
Abstract
The abuse of psychoactive ‘bath salts’ containing cathinones such as 3,4-methylenedioxypyrovalerone (MDPV) is a growing public health concern, yet little is known about their pharmacology. Here, we evaluated the effects of MDPV and related drugs using molecular, cellular, and whole-animal methods. In vitro transporter assays were performed in rat brain synaptosomes and in cells expressing human transporters, while clearance of endogenous dopamine was measured by fast-scan cyclic voltammetry in mouse striatal slices. Assessments of in vivo neurochemistry, locomotor activity, and cardiovascular parameters were carried out in rats. We found that MDPV blocks uptake of [3H]dopamine (IC50=4.1 nM) and [3H]norepinephrine (IC50=26 nM) with high potency but has weak effects on uptake of [3H]serotonin (IC50=3349 nM). In contrast to other psychoactive cathinones (eg, mephedrone), MDPV is not a transporter substrate. The clearance of endogenous dopamine is inhibited by MDPV and cocaine in a similar manner, but MDPV displays greater potency and efficacy. Consistent with in vitro findings, MDPV (0.1–0.3 mg/kg, intravenous) increases extracellular concentrations of dopamine in the nucleus accumbens. Additionally, MDPV (0.1–3.0 mg/kg, subcutaneous) is at least 10 times more potent than cocaine at producing locomotor activation, tachycardia, and hypertension in rats. Our data show that MDPV is a monoamine transporter blocker with increased potency and selectivity for catecholamines when compared with cocaine. The robust stimulation of dopamine transmission by MDPV predicts serious potential for abuse and may provide a mechanism to explain the adverse effects observed in humans taking high doses of ‘bath salts’ preparations.
Similar content being viewed by others

INTRODUCTION
The non-medical use of synthetic designer drugs is increasing on a global scale (Carroll et al, 2012; Hill and Thomas, 2011). In particular, synthetic analogs of the plant-derived stimulant cathinone are being sold online and in retail shops as legal alternatives to illicit drugs like cocaine, amphetamine, and 3,4-methylenedioxymethamphetamine (MDMA; Ross et al, 2011; Spiller et al, 2011). Products containing synthetic cathinones are given innocuous names such as ‘bath salts’ or ‘plant food’ and are often labeled ‘not for human consumption’. Although some cathinone derivatives are legal and can be sold without penalty, three of the most popular drugs of this type—3,4-methylenedioxypyrovalerone (MDPV), 4-methylmethcathinone (mephedrone), and 3,4-methylenedioxymethcathinone (methylone)—have been classified as Schedule I-controlled substances in the United States (DEA, 2011). All three of the Schedule I cathinones, depicted in (Figure 1), have been identified in bath salts products (Ross et al, 2011; Spiller et al, 2011), but MDPV is the chief substance found in the blood and urine from patients exposed to bath salts who are admitted to emergency departments in the United States (Borek and Holstege, 2012; Kyle et al, 2011; Murray et al, 2012; Spiller et al, 2011). The available evidence suggests that MDPV is a primary culprit in mediating adverse effects of bath salts products.

Chemical structures of MDPV and related test drugs.
Anecdotal clinical reports indicate that recreational doses of MDPV produce euphoria and increase alertness (Psychonaut, 2009), whereas high-dose exposure causes serious adverse effects, including agitation, psychosis, tachycardia, and even death (Borek and Holstege, 2012; Kyle et al, 2011; Murray et al, 2012; Ross et al, 2011; Spiller et al, 2011). Despite the widespread use of MDPV, no information is available regarding its molecular mechanism of action. Older literature demonstrates that pyrovalerone, a compound with structural similarity to MDPV, displays amphetamine-like stimulant properties in rodents (Fauquet et al, 1976; Vaugeois et al, 1993) and humans (Goldberg et al, 1973; Holliday et al, 1964). Bonnet and colleagues (Heron et al, 1994; Tidjane Corera et al, 2001) showed that pyrovalerone is a dopamine uptake blocker that interacts with transporter proteins in a manner similar to cocaine. Meltzer et al (2006) confirmed that pyrovalerone and several of its analogs are potent inhibitors of dopamine transporter (DAT) binding and uptake, but the effects of MDPV were not examined. Two recent reports show that MDPV has locomotor-stimulant effects and is readily self-administered in rodents (Huang et al, 2012; Watterson et al, 2012), but neither of those investigations addressed a potential mechanism of action for the drug. Based on the known pharmacology of pyrovalerone, and the findings of Meltzer et al (2006) we used in vitro and in vivo methods to test the hypothesis that MDPV interacts with DATs, norepinephrine transporters (NET), and serotonin transporters (SERT) in brain tissue. Our data demonstrate that MDPV is a catecholamine-selective transporter blocker that displays greater potency than the prototypical transporter blocker cocaine.
MATERIALS AND METHODS
Drugs and Chemicals
Chemical structures of the drugs tested in this study are depicted in Figure 1. MDPV, (±)-4-methylmethcathinone HCl (mephedrone), and (±)-3,4-methylenedioxymethcathinone HCl (methylone) were synthesized in racemic form in our laboratories. Chemical and structural analysis included proton nuclear magnetic resonance, gas chromatography/mass spectrometry, thin layer chromatography, and melting point determination. All data confirmed the expected structures. (+)-Amphetamine HCl (amphetamine) and (−)-cocaine HCl (cocaine) were obtained from the pharmacy at the National Institute on Drug Abuse (NIDA), Intramural Research Program (IRP) in Baltimore, MD, USA. [3H]Transmitters (specific activity ranging from 30–50 Ci/mmol) were purchased from Perkin Elmer (Shelton, CT, USA), whereas [3H]1-methyl-4-phenylpyridinium ([3H]MPP+; specific activity=85 Ci/mmol) was purchased from American Radiolabeled Chemicals (St Louis, MO, USA). All other chemicals and reagents used for the in vitro assays, voltammetry studies, microdialysis methods, and high-pressure liquid chromatography with electrochemical detection (HPLC-ECD) were acquired from Sigma-Aldrich (St Louis, MO, USA), unless otherwise noted.
Animals
Male Sprague-Dawley rats weighing 300–400 g and male CB57/BL6 mice weighing 25–35 g were housed under conditions of controlled temperature (22±2 °C) and humidity (45±5%), with food and water freely available. Rats and mice were maintained in facilities accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, and procedures were carried out in accordance with the Animal Care and Use Committee of the NIDA IRP. Lights were on from 0700–1900 h and experiments were carried out between 0900–1500 h.
In Vitro Uptake and Release Assays in Synaptosomes
Rats were euthanized by CO2 narcosis, and the brains were processed to yield synaptosomes as previously described (Rothman et al, 2001; Rothman et al, 2003b). For uptake inhibition assays, 5 nM [3H]dopamine, 10 nM [3H]norepinephrine, and 5 nM [3H]serotonin were used to assess transport activity at DAT, NET and SERT, respectively. The selectivity of uptake assays was optimized for a single transporter by including unlabeled blockers to prevent the uptake of [3H]transmitter by competing transporters. Uptake inhibition assays were initiated by adding 100 μl of tissue suspension to 900 μl Krebs-phosphate buffer (126 mM NaCl, 2.4 mM KCl, 0.83 mM CaCl2, 0.8 mM MgCl2, 0.5 mM KH2PO4, 0.5 mM Na2SO4, 11.1 mM glucose, 0.05 mM pargyline, 1 mg/ml bovine serum albumin, and 1 mg/ml ascorbic acid, pH 7.4) containing test drug and [3H]transmitter. Uptake inhibition assays were terminated by rapid vacuum filtration through Whatman GF/B filters, and retained radioactivity was quantified by liquid scintillation counting. For release assays, 9 nM [3H]MPP+ was used as the radiolabeled substrate for DAT and NET, while 5 nM [3H]serotonin was used as a substrate for SERT. All buffers used in the release assay methods contained 1 μM reserpine to block vesicular uptake of substrates. The selectivity of release assays was optimized for a single transporter by including unlabeled blockers to prevent the uptake of [3H]MPP+ or [3H]serotonin by competing transporters. Synaptosomes were preloaded with radiolabeled substrate in Krebs-phosphate buffer for 1 h (steady state). Release assays were initiated by adding 850 μl of preloaded synaptosomes to 150 μl of test drug. Release was terminated by vacuum filtration and retained radioactivity was quantified as described for uptake inhibition.
Efflux Assays in Cells Expressing DAT and SERT
HEK293 cells stably expressing either the human DAT or SERT were used for efflux studies (Sucic et al, 2010). The cells were cultivated on 10-cm dishes in medium complemented with fetal calf serum (10%), L-glutamine (1%), glucose (4.5 mg/ml), and gentamycin (50 μg/ml) at 37 °C, 95% humidity, and 5% CO2. For superfusion experiments (Scholze et al, 2000), cells (20 × 105) were seeded on 5-mm glass coverslips coated with poly-D-lysine. Before the experiment, 0.3 μM of the radiolabeled substrate [3H]MPP+ was added to the cells and incubated for 20 min at 37 °C. The coverslips were transferred to small chambers (volume=0.2 ml) and superfused with Krebs Ringer HEPES buffer (120 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 20 mM glucose, 10 mM HEPES, pH 7.4) at room temperature (25 °C) at a rate of 0.7 ml/min for 45 min to establish basal efflux. After basal efflux had stabilized, the experiment was initiated, with the collection of fractions every 2 min. After 6 min, monensin (25 μM) or control buffer was added to the cells. After 14 min, amphetamine (3 μM for HEK293-DAT cells), p-chloroamphetamine (3 μM for HEK293-SERT cells), or MDPV (3 μM for both transporters) was added. Finally, the remaining radioactivity was recovered by superfusing the cells for 6 min with 1% SDS. Drug-induced release of radioactivity at any time point was compared with the amount released in the absence of drugs and is expressed as percentage released.
Fast-Scan Cyclic Voltammetry in Mouse Striatal Slices
Mice were euthanized by cervical dislocation, and the brains were rapidly removed and placed in ice-cold, high-sucrose artificial cerebrospinal fluid. Coronal hemisections (280 μm) containing the striatum were cut using a vibratome (Leica VT1000S, Buffalo Grove, IL, USA). Slices were incubated in standard oxygenated artificial cerebrospinal fluid at 34–35 °C for 20–30 min, then allowed to stabilize at room temperature for 30 min before initiating recordings. During recordings, slices were continuously superfused with artificial cerebrospinal fluid at a rate of 2 ml/min and maintained at 28–30 °C. Fast-scan cyclic voltammetry was performed as described previously (Chen et al, 2008; Good et al, 2011). Carbon fibers (7 μm diameter) were vacuum-aspirated into borosilicate glass pipettes and cut so that ∼25–30 μm of exposed surface protruded from the pipette tip. Pipettes were back filled with a 4 M potassium acetate/150 mM KCl solution and connected to a holder/headstage assembly. Voltammetric scan and stimulation timing protocols were performed using PCI-based A/D boards and custom software (courtesy of Dr Mark Wightman, University of North Carolina). Scans consisted of sweeps from −0.4 to 1.0 V and back to −0.4 V, at a rate of 400 V/s, and were obtained every 100 ms. A 5-s (50 scan) control period preceded each electrically evoked response and was used to obtain a background current that was digitally subtracted from the current obtained during the peak of the response. Currents were converted to concentrations based on in vitro calibration curves for each electrode using 1–5 μM dopamine. Under stereoscopic magnification, carbon fibers were lowered to a depth of ∼100 μm in the dorsal striatum. A bipolar stimulating electrode was positioned ∼75–100 μm from the carbon fiber. In order to construct cumulative concentration-response curves, three predrug baseline signals were first obtained using both single-pulse (1 ms, 150–200 μA) and pulse-train stimulation (5 × 1 ms pulses, delivered at 25 Hz). Single-pulse responses were then monitored every 90 s during drug application. At the end of each drug application period (12–15 min), three post-drug pulse train (25 Hz) responses were obtained. At least four drug concentrations were obtained in each slice.
In Vivo Microdialysis in Rats
Microdialysis sampling was carried out in male rats as described, with minor modifications (Baumann et al, 2012). Rats were anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and received a surgically implanted jugular catheter and an intracranial guide cannula aimed at the nucleus accumbens. Rats were allowed at least 1 week for recovery from surgery. On the evening before an experiment, a 2 × 0.5-mm dialysis probe (CMA/12, Harvard Apparatus, Holliston, MA, USA) was inserted into the guide cannula, and an extension tube was attached to the jugular catheter. Each rat was placed into its own enclosure and connected to a tethering system. Probes were perfused overnight with Ringers’ salt solution pumped at a flow rate of 0.6 μl/min. On the next morning, dialysate samples were collected at 20-min intervals. Samples were immediately assayed for dopamine by HPLC-ECD as described elsewhere (Baumann et al, 2011). Rats were randomly assigned to groups receiving either drug or saline injections. Once three stable baseline samples were obtained, rats received two sequential intravenous (i.v.) injections of drug: one dose at time zero, followed by a threefold higher dose 60 min later. Control rats received sequential i.v. injections of saline (1 ml/kg) according to the same schedule. Microdialysis samples were collected every 20 min throughout the post-injection period of 120 min. At the end of the experiments, rats were euthanized with CO2 and decapitated. The brain sections were examined to verify placement of microdialysis probe tips within the nucleus accumbens. Only those rats with correct placements were included in data analyses.
Locomotor Activity Testing in Rats
Six male rats were used for locomotor activity experiments, which were carried out according to published procedures (Schindler and Carmona, 2002). Six identical locomotor activity monitors (MED Associates, St. Albans, VT, USA) were enclosed in three sound-attenuation chambers (BRS/LVE, Laurel, MD, USA). A smaller 42 × 42 cm Plexiglas chamber was situated inside each locomotor activity monitor. Each monitor consisted of a 16 × 16 infrared photocell array. The monitors were interfaced to a computer that tabulated distanced traveled (in cm) or stereotypy (in counts). Stereotypy was defined as the repeated interruption of the same photocell beam within 2 s. Rats were placed in the activity monitor every weekday for 1 h, at the same time each day, for 7 days before drug testing to allow for habituation to the monitor and to stabilize activity levels. This procedure was carried out to minimize the effects of novelty and light cycle on the results obtained (Halberstadt et al, 2012). Rats were then tested with subcutaneous (s.c.) injections of saline, MDPV (0.1–3.0 mg/kg), or cocaine (3–17 mg/kg) given 5 min before being placed in the activity monitor. Drugs were given no more frequently than two times per week. Rats continued to be placed in the activity monitor for 1 h on other days of the week and at least two non-injection sessions typically separated drug injections. Group results for saline treatment were acquired twice, once during testing of MDPV and once during testing with cocaine.
Telemetric Assessment of Cardiovascular Parameters in Rats
Four male rats were used in the telemetry experiments. Surgery was performed to implant telemetry transmitters (Model PA-C40, Data Sciences International, St. Paul, MN, USA) for the measurement of heart rate and blood pressure as described elsewhere (Tella et al, 1999). Briefly, under isoflurane anesthesia, a 4-cm incision was made along the midline of the abdomen and the descending aorta was exposed. The catheter of the transmitter was inserted about 2 cm into the aorta, the area was dried, and a drop of adhesive was applied to the catheter entry point. The transmitter was sutured to the abdominal musculature, then the abdominal incision and the skin were closed. Rats were singly housed post-operatively and given 2 weeks to recover. All testing occurred in a room separate from the housing room. Testing was typically performed 5 days per week (Monday–Friday) at approximately the same time each day for any individual animal. During the experimental session, the home cage (with food and water removed) was placed on top of the telemetry receiver. Three telemetry receivers were located in three separate but identical sound-attenuation chambers. Heart rate and mean arterial blood pressure were sampled every minute and were monitored for 2 h. Pre-experiment habituation testing was carried out until control cardiovascular parameters were stable from session to session. Animals were then given an s.c. injection of saline just before placement of the cage on the telemetry receiver, at least 2 times per week, until cardiovascular parameters following saline injection remained stable. The cardiovascular response to the injection of saline was indistinguishable from that observed when the animals were placed in the telemetry chamber with no previous injection. Testing then began with MDPV or cocaine, with test drugs given s.c. no more frequently than 2 times per week, usually on Tuesdays and Fridays, with control saline injections given on Thursdays.
Data Analysis and Statistics
All statistical analyses were carried out using GraphPad Prism (v. 5.0; GraphPad Scientific, San Diego, CA, USA). For synaptosome assays, IC50 values for inhibition of uptake and EC50 values for stimulation of release were calculated based on non-linear regression analysis. Time course data from the superfusion experiments, comparing effects of monensin with control, were evaluated by two-way analysis of variance (dose × time) followed by Bonferroni’s post hoc test. For the voltammetry experiments, drug-evoked responses for each drug concentration were analyzed by subtracting the control predrug curve from the postdrug curve (DAdrug−DAcontrol), and performing an area-under-the-curve (AUC) analysis. In this way, changes in both the peak amplitude and time course of the responses could be normalized across all recordings. The dose–response AUC data for MDPV and cocaine were compared by non-linear regression. Neurochemical data from microdialysis experiments were normalized to percentage baseline (based on three preinjection samples) and evaluated using two-way analysis of variance (dose × time) followed by Bonferroni’s post hoc test. For the locomotor studies, the total distance traveled and the number of stereotypic movements were summed across the 1-h testing period to construct dose–effect curves for MPDV and cocaine. One-factor analysis of variance (dose) followed by Newman–Keuls post hoc test was used to determine significant effects of each drug as compared with saline control. The telemetry data were collapsed across the 2-h sampling period to yield mean heart rate (beats/min) and mean blood pressure (mm Hg) at each drug dose. These parameters were used to construct dose–effect curves, which were analyzed by one-factor analysis of variance (dose) followed by Newman–Keuls post hoc test. Differences between peak effects of MDPV vs cocaine were assessed by unpaired t-tests. P<0.05 was the minimum criterion for statistical significance.
RESULTS
MDPV is a Potent Catecholamine-Selective Transporter Blocker
We examined the ability of MDPV, cocaine, and amphetamine to block uptake of [3H]dopamine, [3H]norepinephrine, and [3H]serotonin by their respective transporters—DAT, NET, and SERT. The designer cathinones, mephedrone and methylone, were tested for comparison. Figure 2a, c, and e illustrate that MDPV is a potent uptake blocker at DAT and NET, with weaker effects at SERT. When compared with cocaine, MDPV is 50-times and 10-times more potent as an uptake blocker at DAT and NET, respectively (see Table 1). MDPV and other test drugs are fully efficacious as uptake blockers at all three transporters. MDPV and amphetamine display high selectivity for inhibition of catecholamine uptake, whereas cocaine, mephedrone, and methylone are non-selective.

Effects of MDPV and comparison test drugs on inhibition of uptake and stimulation of release at DAT, NET, and SERT in rat brain synaptosomes. For uptake assays (a, c, and e), synaptosomes were incubated with different concentrations of MDPV, cocaine, amphetamine, or mephedrone in the presence of 5 nM [3H]dopamine (a, for DAT), 10 nM [3H]norepinephrine (c, for NET), or 5 nM [3H]serotonin (e, for SERT). Data are percentage of [3H]transmitter uptake expressed as mean±s.e.m. for N=3–4 experiments. For release assays (b, d, and f), synaptosomes were preloaded with 9 nM [3H]MPP+ for DAT (b) and NET (d), or 5 nM [3H]serotonin for SERT (f), then incubated with different concentrations of test drugs to evoke release via reverse transport. Data are percentage of [3H]substrate release expressed as mean±s.e.m. for N=3–4 experiments.
MDPV is Not a Transporter Substrate
It is well established that traditional uptake inhibition assays cannot distinguish between drugs acting as transporter blockers vs those acting as substrates (ie, releasers; Rothman et al, 2001; Rothman et al, 2003b). Therefore, we performed additional experiments to evaluate the effects of MDPV and related test compounds in transporter-release assays that can distinguish between blockers and substrates. In the release experiments, [3H]substrates are ‘preloaded’ into synaptosomes before addition of test drugs, which then evoke non-exocytotic release via reverse transport. Figure 2b, d, and f illustrate the dose-dependent effects of test drugs on release of [3H]MPP+ via DAT and NET, and release of [3H]serotonin via SERT. MDPV causes ‘partial’ low-efficacy (ie,<30% of maximal) release of [3H]MPP+ from DAT and NET but has no activity at SERT. The uptake blocker cocaine evokes a similar profile of low-efficacy release of [3H]MPP+, whereas amphetamine, mephedrone, and methylone are fully efficacious releasers at all transporters (Table 1). With regard to transporter selectivity, amphetamine is a highly selective releaser at DAT and NET, whereas mephedrone and methylone are relatively non-selective releasers.
We surmised that the low-efficacy releasing activity of MDPV is actually related to uptake blockade. To test this hypothesis, we examined the effects of MDPV on efflux of [3H]MPP+ in HEK cells stably expressing human DAT or SERT. In the superfusion experiments, cells are pretreated with the Na+/H+ ionophore monensin, which alters ionic gradients to increase cytoplasmic Na+ (Mollenhauer et al, 1990; Turetta et al, 2004). Monensin is known to facilitate reversal of normal transporter flux, thereby markedly enhancing the releasing capability of transporter substrates but not uptake blockers (Scholze et al, 2000). Figure 3a demonstrates that MDPV produces a modest low-efficacy efflux of [3H]MPP+ in HEK cells expressing DAT, similar to effects of the drug on release of [3H]MPP+ in synaptosomes. Importantly, monensin pretreatment does not augment MDPV-induced efflux of [3H]MPP+ (F[1,12]=0.9519, P=0.49; Figure 3a), whereas the ionophore dramatically enhances efflux induced by the transporter substrate amphetamine (F[1,12]=14.19, P<0.001; Figure 3b). Analogous results are found in HEK cells expressing SERT, where monensin has no effect on MDPV-induced efflux of [3H]MPP+ but enhances the efflux produced by the SERT substrate p-chloroamphetamine (data not shown). The findings from HEK cells provide decisive evidence that effects of MDPV on efflux are due to uptake blockade rather than release.

Effects of MDPV and comparison drugs on efflux of [3H]MPP+ in HEK239 cells expressing human DAT and clearance of endogenous dopamine in mouse striatal slices. For efflux assays (a, b), control buffer or 25 μM monensin was added to the medium after 6 min of baseline superfusion (see 6-min arrows), followed by addition of 3 μM MDPV (a) or amphetamine (b) at 14 min (see 14-min arrows). Data are percentage of [3H]MPP+ efflux expressed as mean±s.e.m. for N=3–9 experiments. *P<0.05 compared with control at a particular time point (Bonferroni’s test). For voltammetry measurements (c, d), endogenous dopamine (DA) release was evoked by electrical stimulation (5 × 1 ms pulses, 25 Hz; see arrows) in the presence or absence of different concentrations of MDPV (c) or cocaine (d), and DA signal was measured thereafter. Data in (c) and (d) represent μM concentrations of DA determined in individual slices. Dose–response curves for DA area-under-the-curve (AUC) (e) and percentage of maximal response (f) are expressed as mean±s.e.m. for N=3–5 experiments.
MDPV Inhibits Dopamine Clearance in Mouse Striatal Slices
Once we were confident that MDPV acts as a pure uptake blocker, it seemed prudent to carry out further experiments comparing effects of MDPV to those produced by the classic uptake blocker cocaine. To this end, we assessed effects of MPDV and cocaine on dopamine clearance using fast-scan cyclic voltammetry in mouse striatal slices (Chen et al, 2008; Good et al, 2011). In these experiments, release of endogenous dopamine is elicited by trains of stimulation (5 × 1 ms pulses, 25 Hz) designed to mimic burst firing of dopamine neurons (Hyland et al, 2002). After obtaining stable baseline responses, increasing concentrations of MDPV or cocaine are applied while continuing to monitor the response every 90 s. Representative examples of raw voltammetric signals are shown in Figure 3c and d, while dose–response curves comparing overall effects of MDPV and cocaine on dopamine clearance are depicted in Figure 3e and f. Both MDPV and cocaine increase the amplitude of the dopamine signal and slow its decay. However, MDPV produces a much more robust shift in the shape of the curve than cocaine, resulting in a significantly greater amount of dopamine detected by the carbon fiber electrode (Figure 3e). When comparing AUC values of normalized voltammetric data, MDPV (EC50=115±17 nM) is significantly more potent than cocaine (EC50=308±75 nM) at inhibiting dopamine clearance (F[1,55]=15.32, P<0.001; Figure 3f).
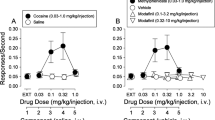
MDPV is More Potent and Efficacious than Cocaine In Vivo
Given the potent effects of MDPV on dopamine clearance, we predicted that the drug would have powerful cocaine-like actions in vivo. Using microdialysis sampling in conscious rats (Baumann et al, 2012), we compared the effects of MDPV and cocaine on extracellular dopamine levels in nucleus accumbens. Figure 4a shows that i.v. MDPV significantly increases dialysate dopamine in a dose-related manner (F[1,8]=87.12, P<0.0001), and Figure 4b shows that i.v. cocaine has a similar effect (F[1,8]=65.89, P<0.0001). However, MDPV is 10-fold more potent than cocaine in its ability to increase extracellular dopamine, as the lowest effective dose of MDPV is 0.1 mg/kg (P<0.05, Bonferroni’s) compared with 1.0 mg/kg for cocaine (P<0.05, Bonferroni’s). The rise in extracellular dopamine produced by 0.3 mg/kg MDPV remains significantly elevated above saline control for at least 60 min post-injection (P<0.05, Bonferroni’s), whereas the response to a 3.0-mg/kg cocaine returns to baseline by 40 min post-injection. Figure 4c and d illustrate the effect of MDPV and cocaine on locomotor activity in rats. In these experiments, rats receive s.c. injection of drug or saline (zero dose) and are placed into enclosures equipped with photobeam sensors that detect movements (Schindler and Carmona, 2002). Figure 4c shows that MDPV elicits dose-related stimulation of forward locomotion (ie, distance traveled; F[4,25]=15.32, P<0.0001) and cocaine has a similar effect (F[3,20]=4.14, P<0.01). MDPV is more potent than cocaine, as the threshold dose of MDPV that stimulates significant forward locomotion is 0.3 mg/kg (P<0.05, Newman–Keuls), compared with 10 mg/kg for cocaine (P<0.05, Newman–Keuls). Figure 4d demonstrates that stereotypy is significantly increased by administration of MDPV (F[4,25]=26.31, P<0.0001) and cocaine (F[3,20]=7.68, P<0.001). The threshold dose of MDPV capable of stimulating significant stereotypy is 0.3 mg/kg (P<0.05, Newman–Keuls), while the threshold dose for cocaine is 3 mg/kg (P<0.05, Newman–Keuls). It is important to note that MDPV is more efficacious than cocaine in both locomotor assays. Specifically, MDPV induces significantly greater peak effects than cocaine with regard to distance traveled (P<0.001, t-test; effect of 3.0 mg/kg MDPV vs 10 mg/kg cocaine) and stereotypy (P<0.001, t-test; effect of 3.0 mg/kg MDPV vs 10 mg/kg cocaine). We assessed cardiovascular actions of MDPV in conscious rats bearing surgically implanted telemetric devices (Tella et al, 1999). The data in Figure 4e and f demonstrate that s.c. injection of MDPV induces tachycardia (F[4,25]=32.61, P<0.0001) and hypertension (F[4,25]=3.05, P<0.05). By comparison, cocaine significantly increases heart rate (F[3,20]=6.15, P<0.01) but not blood pressure (F[3,20]=1.10, P=0.37). MDPV elicits a significantly greater peak effect than cocaine with respect to heart rate (P<0.01, t-test; effect of 3.0 mg/kg MDPV vs 10 mg/kg cocaine) but not blood pressure (P=0.23, t-test; effect of 3.0 mg/kg MDPV vs 10 mg/kg cocaine).

Effects of MDPV and cocaine on extracellular dopamine in nucleus accumbens, locomotor activity and cardiovascular parameters in conscious rats. For microdialysis studies (a, b), rats received i.v. injection of MDPV (a) or cocaine (b) at time zero (see t=0-min arrows), followed by a threefold higher dose 60 min later (see 60-min arrows). Vehicle controls received i.v. saline injections on the same time schedule. Dialysate DA data are percentage of basal expressed as mean±s.e.m. for N=6–7 rats/group. *P<0.05 compared with saline vehicle at a particular time point (Bonferroni’s test). For locomotor studies (c, d), rats received s.c. injection of MDPV, cocaine, or saline vehicle and were placed into chambers equipped with photobeams. Distance traveled (c) and stereotypic movements (d) were measured for 1 h post-injection. Data are mean±s.e.m. for N=6–7 test sessions/dose. *P<0.05 compared with saline-treated control (Newman–Keuls test). For the cardiovascular measurements (e, f), rats bearing surgically implanted telemetric sensors received s.c. injection of MDPV, cocaine, or saline vehicle. Heart rate (e) and blood pressure (f) were measured for 2 h post-injection. Data are mean±s.e.m. for N=6–7 test sessions/dose. *P<0.05 compared with saline-treated control (Newman–Keuls test).
DISCUSSION
The primary goal of the present study was to provide a systematic evaluation of the mechanism and pharmacological effects of MDPV, a primary constituent of psychoactive bath salts sold on the internet and in retail shops (Ross et al, 2011; Spiller et al, 2011). A variety of designer cathinone derivatives and other substances (eg, caffeine) can be found in bath salts products, but MDPV is the main compound detected in biological fluids from patients admitted to emergency departments for bath salts overdose in the United States (Borek and Holstege, 2012; Kyle et al, 2011; Murray et al, 2012; Spiller et al, 2011). We found that MDPV is a catecholamine-selective transporter blocker that is much more potent than cocaine when assessed using in vitro transporter assays. In contrast to other bath salts compounds like mephedrone and methylone, MDPV is not a transporter substrate. Thus, MDPV displays a profile of monoamine transporter activity that is similar to the structurally related compound pyrovalerone (Meltzer et al, 2006; Tidjane Corera et al, 2001; Vaugeois et al, 1993). Consistent with the potent blockade of dopamine uptake, MDPV inhibits dopamine clearance in mouse striatal slices and elevates dialysate dopamine concentrations in rat nucleus accumbens. Importantly, MDPV is at least 10 times more potent than cocaine in assays measuring motor hyperactivity and cardiovascular stimulation in vivo.
A critical secondary aim of the present study was to ascertain whether MDPV functions as a monoamine transporter substrate (ie, like amphetamine) or blocker (ie, like cocaine). This distinction in molecular mechanism is essential to determine for at least three reasons: (1) transporter substrates, but not blockers, are translocated into cells and evoke non-exocytotic release of transmitters via reverse transport (Rothman and Baumann, 2003a; Sitte, Freissmuth, 2010); (2) transporter substrates, but not blockers, are known to produce long-term deficits in monoamine neurons that are often viewed as neurotoxicity (Baumann et al, 2007; Fleckenstein et al, 2007); and (3) there are conflicting reports in the literature regarding the precise interactions of designer cathinones with monoamine transporters (Baumann et al, 2012; Cozzi et al, 1999; Hadlock et al, 2011; Martinez-Clemente et al, 2011; Nagai et al, 2007; Sogawa et al, 2011). Our results from in vitro experiments in synaptosomes confirm that uptake inhibition assays can identify drugs which interact with transporter proteins. However, uptake assays cannot discriminate between drugs which act as blockers or substrates because both types of drugs are fully efficacious in their ability to reduce the accumulation of [3H]transmitter in synaptosomes (see Figure 2a, c, and e). Results from the release assays, on the other hand, uncover obvious differences among the various drugs tested (see Figure 2b, d, and f). In particular, MDPV and cocaine exhibit low-efficacy ‘partial’-releasing actions at DAT and NET (ie,<30% of maximal release), whereas amphetamine, mephedrone, and methylone are fully efficacious in their ability to evoke release of preloaded [3H]MPP+ and [3H]serotonin (ie, 100% of maximal release). The data with mephedrone and methylone agree with the published findings showing that these bath salts compounds are transporter substrates and not blockers (Baumann et al, 2012; Nagai et al, 2007; Sogawa et al, 2011).
Based on previous observations, we surmised that the apparent releasing effects of MDPV and cocaine are secondary to uptake blockade, rather than substrate activity per se. For example, we have shown that established uptake blockers like GBR12909 and indatraline display low-efficacy releasing effects in synaptosomes (Rothman and Baumann, 2003a; Rothman et al, 2001). The partial releasing activity of uptake blockers is presumably related to the passive ‘leak’ of radiolabeled substrates from synaptosomes, a phenomenon that is normally counteracted by ongoing transporter-mediated uptake. In the presence of transporter blockers, the leak is unmasked and gives the illusion of drug-induced release. In support of this notion, Scholze et al (2000) demonstrated that the serotonin uptake blocker imipramine evokes low-efficacy efflux of [3H]serotonin from HEK cells expressing human SERT. In that same publication, pretreatment with the Na+/H+ ionophore monensin markedly enhanced the efflux of [3H]MPP+ produced by the SERT substrate p-chloroamphetamine without influencing the effects of SERT blockers. Here we report that monensin dramatically augments amphetamine-induced efflux of [3H]MPP+ from DAT-expressing cells, whereas the ionophore has no effect on MDPV-induced efflux under identical conditions (compare Figure 3a and b). Thus, monensin clearly differentiates the mechanism of efflux produced by amphetamine from that produced by MDPV. Taken together, the in vitro findings provide compelling evidence that MDPV functions as a ‘pure’ transporter blocker rather than a substrate. The present results also highlight the utility of monensin as a reliable tool to discriminate transporter substrates from blockers in cells stably expressing transporter proteins.
Our in vitro data reveal a clear mechanistic dichotomy among the most common bath salts constituents: namely, ring-substituted analogs of methcathinone like mephedrone and methylone act as transporter substrates, whereas pyrrolidinophenones like MDPV act as transporter blockers. There are notable differences in transporter selectivity across this group of compounds as well. Mephedrone and methylone are non-selective transporter substrates that produce neurochemical effects akin to those produced by MDMA (Baumann et al, 2012; Kehr et al, 2011). By contrast, MDPV is a catecholamine-selective uptake blocker. The presence of a 3,4-methylenedioxy ring-substitution in MDPV does not confer serotonergic activity as observed with MDMA and methylone (Baumann et al, 2012; Baumann et al, 2007). Indeed, MDPV displays more than 100-fold greater potency at DAT and NET when compared with SERT. Such findings suggest that ring-substituted analogs of methcathinone will produce MDMA-like behavioral effects, while pyrrolidinophenones will be more stimulant-like in their profile of actions. In agreement with this proposal, recent findings in rats have shown that wheel-running behavior produced by mephedrone is MDMA-like, while that produced by MDPV is more methamphetamine-like (Huang et al, 2012). Additional investigations are warranted to compare the effects of ring-substituted cathinones and pyrrolidinophenones in animal models and in human subjects under controlled laboratory conditions.
Using fast-scan cyclic voltammetry, we found that MDPV exhibits powerful dose-related effects on dopamine clearance. Specifically, MDPV and cocaine inhibit dopamine clearance in a manner consistent with the dopamine uptake blockers (John and Jones, 2007), but MPDV is more potent and efficacious (Figure 3c–f). As is typically observed in cyclic voltammetry experiments, uptake inhibition results in a large increase in the magnitude of evoked dopamine signal and a profound broadening of the dopamine decay curve (Schmitz et al, 2001). MDPV causes a much greater increase in the dopamine AUC response when compared with cocaine (Figure 3e). The mechanistic explanation for the heightened efficacy of MDPV in the slice preparation is not known, but we hypothesize that the potency of MDPV might be a critical factor. Owing to its high potency at DAT, MDPV may display a slow dissociation from the site (ie, persistent binding), thereby augmenting and extending its pharmacological effects. In fact, previous voltammetric studies have shown that high-affinity dopamine uptake blockers like nomifensine and GBR12909 produce marked inhibition of dopamine clearance that mirrors the effects of MDPV shown here (Bull et al, 1990; Schmitz et al, 2001). It also remains possible that MDPV interacts with non-transporter sites of action, which further enhance its effects on dopamine clearance, and this proposal warrants examination.
In vivo microdialysis methods were used to compare the effects of MDPV and cocaine on extracellular dopamine in the nucleus accumbens, a brain region implicated in addictive properties of drugs (Bonci et al, 2003; Willuhn et al, 2010). As predicted from the in vitro findings, both drugs elicit dose-related increases in dialysate dopamine, but MDPV is at least 10 times more potent than cocaine in this regard (Figure 4a and b). Furthermore, the rise in extracellular dopamine associated with MDPV is sustained when compared with the short-lived effects of cocaine. It is well established that drug-induced elevations in extracellular dopamine in the nucleus accumbens are involved with locomotor-activating properties of stimulant drugs (Ikemoto, 2002; Pennartz et al, 1994). Our previous experiments examining stimulant drug effects in rats have shown strong positive correlations between dialysate dopamine responses and the extent of hyperactivity (Baumann et al, 2011; Zolkowska et al, 2009). The present data show that MDPV produces robust dose-related forward locomotion and stereotypy. Consistent with the voltammetric findings, locomotor effects of MDPV are much greater in magnitude than the comparable effects of cocaine (see Figure 4c and d). Collectively, the potent and efficacious actions of MDPV on extracellular dopamine and motor activity suggest a high potential for abuse. The fact that MDPV has weak effects on SERT may further enhance its reinforcing properties, as substantial evidence indicates that elevations in synaptic 5-HT can dampen stimulant effects mediated by DA (Wee et al, 2005; Baumann et al, 2011). A recent study by Watterson et al (2012) demonstrates that MDPV is readily self-administered by rats, and the range of self-injected doses (0.05–0.20 mg/kg, i.v.) agrees well with the i.v. doses shown here to elevate dialysate dopamine. Two of the most serious side-effects of bath salts ingestion in human users are acute tachycardia and hypertension (Ross et al, 2011; Spiller et al, 2011). Here we compared the effects of MDPV and cocaine on cardiovascular parameters in conscious rats. Similar to its effects on other in vivo endpoints, MDPV is more potent than cocaine in its ability to increase heart rate and blood pressure.
Our preclinical findings may have important implications for people who take bath salts products for recreational purposes. The potent blockade of dopamine uptake caused by MDPV predicts that the drug has a high risk for abuse, whereas the potent blockade of norepinephrine uptake portends dangerous cardiovascular stimulation. Patients admitted to emergency departments for bath salts overdose, who have toxicological verification of MDPV consumption, display symptoms, including agitation, psychosis, violent behavior, hyperthermia, and tachycardia (Borek and Holstege, 2012; Kyle et al, 2011; Murray et al, 2012; Spiller et al, 2011). The constellation of adverse effects produced by high-dose MDPV resembles the life-threatening excited delirium syndrome associated with acute cocaine toxicity (Mash et al, 2009; Ruttenber et al, 1997). Accordingly, the preclinical findings in the present study, demonstrating powerful cocaine-like actions of MDPV, suggest that efforts to remediate symptoms of bath salts overdose should aim to manage excessive dopaminergic and noradrenergic stimulation. It is noteworthy that new legal cathinone-related compounds are being marketed to replace Schedule I drugs like MDPV, mephedrone, and methylone (Shanks et al, 2012). Given the accumulating evidence that various bath salts constituents display unique interactions with monoamine transporters and possibly other targets, it seems reasonable to assume that determining the pharmacology of ‘second-generation’ replacement cathinones will require empirical investigations on a case-by-case basis.
References
Baumann MH, Ayestas MA, Partilla JS, Sink JR, Shulgin AT, Daley PF et al (2012). The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37: 1192–1203.
Baumann MH, Clark RD, Woolverton WL, Wee S, Blough BE, Rothman RB (2011). In vivo effects of amphetamine analogs reveal evidence for serotonergic inhibition of mesolimbic dopamine transmission in the rat. J Pharmacol Exp Ther 337: 218–225.
Baumann MH, Wang X, Rothman RB (2007). 3,4-Methylenedioxymethamphetamine (MDMA) neurotoxicity in rats: a reappraisal of past and present findings. Psychopharmacology (Berl) 189: 407–424.
Bonci A, Bernardi G, Grillner P, Mercuri NB (2003). The dopamine-containing neuron: maestro or simple musician in the orchestra of addiction? Trends Pharmacol Sci 24: 172–177.
Borek HA, Holstege CP (2012). Hyperthermia and multiorgan failure after abuse of ‘bath salts’ containing 3,4-methylenedioxypyrovalerone. Ann Emerg Med 60: 103–105.
Bull DR, Palij P, Sheehan MJ, Millar J, Stamford JA, Kruk ZL et al (1990). Application of fast cyclic voltammetry to measurement of electrically evoked dopamine overflow from brain slices in vitro. J Neurosci Methods 32: 37–44.
Carroll FI, Lewin AH, Mascarella SW, Seltzman HH, Reddy PA (2012). Designer drugs: a medicinal chemistry perspective. Ann NY Acad Sci 1248: 18–38.
Chen YH, Harvey BK, Hoffman AF, Wang Y, Chiang YH, Lupica CR (2008). MPTP-induced deficits in striatal synaptic plasticity are prevented by glial cell line-derived neurotrophic factor expressed via an adeno-associated viral vector. FASEB J 22: 261–275.
Cozzi NV, Sievert MK, Shulgin AT, Jacob P, Ruoho AE (1999). Inhibition of plasma membrane monoamine transporters by beta-ketoamphetamines. Eur J Pharmacol 381: 63–69.
Drug Enforcement Administration (2011). Schedules of controlled substances: temporary placement of three synthetic cathinones in Schedule I. Final Order. In: Fed Regist Vol. 76 2011/10/25 edn, pp 65371–65375.
Fauquet JP, Morel E, Demarty C, Rapin JR (1976). [Role of central catecholamines in the psychostimulant activity of pyrovalerone]. Arch Int Pharmacodyn Ther 224: 325–337.
Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR (2007). New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol 47: 681–698.
Goldberg J, Gardos G, Cole JO (1973). A controlled evaluation of pyrovalerone in chronically fatigued volunteers. Int Pharmacopsychiatry 8: 60–69.
Good CH, Hoffman AF, Hoffer BJ, Chefer VI, Shippenberg TS, Backman CM et al (2011). Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson's disease. FASEB J 25: 1333–1344.
Hadlock GC, Webb KM, McFadden LM, Chu PW, Ellis JD, Allen SC et al (2011). 4-Methylmethcathinone (mephedrone): neuropharmacological effects of a designer stimulant of abuse. J Pharmacol Exp Ther 339: 530–536.
Halberstadt AL, Buell MR, Price DL, Geyer MA (2012). Differences in the locomotor-activating effects of indirect serotonin agonists in habituated and non-habituated rats. Pharmacol Biochem Behav 102: 88–94.
Heron C, Costentin J, Bonnet JJ (1994). Evidence that pure uptake inhibitors including cocaine interact slowly with the dopamine neuronal carrier. Eur J Pharmacol 264: 391–398.
Hill SL, Thomas SH (2011). Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila) 49: 705–719.
Holliday AR, Morris RB, Sharpley RP (1964). Compound 84/F 1983 compared with D-amphetamine and placebo in regard to effects on human performance. Psychopharmacologia 6: 192–200.
Huang PK, Aarde SM, Angrish D, Houseknecht KL, Dickerson TJ, Taffe MA (2012). Contrasting effects of d-methamphetamine, 3,4-methylenedioxymethamphetamine, 3,4-methylenedioxypyrovalerone, and 4-methylmethcathinone on wheel activity in rats. Drug Alcohol Depend (in press).
Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R (2002). Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 114: 475–492.
Ikemoto S (2002). Ventral striatal anatomy of locomotor activity induced by cocaine, D-amphetamine, dopamine and D1/D2 agonists. Neuroscience 113: 939–955.
John CE, Jones SR (2007). Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology 52: 1596–1605.
Kehr J, Ichinose F, Yoshitake S, Goiny M, Sievertsson T, Nyberg F et al (2011). Mephedrone, compared with MDMA (ecstasy) and amphetamine, rapidly increases both dopamine and 5-HT levels in nucleus accumbens of awake rats. Br J Pharmacol 164: 1949–1958.
Kyle PB, Iverson RB, Gajagowni RG, Spencer L (2011). Illicit bath salts: not for bathing. J Miss State Med Assoc 52: 375–377.
Martinez-Clemente J, Escubedo E, Pubill D, Camarasa J (2011). Interaction of mephedrone with dopamine and serotonin targets in rats. Eur Neuropsychopharmacol 22: 231–236.
Mash DC, Duque L, Pablo J, Qin Y, Adi N, Hearn WL et al (2009). Brain biomarkers for identifying excited delirium as a cause of sudden death. Forensic Sci Int 190: e13–e19.
Meltzer PC, Butler D, Deschamps JR, Madras BK (2006). 1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (Pyrovalerone) analogues: a promising class of monoamine uptake inhibitors. J Med Chem 49: 1420–1432.
Mollenhauer HH, Morre DJ, Rowe LD (1990). Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochim Biophys Acta 1031: 225–246.
Murray BL, Murphy CM, Beuhler MC (2012). Death following recreational use of designer drug ‘bath salts’ containing 3,4-Methylenedioxypyrovalerone (MDPV). J Med Toxicol 8: 69–75.
Nagai F, Nonaka R, Satoh Hisashi Kamimura K (2007). The effects of non-medically used psychoactive drugs on monoamine neurotransmission in rat brain. Eur J Pharmacol 559: 132–137.
Pennartz CM, Groenewegen HJ, Lopes da Silva FH (1994). The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioural, electrophysiological and anatomical data. Prog Neurobiol 42: 719–761.
Psychonaut Web Mapping Group (2009) MDPV Report edn Institute of Psychiatry, Kings College, London: London, UK.
Ross EA, Watson M, Goldberger B (2011). ‘Bath salts’ intoxication. N Engl J Med 365: 967–968.
Rothman RB, Baumann MH (2003a). Monoamine transporters and psychostimulant drugs. Eur J Pharmacol 479: 23–40.
Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI et al (2001). Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 39: 32–41.
Rothman RB, Vu N, Partilla JS, Roth BL, Hufeisen SJ, Compton-Toth BA et al (2003b). In vitro characterization of ephedrine-related stereoisomers at biogenic amine transporters and the receptorome reveals selective actions as norepinephrine transporter substrates. J Pharmacol Exp Ther 307: 138–145.
Ruttenber AJ, Lawler-Heavner J, Yin M, Wetli CV, Hearn WL, Mash DC (1997). Fatal excited delirium following cocaine use: epidemiologic findings provide new evidence for mechanisms of cocaine toxicity. J Forensic Sci 42: 25–31.
Schindler CW, Carmona GN (2002). Effects of dopamine agonists and antagonists on locomotor activity in male and female rats. Pharmacol Biochem Behav 72: 857–863.
Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D (2001). Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci 21: 5916–5924.
Scholze P, Zwach J, Kattinger A, Pifl C, Singer EA, Sitte HH (2000). Transporter-mediated release: a superfusion study on human embryonic kidney cells stably expressing the human serotonin transporter. J Pharmacol Exp Ther 293: 870–878.
Shanks KG, Dahn T, Behonick G, Terrell A (2012). Analysis of first and second generation legal highs for synthetic cannabinoids and synthetic stimulants by ultra-performance liquid chromatography and time of flight mass spectrometry. J Anal Toxicol 36: 360–371.
Sitte HH, Freissmuth M (2010). The reverse operation of Na(+)/Cl(-)-coupled neurotransmitter transporters--why amphetamines take two to tango. J Neurochem 112: 340–355.
Sogawa C, Sogawa N, Ohyama K, Kikura-Hanajiri R, Goda Y, Sora I et al (2011). Methylone and monoamine transporters: correlation with toxicity. Curr Neuropharmacol 9: 58–62.
Spiller HA, Ryan ML, Weston RG, Jansen J (2011). Clinical experience with and analytical confirmation of ‘bath salts’ and ‘legal highs’ (synthetic cathinones) in the United States. Clin Toxicol (Phila) 49: 499–505.
Sucic S, Dallinger S, Zdrazil B, Weissensteiner R, Jorgensen TN, Holy M et al (2010). The N terminus of monoamine transporters is a lever required for the action of amphetamines. J Biol Chem 285: 10924–10938.
Tella SR, Schindler CW, Goldberg SR (1999). Cardiovascular responses to cocaine self-administration: acute and chronic tolerance. Eur J Pharmacol 383: 57–68.
Tidjane Corera A, Do-Rego JC, Costentin J, Bonnet JJ (2001). Differential sensitivity to NaCl for inhibitors and substrates that recognize mutually exclusive binding sites on the neuronal transporter of dopamine in rat striatal membranes. Neurosci Res 39: 319–325.
Turetta L, Donella-Deana A, Folda A, Bulato C, Deana R (2004). Characterisation of the serotonin efflux induced by cytosolic Ca2+ and Na+ concentration increase in human platelets. Cell Physiol Biochem 14: 377–386.
Vaugeois JM, Bonnet JJ, Duterte-Boucher D, Costentin J (1993). In vivo occupancy of the striatal dopamine uptake complex by various inhibitors does not predict their effects on locomotion. Eur J Pharmacol 230: 195–201.
Watterson LR, Kufahl PR, Nemirovsky NE, Sewalia K, Grabenauer M, Thomas BF et al (2012). Potent rewarding and reinforcing effects of the synthetic cathinone 3,4-methylenedioxypyrovalerone (MDPV). Addict Biol (in press).
Wee S, Anderson KG, Baumann MH, Rothman RB, Blough BE, Woolverton WL (2005). Relationship between the serotonergic activity and reinforcing effects of a series of amphetamine analogs. J Pharmacol Exp Ther 313: 848–854.
Willuhn I, Wanat MJ, Clark JJ, Phillips PE (2010). Dopamine signaling in the nucleus accumbens of animals self-administering drugs of abuse. Curr Top Behav Neurosci 3: 29–71.
Zolkowska D, Jain R, Rothman RB, Partilla JS, Roth BL, Setola V et al (2009). Evidence for the involvement of dopamine transporters in behavioral stimulant effects of modafinil. J Pharmacol Exp Ther 329: 738–746.
Acknowledgements
We thank Mario A. Ayestas Jr. for helpful technical assistance. This research was generously supported by the Intramural Research Program of the National Institute on Drug Abuse, National Institutes of Health, USA, and by the Austrian Science Fund/FWF, SFB3506. The views expressed herein are those of the authors and do not necessarily represent the views of the Drug Enforcement Administration, the United States Department of Justice or an officer or entity of the United States.
Author contributions
MHB designed and supervised the project, analyzed data, and wrote the first draft of the manuscript. NVC and SDB synthesized and characterized the identity and purity of MDPV, mephedrone, and methylone. JSP, MHB, and RBR designed, carried out, and analyzed the uptake and release experiments. MH and HHS designed and performed efflux experiments in HEK cells. AFH and CRL designed, carried out, and analyzed the cyclic voltammetry studies. KRL and MHB designed and performed microdialysis experiments. EFT, CWS, and SRG designed, carried out, and analyzed the locomotor activity studies. EFT, SRT, and CWS designed, performed, and analyzed the telemetry experiments. All authors contributed significantly to the writing of the final version of the article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Baumann, M., Partilla, J., Lehner, K. et al. Powerful Cocaine-Like Actions of 3,4-Methylenedioxypyrovalerone (MDPV), a Principal Constituent of Psychoactive ‘Bath Salts’ Products. Neuropsychopharmacol 38, 552–562 (2013). https://doi.org/10.1038/npp.2012.204
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.204
Keywords
This article is cited by
-
Cocaine hydrochloride, cocaine methiodide and methylenedioxypyrovalerone (MDPV) cause distinct alterations in the structure and composition of the gut microbiota
Scientific Reports (2023)
-
Interactions of neuroimmune signaling and glutamate plasticity in addiction
Journal of Neuroinflammation (2021)
-
MDPV “high-responder” rats also self-administer more oxycodone than their “low-responder” counterparts under a fixed ratio schedule of reinforcement
Psychopharmacology (2021)
-
MDPV self-administration in female rats: influence of reinforcement history
Psychopharmacology (2021)
-
An updated review on synthetic cathinones
Archives of Toxicology (2021)
