
Abstract
One of the most exciting discoveries in the learning and memory field in the past two decades is the observation that active regulation of gene expression is necessary for experience to trigger lasting functional and behavioral change, in a wide variety of species, including humans. Thus, as opposed to the traditional view of ‘nature’ (genes) being separate from ‘nurture’ (environment and experience), it is now clear that experience actively drives alterations in central nervous system (CNS) gene expression in an ongoing fashion, and that the resulting transcriptional changes are necessary for experience to trigger altered long-term behavior. In parallel over the past decade, epigenetic mechanisms, including regulation of chromatin structure and DNA methylation, have been shown to be potent regulators of gene transcription in the CNS. In this review, we describe data supporting the hypothesis that epigenetic molecular mechanisms, especially DNA methylation and demethylation, drive long-term behavioral change through active regulation of gene transcription in the CNS. Specifically, we propose that epigenetic molecular mechanisms underlie the formation and stabilization of context- and cue-triggered fear conditioning based in the hippocampus and amygdala, a conclusion reached in a wide variety of studies using laboratory animals. Given the relevance of cued and contextual fear conditioning to post-traumatic stress, by extension we propose that these mechanisms may contribute to post-traumatic stress disorder (PTSD) in humans. Moreover, we speculate that epigenetically based pharmacotherapy may provide a new avenue of drug treatment for PTSD-related cognitive and behavioral function.
Similar content being viewed by others
INTRODUCTION
A long-standing aim in the study of learning and memory has been to uncover the biological mechanisms that support the maintenance of memories over a lifetime despite of the constant turnover of the molecules that underlie memory storage (Day and Sweatt, 2010). In recent years, the discovery that epigenetic processes regulate experience-induced plasticity in the neural networks that support memory formation and maintenance has produced a fundamental shift in our understanding of the molecular mechanisms that underlie information storage in the central nervous system (CNS). Epigenetic mechanisms have classically been studied for their role in the regulation of cellular differentiation during development and in the maintenance of cellular identity over the lifespan. In the present discussion, we use the term epigenetics to refer to chromatin-regulating molecular mechanisms in non-dividing neuronal cells, rather than the traditional role of epigenetic mechanisms in driving the transmission of heritable changes in gene expression during cell division (reviewed in Levenson and Sweatt, 2005; Sweatt, 2009; and Wood et al, 2006). This review addresses the broad hypothesis that epigenetic marking of chromatin is modified in response to an organism’s experience, and has a role in dynamically regulating the gene transcription that supports synaptic plasticity and long-term behavioral change (Graff and Mansuy, 2008; Jiang et al, 2008; Sweatt, 2009). Much of the work devoted to testing the idea that epigenetic mechanisms regulate gene transcription necessary for memory function has focused on post-translational histone modifications, especially histone acetylation (Barrett and Wood, 2008; Graff and Mansuy, 2008; Jiang et al, 2008; Sweatt, 2009), and this topic was discussed by Ted Abel in another review in this special issue of Neuropsychopharmacology Reviews. DNA methylation is another epigenetic mechanism potentially important for synaptic plasticity and fear memory, and in this review, we will discuss this mechanism in detail. Further, we extend our discussion to the emerging hypothesis that the same mechanisms that underlie normal fear learning also operate in the establishment and persistence of the intense pathological fear that is characteristic of post-traumatic stress disorder (PTSD).
DNA METHYLATION AS A CANDIDATE STABLE MOLECULAR MARK IN CELLS
DNA methylation and histone modifications are the two most extensively investigated epigenetic mechanisms (Figure 1). Until recently, it was thought that once laid down, DNA methylation remained unchanged for the lifetime of the organism, although recent studies have challenged this view (discussed below). DNA methylation is an epigenetic mark that is most often associated with transcriptional silencing, although there are instances in which DNA methylation can have an activating role (Chahrour et al, 2008). DNA methylation occurs in regions of the genome rich in cytosine-guanine (CpG) dinucleotides (ie, CpG islands), which are often found in the promoter region of genes. The family of enzymes that carry out DNA methylation, the DNMTs, come in two variants, maintenance DNMTs, including DNMT1, and de novo DNMTs, including DNMT3a and 3b. The function of de novo DNMTs is to methylate previously unmethylated CpG sites, whereas the maintenance DNMTs methylate hemi-methylated DNA.

Schematic representation of epigenetic marks. (a) DNA is condensed within the nucleus through interactions with histones. The DNA–protein complex is referred to as chromatin. (b) The N-terminal tail of a histone has several sites for epigenetic marking that can promote or repress gene transcription. (c) Methylation of DNA in which a methyl group (red diamonds) is transferred to cytosines in genomic regions in and around gene promoters rich in cytosine-guanine nucleotides (CpG islands). From Jiang et al (2008).
DNA methylation and attendant changes in chromatin structure are capable of self-regeneration and self-perpetuation. Self-perpetuation of DNA methylation is accomplished, in part, by the action of maintenance DNMTs. DNMTs can recognize a hemi-methylated CpG dinucleotide (ie, methylated on only one strand of the DNA) and convert the complementary CpG on the opposite strand into a methylated CpG. Through this mechanism, DNA can be perpetually methylated in the face of ongoing turnover of molecular marks, making it an ideal candidate for the perpetuation of memory. Griffith and Mahler (1969) were the first to suggest that DNA methylation or demethylation may be involved in memory formation through a ‘ticketing’ mechanism that has since been proven inaccurate in its details. Years later, Crick (1984) proposed that a self-perpetuating biochemical autoconversion of methylated DNA might serve as a memory mechanism at the molecular level. Crick’s idea has lain largely unaddressed since then, with the exception of one further paper discussing the theoretical concept published by Holliday (1999), one of the founding fathers of the epigenetics field, and a more recent treatment by Tollefsbol (Liu et al, 2009). These earlier discussions lay the foundation for our studies that provided the first empirical evidence in support of a role for DNA methylation in the formation and stabilization of fear memories.
FEAR LEARNING AND MEMORY IN RODENTS
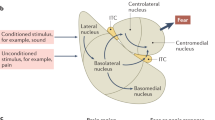
Memory formation is not a unitary process. In order for newly acquired information to develop into a stable memory, it must be consolidated through a complex set of molecular changes in distinct brain regions that support discrete forms of associative learning. Fear conditioning is a commonly used model of associative learning in rodents, in which memory for a context-shock association is stabilized via hippocampal-dependent consolidation processes, and memory for a cue-shock association is stabilized through amygdala-dependent consolidation processes (see Box 1 and Johnson et al, 2012). Previously consolidated memories can become labile upon recall, wherein an additional set of molecular changes are required for the recalled memory to become updated or re-stabilized in a process that has been termed reconsolidation (McKenzie and Eichenbaum, 2011). Over time, recently acquired contextual fear memories are downloaded from the temporary storage site in the hippocampus to the permanent site of remote memory storage in the cortex (Frankland et al, 2004). There is now evidence to suggest that DNA methylation is involved in all stages (ie, consolidation, reconsolidation and maintenance) of memory formation for distinct associations (ie, cue or context), indicating that DNA methylation is an essential and a broadly utilized mechanism for memory formation and maintenance.
EPIGENETIC MECHANISMS IN HIPPOCAMPUS-DEPENDENT FEAR CONSOLIDATION
As our first step in pursuing the idea that epigenetic molecular mechanisms might be involved in memory, we investigated whether mechanisms regulating chromatin structure were activated during the early stages of long-term memory formation in the hippocampus (Levenson et al, 2004b). Specifically, we investigated hippocampal histone acetylation during the initial stages of consolidation of long-term association memories using a contextual fear conditioning paradigm (see Box 1). We found that acetylation of histone H3 in the area CA1 of the hippocampus was regulated in contextual fear conditioning and that pharmacological enhancement of histone acetylation with histone deacetylase (HDAC) inhibitors improved memory formation in normal adult animals (Levenson et al, 2004b). This finding has been replicated and greatly extended in a variety of laboratories, using various behavioral paradigms such as cued fear conditioning and contextual fear conditioning and extinction (eg, Bredy and Barad, 2008; Lattal et al, 2007; Lubin et al, 2008; Maddox and Schafe, 2011; Miller et al, 2008; Monsey et al, 2011; Stafford et al, 2012).
Several pieces of evidence are now available that support the idea that DNA methylation is also critically involved in hippocampal memory function in the adult CNS. Levenson et al. (2006) demonstrated that inhibitors of DNMT activity reduced DNA methylation of the plasticity-promoting genes reelin and BDNF (brain-derived neurotrophic factor) in hippocampal slices. Additional studies demonstrated that hippocampal DNMT 3a and 3b expression was upregulated in response to contextual fear conditioning (Miller and Sweatt, 2007). This upregulation is functionally relevant, as blocking hippocampal DNMT activity pharmacologically (Lubin et al, 2008; Miller and Sweatt, 2007; Figure 2), or through a forebrain-specific DNMT1 and 3a knockout (Feng et al, 2010), impaired contextual fear conditioning. Moreover, contextual fear conditioning is associated with rapid (within 1 h of training) methylation and transcriptional silencing of the memory suppressor gene protein phosphatase 1 and demethylation and transcriptional activation of the plasticity gene reelin (Miller and Sweatt, 2007). These findings suggest that DNA methylation is an important regulator of experience-induced changes in gene expression and behavior. DNA methylation and mRNA expression levels of these genes returned to baseline levels within 24 h of fear conditioning, indicating that the covalent modification of DNA in response to associative learning is both rapid and transient. These findings have the surprising implication that both active DNA methylation and active demethylation might be involved in long-term memory consolidation in the adult CNS.

DNMT inhibition blocks long-term memory formation. (a) DNMT inhibitors infused bilaterally into the hippocampus immediately after contextual fear conditioning impaired the consolidation of long-term memory, as assessed by freezing behavior 24 h later. DNMT inhibitors were not effective when given outside of the initial consolidation window, 6 h after training. (b) DNMT inhibitors (three injections, 12 h apart) administered into the anterior cingulate cortex (ACC) 24 h after fear conditioning had no effect on remote memory recall, as assessed by freezing behavior 30 days after training. The same injection protocol administered 29 days after training impaired freezing behavior at 30 days, indicating that DNA methylation in the ACC is critical for memory maintenance, but not for memory formation.
A recent series of studies found that the brain-derived neurotrophic factor BDNF gene locus, which is associated with learning and memory in general and with fear memory in particular (Alonso et al, 2005; Bekinschtein et al, 2007; Bekinschtein et al, 2008a; Bekinschtein et al, 2008b), is also subject to memory-associated changes in DNA methylation. Specifically, contextual fear conditioning was associated with decreased methylation and increased expression of BDNF exon IV, and this effect was regulated by the NMDA receptor (Lubin et al, 2008 and Figure 3). Further, we demonstrated dynamic regulation of DNA methylation at specific CpG islands around the transcription initiation site of BDNF exon IV in rats exposed to contextual fear conditioning (Lubin et al, 2008). We also found that a change in DNA methylation at specific BDNF promoters could affect exon-specific BDNF gene mRNA levels in the hippocampus. These results suggest differential epigenetic regulation (ie, DNA methylation) of specific BDNF gene promoters in the adult CNS and implicate DNA methylation as a potential mechanism for influencing alternative splicing in response to experience.

Alterations in DNA methylation and gene transcription with contextual fear conditioning. (a) Fear conditioning induces rapid and reversible gene-specific changes in DNA methylation and transcription in the hippocampus. (b) Fear conditioning induces delayed and persistent changes in DNA methylation and expression of the calcineurin gene in the medial prefrontal cortex (mPFC). This effect is gene specific, as reelin methylation in the mPFC is rapidly induced and returns to baseline by 30 days.
Additional work has also begun to investigate the mechanistic interplay between histone acetylation and DNA methylation (Miller and Sweatt, 2007). Specifically, contextual fear conditioning results in increased levels of acetylated histone H3 at specific BDNF promoters (Lubin et al, 2008) and inhibition of DNMT activity blocked global increases in learning-induced histone acetylation, along with memory consolidation, as previously reported (Levenson et al, 2006; Miller and Sweatt, 2007). These deficits, however, were rescued by pharmacologically increasing histone acetylation before DNMT inhibition, indicating that these modifications interact to regulate memory formation. Together, these findings lend strong support to the idea that DNMT activity is not only necessary for the establishment of fear memory, but that DNA methylation and histone modifications work together to regulate memory formation in the adult hippocampus. It is important to note that these findings suggest that memory formation involves both increased methylation at memory-suppressor genes and decreased methylation at memory-promoting genes. Thus, long-term functional change might be driven by either hypermethylation or hypomethylation.
EPIGENETICS IN AMYGDALA-DEPENDENT FEAR LEARNING
Recent studies of cued fear conditioning (see Box 1) have begun to implicate DNA methylation and histone acetylation in amygdala-dependent learning and memory. Specifically, Glen Schafe’s group has shown that cued fear conditioning is associated with increased expression of DNMT3A and acetylated histone H3 in the lateral amygdala (Monsey et al, 2011). As in the hippocampus, administration of DNMT inhibitors into the amygdala impaired the formation of a cue-shock association and this effect was rescued by pretreatment with an intra-amygdala injection of an HDAC inhibitor after training (Monsey et al, 2011). In a separate set of experiments, the same group showed that DNMT inhibitors administered into the lateral amygdala impaired memory reconsolidation and that this deficit was reversed by pre-treatment with an HDAC inhibitor immediately after recall (Maddox and Schafe, 2011). These findings are in conflict with the typically observed synergistic effect of DNMT and HDAC inhibitors in studies of cancer, which are attributed to the repressive effects of both DNMTs and HDACs on transcription (eg, Arzenani et al, 2011). A number of explanations are possible for this discrepancy. First, HDAC inhibitors can induce activation of methylated gene promoters without causing changes in DNA methylation (Zhou et al, 2007), indicating that altered DNA methylation may not be the solitary mechanism of memory disruption. Alternatively, although DNA methylation is predominantly associated with transcriptional repression, DNA methylation can also activate transcription through association with the methyl-CpG–binding protein 2 (MeCP2) and CREB (Chahrour et al, 2008). These findings suggest that inhibition of DNA methylation may actually reduce the expression of specific genes, which may explain reduced H3 acetylation that Glen Schafe’s group observed after DNMT inhibition (Monsey et al, 2011). In addition, DNA methylation-independent effects of DNMTs have been noted (Milutinovic et al, 2004), as have direct interactions between DNMTs and HDACs (Robertson, 2002). Thus, further studies of gene-specific changes in DNA methylation are required to strengthen the conclusion that memory impairment caused by DNMT inhibitors can be attributed to changes in DNA methylation.
Maddox and Schafe (2011) further demonstrated that DNMT inhibition impaired memory rather than simply enhancing extinction, indicating that DNA methylation is required for the persistence of amygdala-dependent memory for cued fear conditioning. These studies did not examine changes in DNA methylation at specific genes and the duration of epigenetic changes in the amygdala remains to be determined. Specifically, the upregulation of histone acetylation and DNMT expression was evident only at the latest time point tested (90 min), thus it is not clear whether these changes are transient in the amygdala as they are in the hippocampus. This point is of particular relevance in the amygdala because this region has been implicated in the initial memory formation and in the maintenance of remote memories for cued fear. The specific nature of epigenetic marks established after cued fear conditioning, and the potential persistence of these marks at remote time points remains to be determined.
EPIGENETICS IN CORTEX-DEPENDENT MEMORY MAINTENANCE
Although the time course of epigenetic changes in the amygdala is not clear, methylation of plasticity-associated genes in the hippocampus is rapidly induced and reversed to baseline levels within 24 h (Miller and Sweatt, 2007). This transience challenged our initial hypothesis that self-perpetuation of DNA methylation in the hippocampus would support memory maintenance over prolonged time periods. However, the rapid reversal of epigenetic changes in the hippocampus is consistent with the systems consolidation proposition that recently consolidated memories are downloaded from the hippocampus to the cortex for long-term storage and maintenance (Ding et al, 2008; Frankland and Bontempi, 2005; Frankland et al, 2004). Based on evidence in support of a temporal and regional shift in memory storage (Frankland and Bontempi, 2005), we reasoned that persistent, self-perpetuating DNA methylation in the cortex, and not in the hippocampus, supports the storage and maintenance of remote memory.
Indeed, Miller et al (2010) found that contextual fear conditioning induced a delayed and long-lasting change in methylation of the memory-suppressor gene calcineurin in the medial prefrontal cortex (mPFC) that emerged 1 day after fear conditioning and persisted for at least 30 days. Increased DNA methylation was associated with decreased calcineurin expression at 30 days and this effect was reversed by intra-cortical injections of DNMT inhibitors. The persisting change in methylation was gene-specific, in that the learning-induced effect on methylation and expression of the reelin gene was evident 1 h after training and gradually returned to baseline levels by 30 days. Moreover, three intra-cortical injections of DNMT inhibitors, starting 29 days after training, blocked the recall of remote memory at 30 days, whereas a similar injection protocol at the recent time point (starting 1 day after fear conditioning) had no effect on memory recall at 30 days. The temporal specificity of DNMT inhibitor effects on remote memory indicates that DNA methylation has temporal- and region-specific effects on memory formation, although the mechanistic basis for these differences is not clear. In contrast to the transient induction in the hippocampus, persistent cortical methylation is more closely aligned with the traditional view of DNA methylation as a long-lasting epigenetic mark. Moreover, these findings suggest that non-dividing neuronal cells have adapted traditional epigenetic processes such that they can be utilized for distinct purposes in brain regions with temporally specific roles in memory formation and maintenance.
Fear extinction is an additional memory process that is largely dependent on the mPFC. Extinction results in the reversal, but not the erasure (see Box 1) of a previously learned association for contextual and cued fear conditioning, through repeated exposure to the conditioning cue in the absence of shock (Quirk et al, 2006). Currently, no study has investigated a potential role for DNA methylation in fear extinction. However, a number of studies have shown that extinction learning is associated with increased levels of histone acetylation, and that pharmacological enhancement of histone acetylation with HDAC inhibitors promotes the extinction of conditioned fear (Bredy et al, 2007; Lattal et al, 2007; Stafford et al, 2012). Specifically, extinction training enhanced histone acetylation in the hippoacmpus and the mPFC (Bredy et al, 2007; Stafford et al, 2012) and post-training treatment with an HDAC inhibitor increased c-fos expression in the hippocampus and the infralimbic mPFC (Stafford et al, 2012). Treatment with HDAC inhibitors systemically, into the hippocampus or into the infralimbic mPFC, enhanced extinction learning for up to 14 days (Itzhak et al, 2012; Lattal et al, 2007; Stafford et al, 2012), whereas HDAC inhibitors administered into the prelimbic cortex were without effect (Stafford et al, 2012). These data suggest that histone acetylation has persistent and brain-region specific effects on extinction of fear learning. Interestingly, one study found that blocking histone acetylation through interference with the histone acetyltransferase enzyme p300 in fact enhanced extinction learning (Marek et al, 2011). One possible explanation for this paradoxical finding is the observation that p300 co-occurs in a protein complex that contains HDAC 1 (Simone et al, 2004), such that interference with p300 may have disrupted the entire complex, including HDAC 1.
Overall, the parallel findings with contextual and cued fear conditioning in the hippocampus, the amygdala, and the cortex suggest that DNA methylation, through interactions with histone acetylation, is an essential and broadly utilized mechanism for all stages of memory formation, including consolidation, reconsolidation, maintenance, and possibly extinction. Importantly, although we limit our discussion to fear learning and memory, these mechanisms have been identified as critical regulators of memory in other learning paradigms that are unrelated to fear (eg, novel object recognition and conditioned place preference; Barros et al, 2011; Munoz et al, 2010), and in mediating long-lasting changes in gene expression and behavior produced by stress and early-life environment (Meaney and Szyf, 2005a, 2005b; Roth et al, 2009). These examples illustrate the pervasive role of DNA methylation in the regulation of gene transcription that supports long-term behavioral change in response to a broad set of experiences.
REGULATION OF DNA DEMETHYLATION
The studies presented thus far challenge the view that DNA methylation is laid down early in life and remains unchanged thereafter, and suggest that DNA methylation can be actively removed in the process of memory consolidation (Lubin et al, 2008; Lubin and Sweatt, 2007; Miller et al, 2008; Miller and Sweatt, 2007; Roth et al, 2009; Roth et al, 2011). The idea of the occurrence of active DNA demethylation has been contentious. Traditional epigenetic mechanisms and studies have posited only passive DNA demethylation as a result of cell division and failure to replicate DNA methylation marks. Active demethylation through direct chemical removal of methyl groups on cytosines (or methylcytosines themselves) has been proposed by several groups, including those of Szyf and Meaney (Meaney and Szyf, 2005a, 2005b; Weaver et al, 2005) and Sweatt and co-workers (Day and Sweatt, 2010; Miller and Sweatt, 2007), but this concept has been controversial (Ooi and Bestor, 2008).
However, in addition to our experiments, other recent publications (Kangaspeska et al, 2008; Metivier et al, 2008) have demonstrated rapid DNA demethylation and re-methylation, referred to as ‘cycling’ of 5-methyl-cytosine (5mC) in cultured cells and skeletal muscle. This demethylation occurs too rapidly to be explained by passive demethylation through cell division and must therefore be due to an active demethylation process. These investigators have also proposed a specific demethylation mechanism: cytosine (C) to thymine (T) conversion of 5mC, followed by base-excision repair of the resulting nucleotide mismatch. Most recently, exciting work from Song and colleagues supported this idea (Engel et al, 2009; Gehring et al, 2009; Ma et al, 2009a; Ma et al, 2009b; Niehrs and Schafer, 2012; Schmitz et al, 2009; Wu and Sun, 2009). These investigators have demonstrated that DNA repair mechanisms are used to demethylate DNA in non-dividing neurons, specifically through base-excision repair mechanisms controlled by the growth and DNA damage 45 (GADD45-beta) regulatory system. This finding demonstrates that demethylation can occur independently of DNA replication, and in a terminally differentiated neuron. Moreover, two new studies (Kriaucionis and Heintz, 2009; Tahiliani et al, 2009) have shown that a novel DNA base, hydroxymethyl-cytosine (5hmC), may uniquely occur in the CNS and serve as a precursor nucleoside for active demethylation through base-excision repair. The conversion of 5mC to 5hmC is catalyzed by TET (ten-eleven-translocation) enzymes, which promote activity-induced demethylation in the mouse dentate gyrus (Guo et al, 2011a; Guo et al, 2011b). In additon, work from Pene de Ortiz's lab indicates that the disruption of DNA recombination and repair processes in the brain interferes with contextual fear conditioning and conditioned taste aversion (Colon-Cesario et al, 2006; Saavedra-Rodriguez et al, 2009). Thus, there is now a substantial body of evidence supporting the idea that active DNA demethylation can occur in non-dividing neurons, findings which make viable the idea that active control of DNA methylation may have a role in activity-dependent processes in the CNS. Thus, it is intriguing to consider the idea of a role for acute regulation of DNA methylation and demethylation in learned fear responses by testing the capacity of DNA methylation to control experience-dependent cortical, hippocampal, and amygdalar gene transcription.
EPIGENETIC MECHANISMS IN PTSD
PTSD is a debilitating anxiety disorder that develops in a subset of people who experience psychological trauma (Yehuda and LeDoux, 2007). PTSD patients are commonly plagued by recurrent frightening thoughts and memories of the aversive experience and suffer from a host of persistent physiological and behavioral sequelae that include altered sympathetic and hypothalamic-pituitary-adrenal (HPA) axis responsivity, chronic anxiety, exaggerated startle response, and cognitive dysfunction (Johnsen and Asbjornsen, 2008; Mittal et al, 2001; Moore, 2009; Nemeroff et al, 2006; Yehuda and LeDoux, 2007). Recent studies have implicated epigenetic mechanisms in the psychopathology of PTSD. For example, Kerry Ressler’s lab has shown that PTSD patients manifest altered DNA methylation in peripheral blood immune cells and identified several immune system-relevant genes that exhibit persisting epigenetic modification (Smith et al, 2011).
The presence of individual differences in the risk for developing PTSD points to at least two interacting factors that determine outcomes associated with adult trauma. The first involves the presence of a pre-existing source of vulnerability, particularly of negative early-life experiences, that ultimately shape the way in which individuals respond to, and cope with, stress and trauma later in life (Perkonigg et al, 2000; Yehuda and Bierer, 2009). The second involves exacerbated negative adaptations in response to the traumatic event that is the proximal cause of PTSD psychopathology (Yehuda and Bierer, 2009). Implicit in these factors is the idea that some biological mechanism must bridge a salient experience (ie, early-life environment or adult trauma) with a persistent behavioral and physiological outcome, particularly with symptoms that involve pathological fear, anxiety, and cognitive impairment. Epigenetic modifications offer an attractive mechanism for explaining the persistent nature of outcomes associated with environmental events that occur during development and in the mature CNS. This idea is supported by a growing literature pointing to an epigenetic bridge between early environment, which in rodents is shaped largely by maternal behavior during the first week of life, and altered reactivity to stress-inducing stimuli in adult rodents (Meaney and Szyf, 2005a, 2005b). Moreover, a role for epigenetic mechanisms in the consolidation and the persistence of traumatic memories experienced in adulthood is supported by evidence for epigenetic regulation of memory in cued and contextual fear conditioning, which are often used as animal models of PTSD (see Box 2). Evidence for a role of epigenetic mechanisms in each of these processes is discussed below.
Work from Michael Meaney's lab has provided evidence demonstrating a role for epigenetic mechanisms in regulating the effects of early environment on adult outcomes through altered expression of stress-related genes. This work has received attention among PTSD researchers because of the parallel findings in humans, which link hypothalamic-pituitary-adrenal axis dysregulation and childhood maltreatment with predisposition to PTSD (Binder et al, 2008; Shea et al, 2005; Yehuda and LeDoux, 2007). Specifically, Meaney’s lab has shown that natural variations in maternal licking and grooming alter DNA methylation and the expression of the glucocorticoid receptor gene, and produce altered patterns of hypothalamic-pituitary-adrenal axis reactivity, anxiety, and cognitive function in adulthood (Francis and Meaney, 1999; Liu et al, 2000; Meaney and Szyf, 2005a; Weaver et al, 2002a; Weaver et al, 2002b; Zaharia et al, 1996). These studies implicate epigenetic mechanisms in providing a critical link between early-life environment and sensitivity to stressors in later life, which is critical for developing a complete model of PTSD that integrates pre-existing risk factors with an exaggerated response to trauma in adulthood.
ROLE OF BDNF IN PTSD
Much of the current work on mechanisms of PTSD is shifting toward identifying candidate genes that can explain the basis for individual differences in susceptibility to PTSD, for the initial formation of an intense fear memory in response to trauma, and finally, for the maintenance of that memory over a prolonged period of time. BDNF is quickly emerging as a gene that meets all of required criteria. BDNF gene product is altered by early-life experiences though epigenetic changes (Roth et al, 2009), is regulated by proximal fear-inducing stimuli in various paradigms of aversive learning (Lubin et al, 2008; Ou and Gean, 2007), and altered levels of the gene product are evident long after the fear-inducing stimulus has passed (Bredy et al, 2007; Takei et al, 2011). Each of these points are discussed in greater detail below.
A recent study from the Sweatt lab has found evidence for long-lasting epigenetic alterations of the BDNF gene in response to early maternal maltreatment, which may predispose individuals to later risk for PTSD. Specifically, Roth et al. (2009) found that exposure to an abusive dam in the first 7 days of life resulted in increased methylation and decreased expression of BDNF exon IV in the prefrontal cortex of adult offspring. This effect was partly reversed by cross-fostering and fully reversed by the administration of DNMT inhibitors in adulthood (Roth et al, 2009). Given the established role for BDNF in fear learning and cognitive function in general (Alonso et al, 2005; Bekinschtein et al, 2008a; Bekinschtein et al, 2008b; Lubin et al, 2008), these results indicate that early-life experiences may increase the susceptibility to PTSD at least in part through epigenetic regulation of the BDNF gene, but additional studies are required to test this hypothesis directly. Overall, these findings, as well as those from Michael Meaney’s lab, are consistent with the emerging unifying hypothesis that the accumulation of aberrant epigenetic marks over the lifespan may be a driver of PTSD-related cellular, cognitive, and physiologic changes.
In addition to potentially linking early-life experience to later risk for PTSD, there is now evidence to suggest that epigenetic mechanisms also have a role in the establishment and persistence of PTSD symptoms produced by proximal traumatic triggers in adulthood. As discussed earlier, epigenetic regulation of the BDNF gene has been implicated in the establishment of contextual fear memories in the hippocampus (Lubin et al, 2008) and changes in BDNF transcript levels have been described in the amygdala during consolidation of cued fear memory (Ou and Gean, 2007). Altered histone acetylation at specific loci on the BDNF gene has also been noted with stress-based models of PTSD (Bredy et al, 2007; Takei et al, 2011; see Box 2), indicating that epigenetic regulation of BDNF expression occurs in response to a range of aversive stimuli. Studies of predator-stress based models of PTSD have also found that BDNF expression is regulated by exposure to traumatic stimuli long after the fear-inducing stimulus has passed (Kozlovsky et al, 2007; Ou and Gean, 2007), indicating that BDNF regulation may be important for the persistence of pathological fear. Indeed, extinction of conditioned fear involves exon-specific regulation of BDNF gene expression in the prefrontal cortex (Bredy et al, 2007) and deletion of the BDNF gene specifically in the hippocampus impaired fear learning and impaired the extinction of conditioned fear (Heldt et al, 2007), implicating BDNF as a potential target for regulating the erasure of maladaptive fear responses.
Given the evidence for altered BDNF expression in animal models of PTSD, a recent study from the Sweatt and Diamond labs evaluated the idea that DNA methylation of the BDNF gene might contribute to learned fear, using an established model of exceptional stress (the rodent–cat exposure model; see Box 2) and PTSD (Zoladz et al, 2008). Adult male Sprague-Dawley rats were given psychosocial stress composed of two acute cat exposures in conjunction with 31 days of daily social instability. In the earlier studies, the Diamond group found these manipulations to produce physiological and behavioral sequelae in rats that are comparable with symptoms observed in traumatized people with PTSD (Zoladz et al, 2008). The cat exposure combined with the psychosocial stress regimen significantly increased methylation of the BDNF gene in the dorsal hippocampus, with the most robust hypermethylation detected in the dorsal CA1 subregion (Roth et al, 2011). Conversely, the psychosocial stress regimen significantly decreased methylation in the ventral hippocampus (CA3). In addition, there were decreased levels of BDNF mRNA in both the dorsal and ventral CA1. These results provide evidence that traumatic stress occurring in adulthood can induce CNS gene methylation, and specifically, support the hypothesis that epigenetic marking of the BDNF gene may underlie hippocampal dysfunction produced by exposure to traumatic events. A recent study has identified another candidate gene, encoding the post-synaptic density-protein disks large-associated protein (DIgap2), which may be involved in PTSD psychopathology. Dlgap2 mRNA expression was associated with behavioral expression of fear in a predator-stress model of PTSD and gene expression was associated with altered DNA methylation in the hippocampus (Chertkow-Deutsher et al, 2010). Overall, this work provides support for the speculative notion that altered hippocampal DNA methylation is a cellular mechanism underlying the persistent cognitive deficits that are the prominent features of the pathophysiology of PTSD.
ROLE OF THE AMYGDALA IN PERSISTING PTSD SYMPTOMS
The biological basis for the development of PTSD is not yet known, but studies of Pavlovian fear conditioning suggest that persistent traumatic memories are likely established through multiple phases that involve a transition from recent to remote memories. Although PTSD has been associated with molecular changes in the hippocampus and the prefrontal cortex, the persistent symptoms of PTSD are most closely associated with alterations in the amygdala (Johnson et al, 2012), a region that has been implicated in the storage of remote memories for cued fear (Dudai, 2004; Gale et al, 2004; Medina et al, 2008). The transition of memories to a stable form is important for the persistence of PTSD and it is thus critical to understand the molecular mechanisms that underlie such memory stability in order to identify potential targets for pharmacological treatment. A variety of recent studies have used fear conditioning to explore the hypothesis that changes in the amygdala support long-lasting memories (Debiec et al, 2011; Gale et al, 2004; Monsey et al, 2011). These findings imply that a tangible persisting molecular mark in the amygdala must underlie the preservation of remote fear memory. There is now evidence for epigenetic changes in the amygdala during consolidation and reconsolidation of cued fear (Maddox and Schafe, 2011; Monsey et al, 2011), but no studies have examined the maintenance phase of cued fear memory. The potential involvement of DNA methylation in the maintenance of remote memory in the amygdala is an important directive for future research.
The advent of sophisticated molecular, genetic, and cellular techniques has lent itself to a relatively deep understanding of how memories are initially formed. By stark contrast, however, is our limited understanding of how these same memories are maintained by the brain (Dudai, 2004; Medina et al, 2008; Sacktor, 2008). While epigenetic blockade can lead to changes in fear learning, future studies need to focus on the mechanisms through which already learned fear responses are stored in order to understand the basis for the persistent fear observed in PTSD. We need to shift toward an understanding of the mechanisms involved in maintaining the pathological fear memory over time.
CANDIDATE GENES IN AMYGDALA-BASED FEAR LEARNING AND PTSD
Most of the available evidence linking epigenetic regulation of specific genes to the establishment and persistence of fear memories has come from studies of contextual fear conditioning-related changes in the hippocampus and the prefrontal cortex (eg, Levenson et al, 2004b; Lubin et al, 2008; Miller et al, 2008; Miller et al, 2010; Miller and Sweatt, 2007). Although recent pharmacologically based evidence implicates DNA methylation in the amygdala in the consolidation and reconsolidation of cued fear (Maddox and Schafe, 2011; Monsey et al, 2011), epigenetic regulation at specific genes in this region is not clear. An advantage of established fear conditioning paradigms is that many of the genes that are critically involved in the establishment and maintenance of fear memories have been identified, which allows for the investigation of epigenetic regulation of relevant candidate genes. The candidate genes need to meet specific criteria. First, the products of selected genes must be altered during memory formation. Second, the transcription of selected genes should be altered in specific fear memory paradigms, such as contextual fear conditioning, in part through epigenetic mechanisms. Using contextual fear conditioning, we have identified a critical role for epigenetic regulation of genes encoding reelin, BDNF, calcineurin, and protein phosphatase 1 in driving memory consolidation and maintenance in the hippocampus and the prefrontal cortex. A report of altered expression of the BDNF gene in the amygdala in response to fear learning (Ou and Gean, 2007) indicates that similar genes may be modified in the amygdala in response to fear conditioning. Moreover, we already provided evidence for epigenetic regulation of the BDNF gene in the prefrontal cortex in response to maternal maltreatment (Roth et al, 2009) and in the hippocampus of adult rodents using a predator-stress model of PTSD (Roth et al, 2011), suggesting that BDNF may be especially relevant for understanding the development of pathological fear. It is also worthwhile to examine potential changes in DNA methylation of DNMT1, DNMT3A, and DNMT3B genes themselves. Epigenetic changes in DNMT genes would provide important insights because an epigenetic alteration at any of the DNMT genes might itself be a mechanism for driving further alterations in DNA methylation in conditioned fear and PTSD. Existing studies in the amygdala have shown that DNA methylation is required for consolidation and reconsolidation of cued fear conditioning, but no study thus far has examined changes in DNA methylation at specific genes.
EPIGENETIC REGULATION OF SYNAPTIC PLASTICITY IN PTSD
PTSD has been associated with persisting changes in the amygdala’s capacity for long-term potentiation (LTP) (Koshibu et al, 2011; Paul et al, 2007; Post et al, 1998), and LTP itself serves as a mediator of learned fear (Rogan et al, 1997). DNMT inhibitors disrupt hippocampal LTP (Levenson et al, 2006; Monsey et al, 2011), and recent studies confirmed the ability of DNMT inhibitors to affect synaptic function in the lateral amygdala (Monsey et al, 2011). In addition, these studies demonstrated that DNMT-inhibitor induced deficits in LTP can be reversed by pretreatment with HDAC inhibitors. The reversal of LTP deficits with HDAC inhibitors indicates that a complete understanding of epigenetic mechanisms of PTSD would involve studies of the cross talk between DNA methylation and histone acetylation.
CLINICAL IMPLICATIONS: MANIPULATING THE EPIGENOME TO IMPROVE FUNCTIONAL RECOVERY
Understanding the role of epigenetic molecular mechanisms in triggering and maintaining persisting behavioral change will have broad relevance in neuroscience and biomedicine. Although behavioral therapy can help alleviate the symptoms of PTSD, no truly effective drug-based adjunct therapy is currently available. We propose that the epigenetic mechanisms that are involved in rodent models of normal fear learning and memory also extend to the development of persistent and pathological fear characteristic of PTSD and that an understanding of these mechanisms will guide new routes of drug development for PTSD. Such an understanding will ultimately lead to the development of new pharmacological agents that will allow for the treatment of PTSD-associated cognitive dysfunction, facilitate therapeutic re-learning, and enhance extinction of conditioned and contextual fear.
EPIGENETIC TREATMENT IN THE CNS
Epigenetically based therapies investigated thus far have shown promise for the treatment of various CNS and peripheral disorders, primarily through manipulations of histone acetylation and DNA methylation (eg, Langley et al, 2005). Histone acetylation is catalyzed by histone acetyltransferase enzymes, including the CREB-binding protein, whereas deacetylation is catalyzed by HDAC enzymes (Alarcon et al, 2004; Levenson et al, 2004a; Lubin et al, 2005; Roberson et al, 1999; Shalin et al, 2006; Vecsey et al, 2007). In an important breakthrough in the past few years, several groups discovered that HDAC inhibitors enhance LTP in vitro and augment memory formation in vivo (eg, Alarcon et al, 2004; Chwang et al, 2007; Chwang et al, 2006; Fischer et al, 2007; Korzus et al, 2004; Vecsey et al, 2007), thereby implicating these agents as potential therapeutic targets for CNS disorders. HDACs are recruited to methylated gene promoters by binding to MeCP2, which results in the reversal of histone acetylation and the repression of gene transcription (Jones et al, 1998). MeCP2 has also been associated with transcriptional activation and histone acetylation through association with CREB and CREB-binding protein (Chahrour et al, 2008). These complexes are the primary targets of epigenetic therapies.
The broad utility of epigenetically based interventions represents the greatest strength and challenge for the development of effective and targeted therapies. That is, the broad treatment potential also poses a risk for producing effects on unintended genetic and cellular targets, as well as non-specific effects on cognitive function (Hirsch and Bonham, 2004; Sandor et al, 2000; Vrana et al, 1999). These concerns emphasize the importance of improving the specificity of epigenetically-based drugs. One way to achieve greater specificity is to develop drugs that target distinct HDAC or DNMT isoforms. There are four classes of HDACs, each containing 18 isoforms. Class I consists of HDACs 1–3, and 8; Class II consists of HDACs 4–7, 9, and 10; Class III consists of sirtuins (Sirt) 1–7; and class IV consists of HDAC 11 (Narayan and Dragunow, 2010). Specificity for sirtuins can be easily attained because of reliance on the cofactor nicotinamide adenine dinuceoteide (NAD+) for their activity, whereas all other classes exhibit zinc (Zn2+) containing active sites (Narayan and Dragunow, 2010). For this reason, most HDAC inhibitors do not target sirtuins (Michan and Sinclair, 2007). However, Sirt1 and Sirt2 selective inhibitors have been identified, including Sirt1 selective inhibitors EX-S27, HR73, 2-Anilincbenzamide, and suramin derivatives; Sirt2 selective inhibitors AGK2 and adenosine mimetics; as well as the Sirt1 and 2 inhibitor cambinol (Alcain and Villalba, 2009a; Itoh et al, 2008). In addition to inhibitors, resveratrol has been identified as a Sirt1 activator (Alcain and Villalba, 2009b; Shakibaei et al, 2011; Villalba et al, 2012), and the development of additional activators is in progress (Villalba et al, 2012). The most commonly studied pan-HDAC inhibitors target class I and II HDACs and include trichostatin A, panobinostat, sodium butyrate, and suberoylanilide hydroxamic acid (SAHA; Varinostat) (Khan et al, 2008), although there is ongoing development of class- and isoform-specific inhibitors. For example, some identified selective inhibitors include the HDAC 1 inhibitors SB429201, bis pyridinium dine, and MS-275; HDAC 1 and 2 inhibitors MGCD0103 and biaryl benzamides; HDAC 6 inhibitors tubacin and thiolate; the HDAC 8 inhibitor SB379278A; Class I selective inhibitors R304634 and valproic acid; and Class II selective inhibitors (aryloxopropenyl) pyrrolyl hydroxyamides (Itoh et al, 2008; Khan et al, 2008).
Similar issues face DNMT inhibitors, as most current inhibitors, including 5-AZA, Zebularine, and RG-108 are not isoform specific, although there is a push for the development of specific DNMT inhibitors (Milutinovic et al, 2004). Some effort has been made to identify DNMT-inhibiting properties of known compounds, which has led to the identification of procainamide as a DNMT1 specific inhibitor (Lee et al, 2005). The progress in epigenetic drug development is occurring rapidly, and it must be met by an improved understanding of the role that specific isoforms play in different CNS conditions.
There is growing evidence that distinct HDAC isoforms are associated with specific cognitive functions and disorders. For example, a protective role of Sirt1 and its activator, resveratrol, has been identified in cell and animal models of Alzheimer's disease (AD) (Kim et al, 2007) and mice lacking Sirt1 exhibit impaired fear conditioning and LTP in the hippocampus (Gao et al, 2010). Moreover, inhibition of Class I HDACs has been associated with the reversal of AD-associated memory deficits (Kilgore et al, 2010) and inhibition of HDAC 2 in particular reversed cognitive, structural, and transcriptional deficits in a mouse model of AD (Graff et al, 2012). In addition, studies in rats have shown that hippocampal DNMT 3a and 3b were selectively upregulated in response to contextual fear conditioning, whereas DNMT1 levels did not change (Miller and Sweatt, 2007). By contrast, only DNMT 3a was upregulated in response to cued fear conditioning in the amygdala (Monsey et al, 2011), indicating that isoform-specific targeting of DNMTs may be useful in manipulating different types of learned fear. A growing understanding of isoform-specific involvement of HDAC and DNMT inhibitors in distinct behavioral outcomes will facilitate the development of more specific therapies.
One promising approach for improving treatment specificity for a particular cognitive function or disorder may be to utilize distinct drug cocktails in combination with specific behavioral therapies. For example, the ability of HDAC inhibitors to enhance object recognition memory is dependent on upstream signaling that includes glucocorticoid receptor-induced activation of PKA (Roozendaal et al, 2010). That is, the ability of HDAC inhibitors to improve memory only in the presence of glucocorticoid receptor activation indicates that histone acetylation must co-occur with a stimulus-specific signaling cascade, which implicates drug co-administration as a potential tool for improving the treatment specificity of HDAC inhibitors. This finding fits with evidence that HDAC inhibition induces specific effects on memory, in that HDAC inhibitors were only effective at influencing memory when administered close in time to the learning or the recall event (eg, Maddox and Schafe, 2011; Monsey et al, 2011). Further efforts to improve target specificity must also take advantage of the growing evidence that DNA methylation is regulated by transcription-factor binding to specific sites on DNA (Brenner et al, 2005), which can be exploited for the development of gene-specific drug targets.
Efforts have also been directed toward developing drugs that can easily cross the blood–brain barrier and that exhibit good biological activity and oral bioavailability for use in humans. Butyrates in particular readily cross the blood–brain barrier (Egorin et al, 1999; Narayan and Dragunow, 2010) and SAHA has been shown to have positive effects on neuronal atrophy and rotarod performance when administered through drinking water in mice (Ferrante et al, 2003; Hockly et al, 2003). The efficacy of existing drugs in the treatment of CNS disorders is evident from studies of Huntington's disease, Rett syndrome, and AD (Fischer et al, 2007; Kilgore et al, 2010; Langley et al, 2005; Vecsler et al, 2010). Huntington's disease is associated with inhibition of the histone acetyltransferase CREB-binding protein, reduced histone acetylation, and cell death (Langley et al, 2005). Cell culture (Steffan et al, 2001), mouse (Ferrante et al, 2003; Hockly et al, 2003), and drosophila (Steffan et al, 2001) models of Huntington's disease have shown that treatment with HDAC inhibitors TSA, SAHA, or sodium butyrate can increase histone acetylation and promote neuronal survival. In Rett syndrome, reduced MeCP2 expression is associated with impaired transcriptional repression and behavioral deficits that include impaired fear conditioning and elevated anxiety (Adachi et al, 2009). Interestingly, knockdown of MeCP2 in human neuroblastoma (SK-NSH) cells resulted in reduced histone acetylation and reduced expression of BDNF and HDACs 2, 5, and 8 (Vecsler et al, 2010). Application of the HDAC inhibitor velproic acid reversed MeCP2 downregulation, as well as the impairment in BDNF expression (Vecsler et al, 2010). The restorative effect of HDAC inhibitors on MeCP2 was specific to the cells with impaired MeCP2 expression, indicating that the baseline levels of MeCP2 may allow for some degree of cell specificity in response to broad HDAC treatment.
In addition, targeted HDAC 2 inhibition (Graff et al, 2012), systemic inhibition of Class I HDACs (Kilgore et al, 2010), and upregulation of Sirt1 (Gao et al, 2010), improved cognitive deficits in mouse models of AD. Studies in humans have also reported improved cognitive and psychiatric symptoms in AD patients by partially reversing DNA hypomethylation through supplementation with methylation-promoting agents (eg, folate, SAM, and vitamin B6) (Narayan and Dragunow, 2010). Overall, these studies suggest that epigenetically based therapies hold promise for the treatment of various CNS disorders and the development of appropriate behavioral, epigenetic, and neural targets is needed to facilitate the development of epigenetic treatments in PTSD.
EPIGENETIC TARGETS IN PTSD
Studies of neural circuits and molecular mechanisms that underlie fear formation, maintenance, expression, and extinction are critical for understanding how these processes are deregulated in PTSD and for identifying ‘drugable’ treatment targets for intervention. Based on their roles in fear conditioning, we suggest that HDAC inhibitors would be useful for facilitating extinction of conditioned fear, whereas DNMT inhibitors can be used to erase a conditioned fear response. There are three basic processes through which HDAC inhibitors or manipulations of the epigenome might prove therapeutically useful in PTSD treatment: (1) through the erasure of fear memory; (2) by promoting extinction of the traumatic memory; and finally, (3) by facilitating learning of a modified response to an earlier fear stimulus. Preclinical proof-of-principle studies are necessary to evaluate the efficacy of HDAC and DNMT inhibitors on fear responses in conditioned animals.
Memory Erasure
Strategies aimed at erasing a previously formed fear memory must focus primarily on targeting memories during the reconsolidation phase. Memories for fear-associated cues and context are reconsolidated in the amygdala and in the hippocampus, respectively (Lee, 2010; Maddox and Schafe, 2011). A recalled memory becomes highly labile during reconsolidation, when it is subject to qualitative modification in response to new information, or to quantitative modification that can weaken or strengthen the memory trace (McKenzie and Eichenbaum, 2011). An advantage of interventions aimed at reconsolidation is that the outcome tends to be specific to the recalled memory and does not affect memories outside of the reconsolidation window (Maddox and Schafe, 2011). Recent evidence has identified DNA methylation and histone acetylation as critical regulators of amygdala- (Maddox and Schafe, 2011) and hippocampus- (Lubin and Sweatt, 2007) dependent reconsolidation, implicating these modifications as promising targets for pharmacological intervention. The reconsolidation process is particularly relevant for PTSD, in which persistent and recurring recall of the traumatic event may strengthen the traumatic memory over time, while also providing an opportunity for recall-based pharmacological intervention. Indeed, evidence from studies of rodents indicates that enhanced noradrenergic signaling, such as that observed in PTSD patients (Geracioti et al, 2001), is associated with enhancement of fear memory during reconsolidation and a subsequent increase in resistance to fear extinction (Debiec et al, 2011).
Most available preclinical studies have focused on identifying strategies that modify the strength of recalled memories through manipulations of cellular processes in the amygdala (for cued memory) and the hippocampus (for contextual memory). However, a caveat to available studies of epigenetic involvement in reconsolidation is that the amygdala and the hippocampus have been identified as the sites of reconsolidation at time points shortly after the initial learning event. Thus, it is not clear how the role of these brain regions might change as memories transition from recent to remote over time, particularly because the hippocampus is thought to have a particularly transient role in the formation of recent memories (Frankland and Bontempi, 2005; Kim and Jung, 2006). The issue of memory redistribution through systems consolidation is particularly relevant to PTSD, given that negative outcomes are associated with the persistence of previously established traumatic memories. Thus, suitable interventions must target the neural systems involved in maintenance of memories over prolonged periods of time. We have found that DNA methylation in the anterior cingulate cortex is critical for the maintenance of remote fear memory and that blocking DNA methylation impaired memory recall (Miller et al, 2010). These results suggest that epigenetic interventions targeted at the anterior cingulate cortex may be beneficial for the erasure of persistent fear memories. Moreover, the excessive fear response in PTSD in humans is associated with impaired ability of the mPFC to regulate and dampen the amygdala-driven fear response (Etkin and Wager, 2007). Chronic stress is associated with decreased dendritic complexity in the mPFC and increased complexity in the amygdala in rodents (Radley et al, 2004; Vyas et al, 2002), indicating that treatments aimed at the mPFC may facilitate the learning of a modified response to the fearful stimulus.
Enhanced Extinction
Fear extinction can also be used as a model for behavioral/experiential clinical treatment for abnormal fear responsiveness. Fear extinction is the decrease in conditioned fear responses that normally occurs when a conditioned stimulus is repeatedly presented in the absence of the aversive unconditioned stimulus. Extinction does not erase the initial conditioned–unconditioned stimulus association, but is thought to form a new memory. After extinction training, extinction memory competes with the conditioned memory for control of fear expression and deficits in fear extinction are thought to contribute to PTSD (Bremner, 2002; Debiec et al, 2011; Rauch et al, 2006; Ursano et al, 2009; Yehuda and LeDoux, 2007). The extinction deficit in PTSD may be associated with the enhanced strength of the traumatic memory that increases its resistance to extinction (Debiec et al, 2011) and with a functional deficit in the mPFC (Bremner, 2002; Debiec et al, 2011; Rauch et al, 2006), a region that is strongly implicated in extinction learning and memory (Quirk et al, 2006). Histone acetylation has been implicated in the extinction of conditioned fear, and pharmacological intervention with HDAC inhibitors delivered to the mPFC or the hippocampus enhances extinction learning (Bredy and Barad, 2008; Bredy et al, 2007; Lattal et al, 2007). Of particular therapeutic relevance is the finding that oral administration of HDAC inhibitors in mice enhances histone acetylation and promotes extinction learning in response to weak extinction protocols that are ineffective when administered on their own (Stafford et al, 2012). The effect of HDACs on extinction persisted for at least 14 days in that study, indicating that this approach may produce lasting alleviation of fear responses in patients with PTSD when HDAC inhibition is combined with exposure therapy to promote extinction learning. A commonly used form of exposure therapy in clinical populations with phobias includes the use of virtual-reality–based simulation of the feared stimulus, such as visualization of a cliff for treatment of acrophobia (fear of heights) (Davis et al, 2006). Previous studies have shown that combining pharmacological approaches with virtual reality produces improvements in phobia symptoms for at least 3 months after exposure (Davis et al, 2006). Thus, a promising avenue for treatment of PTSD is to combine epigenetically based pharmacological intervention with controlled exposure therapy, such as virtual reality. Although more work is needed in this area, the available studies suggest that epigenetically based pharmacological interventions may promote the extinction of conditioned fear, thereby providing a promising intervention strategy for treatment of PTSD.
FUTURE DIRECTIONS
A limitation of the currently available studies is the lack of direct evidence for the mechanism through which DNA methylation and histone modifications at the cellular level get translated into altered circuit and behavioral function. However, the reviewed studies give us mechanistic insights, such as evidence that DNA methylation controls fear memory stability and that changes in DNA methylation in the adult CNS regulate the expression of known fear conditioning-related genes. Future studies need to address the question of the potential involvement of epigenetic mechanisms in controlling remote fear memory storage in the amygdala, given the relevance of this region to fear expression and PTSD.
The reviewed studies serve as a foundation for formulating future specific hypotheses concerning how DNA methylation might control persisting changes in synaptic and neural circuit function at the molecular level, and ultimately understanding how transient or persisting changes in DNA methylation manifest themselves in altered neuronal function. For instance, is methylation altering a neuron’s basal state, thus altering its response to future stimuli? An example of this might be the lowering of a neuron’s firing threshold, which may be accomplished through transcriptional repression of calcineurin, given its importance in long-term depression and interference with LTP (Day and Sweatt, 2010). An additional possibility is that synaptic proteins and signaling pathways downstream of neuronal activation utilize methylation as a mechanism to self-perpetuate through the regulation of their own transcription rate. For example, methylation may support the perpetuation of glutamate receptor exchange at synapses tagged by potentiation (Day and Sweatt, 2010). These possibilities need not be mutually exclusive. Regardless of the specific answers to these questions, the reviewed studies represent first steps toward demonstrating that DNA methylation is a self-perpetuating signal used by the brain to preserve aversive memories, a finding which has important implications for the question of how resulting behavioral changes are maintained.
CONCLUSIONS
Overall, studies of chromatin-regulating/epigenetic mechanisms suggest that DNMT activity is necessary for experience-driven long-term behavioral change, and that DNA methylation may work in concert with histone modifications to regulate memory formation and storage in the adult CNS. The studies presented in this review exhibit a recurring theme, specifically that the stabilization of normal and pathological fear memories involves distinct phases that are dependent on regionally and temporally distinct epigenetic mechanisms. The initial consolidation and reconsolidation of memory are associated with the establishment of rapidly induced and reversible epigenetic changes, whereas memory maintenance involves delayed and enduring changes in DNA methylation. These findings not only implicate epigenetic mechanisms in different phases of memory formation and maintenance, but they also suggest that the functional and mechanistic outcomes of epigenetic marks differ according to the brain region and the stage of memory formation. Further research is required to investigate the mechanistic basis for these distinct effects.
To date, the hypothesis of a role for epigenetic mechanisms in PTSD and amygdala-dependent fear responses has only begun to be tested (Monsey et al, 2011; Roth et al, 2011; Smith et al, 2011). Thus, it is important to begin to assess the role of epigenetics in the amygdala and in PTSD, and the potential therapeutic efficacy of epigenetic drugs in alleviating PTSD.
References
Adachi M, Autry AE, Covington III HE, Monteggia LM (2009). MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. J Neurosci 29: 4218–4227.
Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER et al (2004). Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42: 947–959.
Alcain FJ, Villalba JM (2009a). Sirtuin activators. Expert Opin Ther Pat 19: 403–414.
Alcain FJ, Villalba JM (2009b). Sirtuin inhibitors. Expert Opin Ther Pat 19: 283–294.
Alonso M, Bekinschtein P, Cammarota M, Vianna MR, Izquierdo I, Medina JH (2005). Endogenous BDNF is required for long-term memory formation in the rat parietal cortex. Learn Mem 12: 504–510.
Arzenani MK, Zade AE, Ming Y, Vijverberg SJ, Zhang Z, Khan Z et al (2011). Genomic DNA hypomethylation by histone deacetylase inhibition implicates DNMT1 nuclear dynamics. Mol Cell Biol 31: 4119–4128.
Barrett RM, Wood MA (2008). Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem 15: 460–467.
Barros M, Dempster EL, Illott N, Chabrawi S, Maior RS, Tomaz C et al (2011). Decreased methylation of the NK3 receptor coding gene (TACR3) after cocaine-induced place preference in marmoset monkeys. Addict Biol; e-pub ahead of print 9 November 2011.
Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, Medina JH (2007). Persistence of long-term memory storage requires a late protein synthesis- and BDNF- dependent phase in the hippocampus. Neuron 53: 261–277.
Bekinschtein P, Cammarota M, Izquierdo I, Medina JH (2008a). BDNF and memory formation and storage. Neuroscientist 14: 147–156.
Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A et al (2008b). BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci USA 105: 2711–2716.
Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB et al (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299: 1291–1305.
Bredy TW, Barad M (2008). The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem 15: 39–45.
Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M (2007). Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem 14: 268–276.
Bremner JD (2002). Neuroimaging studies in post-traumatic stress disorder. Curr Psychiatry Rep 4: 254–263.
Brenner C, Deplus R, Didelot C, Loriot A, Vire E, De Smet C et al (2005). Myc represses transcription through recruitment of DNA methyltransferase corepressor. Embo J 24: 336–346.
Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J et al (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320: 1224–1229.
Chertkow-Deutsher Y, Cohen H, Klein E, Ben-Shachar D (2010). DNA methylation in vulnerability to post-traumatic stress in rats: evidence for the role of the post-synaptic density protein Dlgap2. Int J Neuropsychopharmacol 13: 347–359.
Chwang WB, Arthur JS, Schumacher A, Sweatt JD (2007). The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci 27: 12732–12742.
Chwang WB, O’Riordan KJ, Levenson JM, Sweatt JD (2006). ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem 13: 322–328.
Cohen H, Zohar J (2004). An animal model of posttraumatic stress disorder: The use of cut-off behavioral criteria. Ann NY Acad Scie 1032: 167–178.
Colon-Cesario M, Wang J, Ramos X, Garcia HG, Davila JJ, Laguna J et al (2006). An inhibitor of DNA recombination blocks memory consolidation, but not reconsolidation, in context fear conditioning. J Neurosci 26: 5524–5533.
Crick F (1984). Memory and molecular turnover. Nature 312: 101.
Davis M, Myers KM, Chhatwal J, Ressler KJ (2006). Pharmacological treatments that facilitate extinction of fear: relevance to psychotherapy. NeuroRx 3: 82–96.
Day JJ, Sweatt JD (2010). DNA methylation and memory formation. Nat Neurosci 13: 1319–1323.
Debiec J, Bush DE, LeDoux JE (2011). Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats—a possible mechanism for the persistence of traumatic memories in PTSD. Depress Anxiety 28: 186–193.
Ding HK, Teixeira CM, Frankland PW (2008). Inactivation of the anterior cingulate cortex blocks expression of remote, but not recent, conditioned taste aversion memory. Learn Mem 15: 290–293.
Dudai Y (2004). The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol 55: 51–86.
Egorin MJ, Yuan ZM, Sentz DL, Plaisance K, Eiseman JL (1999). Plasma pharmacokinetics of butyrate after intravenous administration of sodium butyrate or oral administration of tributyrin or sodium butyrate to mice and rats. Cancer Chemother Pharmacol 43: 445–453.
Engel N, Tront JS, Erinle T, Nguyen N, Latham KE, Sapienza C et al (2009). Conserved DNA methylation in Gadd45a(-/-) mice. Epigenetics 4: 98–99.
Etkin A, Wager TD (2007). Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry 164: 1476–1488.
Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD et al (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13: 423–430.
Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B et al (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington′s disease mice. J Neurosci 23: 9418–9427.
Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH (2007). Recovery of learning and memory is associated with chromatin remodelling. Nature 447: 178–182.
Francis DD, Meaney MJ (1999). Maternal care and the development of stress responses. Curr Opin Neurobiol 9: 128–134.
Frankland PW, Bontempi B (2005). The organization of recent and remote memories. Nat Rev Neurosci 6: 119–130.
Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ (2004). The involvement of the anterior cingulate cortex in remote contextual fear memory. Science 304: 881–883.
Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR et al (2004). Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci 24: 3810–3815.
Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L et al (2010). A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466: 1105–1109.
Gehring M, Reik W, Henikoff S (2009). DNA demethylation by DNA repair. Trends Genet 25: 82–90.
Geracioti Jr TD, Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB et al (2001). CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry 158: 1227–1230.
Graff J, Mansuy IM (2008). Epigenetic codes in cognition and behaviour. Behav Brain Res 192: 70–87.
Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM et al (2012). An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483: 222–226.
Griffith JS, Mahler HR (1969). DNA ticketing theory of memory. Nature 223: 580–582.
Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA et al (2011a). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 14: 1345–1351.
Guo JU, Su Y, Zhong C, Ming GL, Song H (2011b). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145: 423–434.
Heldt SA, Stanek L, Chhatwal JP, Ressler KJ (2007). Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry 12: 656–670.
Hirsch CL, Bonham K (2004). Histone deacetylase inhibitors regulate p21WAF1 gene expression at the post-transcriptional level in HepG2 cells. FEBS Lett 570: 37–40.
Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E et al (2003). Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington′s disease. Proc Natl Acad Sci USA 100: 2041–2046.
Holliday R (1999). Is there an epigenetic component in long-term memory? J Theor Biol 200: 339–341.
Itoh Y, Suzuki T, Miyata N (2008). Isoform-selective histone deacetylase inhibitors. Curr Pharm Des 14: 529–544.
Itzhak Y, Anderson KL, Kelley JB, Petkov M (2012). Histone acetylation rescues contextual fear conditioning in nNOS KO mice and accelerates extinction of cued fear conditioning in wild type mice. Neurobiol Learn Mem 97: 409–417.
Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH et al (2008). Epigenetics in the nervous system. J Neurosci 28: 11753–11759.
Johnsen GE, Asbjornsen AE (2008). Consistent impaired verbal memory in PTSD: a meta-analysis. J Affect Disord 111: 74–82.
Johnson LR, McGuire J, Lazarus R, Palmer AA (2012). Pavlovian fear memory circuits and phenotype models of PTSD. Neuropharmacology 62: 638–646.
Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N et al (1998). Methylated DNA and MeCP2 recruit histone deacytylase to repress transcription. Nat Genet 19: 187–191.
Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP et al (2008). Transient cyclical methylation of promoter DNA. Nature 452: 112–115.
Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N et al (2008). Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 409: 581–589.
Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD et al (2010). Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer′s disease. Neuropsychopharmacology 35: 870–880.
Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT et al (2007). SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer′s disease and amyotrophic lateral sclerosis. Embo J 26: 3169–3179.
Kim JJ, Jung MW (2006). Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev 30: 188–202.
Korzus E, Rosenfeld MG, Mayford M (2004). CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42: 961–972.
Koshibu K, Graff J, Mansuy IM (2011). Nuclear protein phosphatase-1: an epigenetic regulator of fear memory and amygdala long-term potentiation. Neuroscience 173: 30–36.
Kozlovsky N, Matar MA, Kaplan Z, Kotler M, Zohar J, Cohen H (2007). Long-term down-regulation of BDNF mRNA in rat hippocampal CA1 subregion correlates with PTSD-like behavioural stress response. Int J Neuropsychopharmacol 10: 741–758.
Kriaucionis S, Heintz N (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324: 929–930.
Langley B, Gensert JM, Beal MF, Ratan RR (2005). Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord 4: 41–50.
Lattal KM, Barrett RM, Wood MA (2007). Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci 121: 1125–1131.
Lee BH, Yegnasubramanian S, Lin X, Nelson WG (2005). Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem 280: 40749–40756.
Lee JL (2010). Memory reconsolidation mediates the updating of hippocampal memory content. Front Behav Neurosci 4: 168.
Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC et al (2004a). A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel. J Neurosci 24: 3933–3943.
Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD (2004b). Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 279: 40545–40559.
Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P et al (2006). Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 281: 15763–15773.
Levenson JM, Sweatt JD (2005). Epigenetic mechanisms in memory formation. Nat Rev Neurosci 6: 108–118.
Liu D, Diorio J, Day JC, Francis DD, Meaney MJ (2000). Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci 3: 799–806.
Liu L, van Groen T, Kadish I, Tollefsbol TO (2009). DNA methylation impacts on learning and memory in aging. Neurobiol Aging 30: 549–560.
Lubin FD, Johnston LD, Sweatt JD, Anderson AE (2005). Kainate mediates nuclear factor-kappa B activation in hippocampus via phosphatidylinositol-3 kinase and extracellular signal-regulated protein kinase. Neuroscience 133: 969–981.
Lubin FD, Roth TL, Sweatt JD (2008). Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci 28: 10576–10586.
Lubin FD, Sweatt JD (2007). The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron 55: 942–957.
Ma DK, Guo JU, Ming GL, Song H (2009a). DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle 8: 1526–1531.
Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N et al (2009b). Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323: 1074–1077.
Maddox SA, Schafe GE (2011). Epigenetic alterations in the lateral amygdala are required for reconsolidation of a Pavlovian fear memory. Learn Mem 18: 579–593.
Marek R, Coelho CM, Sullivan RK, Baker-Andresen D, Li X, Ratnu V et al (2011). Paradoxical enhancement of fear extinction memory and synaptic plasticity by inhibition of the histone acetyltransferase p300. J Neurosci 31: 7486–7491.
McKenzie S, Eichenbaum H (2011). Consolidation and reconsolidation: two lives of memories? Neuron 71: 224–233.
Meaney MJ, Szyf M (2005a). Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci 7: 103–123.
Meaney MJ, Szyf M (2005b). Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci 28: 456–463.
Medina JH, Bekinschtein P, Cammarota M, Izquierdo I (2008). Do memories consolidate to persist or do they persist to consolidate? Behav Brain Res 192: 61–69.
Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP et al (2008). Cyclical DNA methylation of a transcriptionally active promoter. Nature 452: 45–50.
Michan S, Sinclair D (2007). Sirtuins in mammals: insights into their biological function. Biochem J 404: 1–13.
Miller CA, Campbell SL, Sweatt JD (2008). DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem 89: 599–603.
Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR et al (2010). Cortical DNA methylation maintains remote memory. Nat Neurosci 13: 664–666.
Miller CA, Sweatt JD (2007). Covalent modification of DNA regulates memory formation. Neuron 53: 857–869.
Milutinovic S, Brown SE, Zhuang Q, Szyf M (2004). DNA methyltransferase 1 knock down induces gene expression by a mechanism independent of DNA methylation and histone deacetylation. J Biol Chem 279: 27915–27927.
Mittal D, Torres R, Abashidze A, Jimerson N (2001). Worsening of post-traumatic stress disorder symptoms with cognitive decline: case series. J Geriatr Psychiatry Neurol 14: 17–20.
Monsey MS, Ota KT, Akingbade IF, Hong ES, Schafe GE (2011). Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS One 6: e19958.
Moore SA (2009). Cognitive abnormalities in posttraumatic stress disorder. Curr Opin Psychiatry 22: 19–24.
Munoz PC, Aspe MA, Contreras LS, Palacios AG (2010). Correlations of recognition memory performance with expression and methylation of brain-derived neurotrophic factor in rats. Biol Res 43: 251–258.
Narayan P, Dragunow M (2010). Pharmacology of epigenetics in brain disorders. Br J Pharmacol 159: 285–303.
Nemeroff CB, Bremner JD, Foa EB, Mayberg HS, North CS, Stein MB (2006). Posttraumatic stress disorder: a state-of-the-science review. J Psychiatr Res 40: 1–21.
Niehrs C, Schafer A (2012). Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol 22: 220–227.
Ooi SK, Bestor TH (2008). The colorful history of active DNA demethylation. Cell 133: 1145–1148.
Ou LC, Gean PW (2007). Transcriptional regulation of brain-derived neurotrophic factor in the amygdala during consolidation of fear memory. Mol Pharmacol 72: 350–358.
Paul S, Olausson P, Venkitaramani DV, Ruchkina I, Moran TD, Tronson N et al (2007). The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biol Psychiatry 61: 1049–1061.
Perkonigg A, Kessler RC, Storz S, Wittchen HU (2000). Traumatic events and post-traumatic stress disorder in the community: prevalence, risk factors and comorbidity. Acta Psychiatr Scand 101: 46–59.
Post RM, Weiss SR, Li H, Smith MA, Zhang LX, Xing G et al (1998). Neural plasticity and emotional memory. Dev Psychopathol 10: 829–855.
Quirk GJ, Garcia R, Gonzalez-Lima F (2006). Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry 60: 337–343.
Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR et al (2004). Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience 125: 1–6.
Rauch SL, Shin LM, Phelps EA (2006). Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research—past, present, and future. Biol Psychiatry 60: 376–382.
Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD (1999). The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19: 4337–4348.
Robertson KD (2002). DNA methylation and chromatin - unraveling the tangled web. Oncogene 21: 5361–5379.
Rogan MT, Staubli UV, LeDoux JE (1997). Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390: 604–607.
Roozendaal B, Hernandez A, Cabrera SM, Hagewoud R, Malvaez M, Stefanko DP et al (2010). Membrane-associated glucocorticoid activity is necessary for modulation of long-term memory via chromatin modification. J Neurosci 30: 5037–5046.
Roth TL, Lubin FD, Funk AJ, Sweatt JD (2009). Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry 65: 760–769.
Roth TL, Zoladz PR, Sweatt JD, Diamond DM (2011). Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J Psychiatr Res 45: 919–926.
Saavedra-Rodriguez L, Vazquez A, Ortiz-Zuazaga HG, Chorna NE, Gonzalez FA, Andres L et al (2009). Identification of flap structure-specific endonuclease 1 as a factor involved in long-term memory formation of aversive learning. J Neurosci 29: 5726–5737.
Sacktor TC (2008). PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res 169: 27–40.
Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV et al (2000). P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer 83: 817–825.
Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schafer A, Grummt I, Mayer C (2009). TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell 33: 344–353.
Shakibaei M, Buhrmann C, Mobasheri A (2011). Resveratrol-mediated SIRT-1 interactions with p300 modulate receptor activator of NF-kappaB ligand (RANKL) activation of NF-kappaB signaling and inhibit osteoclastogenesis in bone-derived cells. J Biol Chem 286: 11492–11505.
Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD (2006). Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron 50: 765–779.
Shea A, Walsh C, Macmillan H, Steiner M (2005). Child maltreatment and HPA axis dysregulation: relationship to major depressive disorder and post traumatic stress disorder in females. Psychoneuroendocrinology 30: 162–178.
Simone C, Stiegler P, Forcales SV, Bagella L, De Luca A, Sartorelli V et al (2004). Deacetylase recruitment by the C/H3 domain of the acetyltransferase p300. Oncogene 23: 2177–2187.
Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B et al (2011). Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B Neuropsychiatr Genet 156B: 700–708.
Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM (2012). Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol Psychiatry; e-pub ahead of print 28 January 2012.
Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL et al (2001). Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413: 739–743.
Sweatt JD (2009). Experience-dependent epigenetic modifications in the central nervous system. Biol Psychiatry 65: 191–197.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935.
Takei S, Morinobu S, Yamamoto S, Fuchikami M, Matsumoto T, Yamawaki S (2011). Enhanced hippocampal BDNF/TrkB signaling in response to fear conditioning in an animal model of posttraumatic stress disorder. J Psychiatr Res 45: 460–468.
Ursano RJ, Zhang L, Li H, Johnson L, Carlton J, Fullerton CS et al (2009). PTSD and traumatic stress from gene to community and bench to bedside. Brain Res 1293: 2–12.
Van der Staay FJ, Arndt SS, Nordquist RE (2009). Evaluation of animal models of neurobehavioral disorders. Behav Brain Funct 5: 11–33.
Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA et al (2007). Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci 27: 6128–6140.
Vecsler M, Simon AJ, Amariglio N, Rechavi G, Gak E (2010). MeCP2 deficiency downregulates specific nuclear proteins that could be partially recovered by valproic acid in vitro. Epigenetics 5: 61–67.
Villalba JM, de Cabo R, Alcain FJ (2012). A patent review of sirtuin activators: an update. Expert Opin Ther Pat 22: 355–367.
Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM et al (1999). Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 18: 7016–7025.
Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S (2002). Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 22: 6810–6818.
Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ et al (2005). Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci 25: 11045–11054.
Weaver IC, Grant RJ, Meaney MJ (2002a). Maternal behavior regulates long-term hippocampal expression of BAX and apoptosis in the offspring. J Neurochem 82: 998–1002.
Weaver IC, Szyf M, Meaney MJ (2002b). From maternal care to gene expression: DNA methylation and the maternal programming of stress responses. Endocr Res 28: 699.
Wood MA, Hawk JD, Abel T (2006). Combinatorial chromatin modifications and memory storage: a code for memory? Learn Mem 13: 241–244.
Wu H, Sun YE (2009). Reversing DNA methylation: new insights from neuronal activity-induced Gadd45b in adult neurogenesis. Sci Signal 2: pe17.
Yehuda R, Bierer LM (2009). The relevance of epigenetics to PTSD: implications for the DSM-V. J Trauma Stress 22: 427–434.
Yehuda R, LeDoux J (2007). Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron 56: 19–32.
Zaharia MD, Kulczycki J, Shanks N, Meaney MJ, Anisman H (1996). The effects of early postnatal stimulation on Morris water-maze acquisition in adult mice: genetic and maternal factors. Psychopharmacology (Berl) 128: 227–239.
Zhou Q, Atadja P, Davidson NE (2007). Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol Ther 6: 64–69.
Zoladz PR, Conrad CD, Fleshner M, Diamond DM (2008). Acute episodes of predator exposure in conjunction with chronic social instability as an animal model of post-traumatic stress disorder. Stress 11: 259–281.
Acknowledgements
Our work is supported by NIH grants MH091122, MH57014, NS 057098, AG031722, and NR012686 (JDS) and NSERC-PDF (IZ).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Zovkic, I., Sweatt, J. Epigenetic Mechanisms in Learned Fear: Implications for PTSD. Neuropsychopharmacol 38, 77–93 (2013). https://doi.org/10.1038/npp.2012.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.79
Keywords
This article is cited by
-
Chromatin Remodeling Factor SMARCA5 is Essential for Hippocampal Memory Maintenance via Metabolic Pathways in Mice
Neuroscience Bulletin (2023)
-
Long non-coding RNA LINC00926 regulates WNT10B signaling pathway thereby altering inflammatory gene expression in PTSD
Translational Psychiatry (2022)
-
An epigenetic mechanism for over-consolidation of fear memories
Molecular Psychiatry (2022)
-
Npas4 impairs fear memory via phosphorylated HDAC5 induced by CGRP administration in mice
Scientific Reports (2021)
-
A gene expression atlas for different kinds of stress in the mouse brain
Scientific Data (2020)
