Neuroscience (2024)
-
The Perceived Causes of Problems with Substance Use, Gambling, and Other Behavioural Addictions from the Perspective of People with Lived Experience: a Mixed-Methods Investigation
International Journal of Mental Health and Addiction (2024)
-
Changes in distinct brain systems identified with fMRI during smoking cessation treatment with varenicline: a review
Psychopharmacology (2024)
-
Cebranopadol for the Treatment of Chronic Pain
Current Pain and Headache Reports (2023)
-
Food Addiction Features Are Related to Worse Academic Performance in Adolescents
International Journal of Mental Health and Addiction (2023)
