
Abstract
Drug addiction is a chronically relapsing disorder that has been characterized by (1) compulsion to seek and take the drug, (2) loss of control in limiting intake, and (3) emergence of a negative emotional state (eg, dysphoria, anxiety, irritability) reflecting a motivational withdrawal syndrome when access to the drug is prevented. Drug addiction has been conceptualized as a disorder that involves elements of both impulsivity and compulsivity that yield a composite addiction cycle composed of three stages: ‘binge/intoxication’, ‘withdrawal/negative affect’, and ‘preoccupation/anticipation’ (craving). Animal and human imaging studies have revealed discrete circuits that mediate the three stages of the addiction cycle with key elements of the ventral tegmental area and ventral striatum as a focal point for the binge/intoxication stage, a key role for the extended amygdala in the withdrawal/negative affect stage, and a key role in the preoccupation/anticipation stage for a widely distributed network involving the orbitofrontal cortex–dorsal striatum, prefrontal cortex, basolateral amygdala, hippocampus, and insula involved in craving and the cingulate gyrus, dorsolateral prefrontal, and inferior frontal cortices in disrupted inhibitory control. The transition to addiction involves neuroplasticity in all of these structures that may begin with changes in the mesolimbic dopamine system and a cascade of neuroadaptations from the ventral striatum to dorsal striatum and orbitofrontal cortex and eventually dysregulation of the prefrontal cortex, cingulate gyrus, and extended amygdala. The delineation of the neurocircuitry of the evolving stages of the addiction syndrome forms a heuristic basis for the search for the molecular, genetic, and neuropharmacological neuroadaptations that are key to vulnerability for developing and maintaining addiction.
Similar content being viewed by others
CONCEPTUAL FRAMEWORK
Addiction Definitions: Drug Use, Abuse, and Dependence Addiction Cycle
Drug addiction is a chronically relapsing disorder that has been characterized by (1) compulsion to seek and take the drug, (2) loss of control in limiting intake, and (3) emergence of a negative emotional state (eg, dysphoria, anxiety, irritability) reflecting a motivational withdrawal syndrome when access to the drug is prevented (defined as Substance Dependence by the Diagnostic and Statistical Manual of Mental Disorders [DSM] of the American Psychiatric Association; Koob and Le Moal, 1997; Table 1). The occasional but limited use of an abusable drug is clinically distinct from escalated drug use, loss of control over limiting drug intake, and the emergence of chronic compulsive drug-seeking that characterizes addiction. The critical nature of the distinction between drug use, abuse, and dependence has been illuminated by data showing that approximately 15.6% (29 million) of the US adult population will go on to engage in nonmedical or illicit drug use at some time in their lives, with approximately 2.9% (5.4 million) going on to substance dependence on illicit drugs (Grant and Dawson, 1998; Grant et al, 2004). For alcohol, 51% (120 million) of people over the age of 12 were current users, and of these current users, 7.7% (18 million) met the criteria for Substance Abuse or Dependence on Alcohol. For nicotine, in 2007, approximately 28.6% (70.9 million) Americans aged 12 or older were current (past month) users of a tobacco product, and of these current users, 24.2% (60.1 million) were current cigarette smokers; 5.4% (13.3 million) smoked cigars; 3.2% (8.1 million) used smokeless tobacco; and 0.8% (2.0 million) smoked tobacco in pipes (Substance Abuse and Mental Health Services Administration, 2008).
Although much of the initial study of the neurobiology of drug addiction focused on the acute impact of drugs of abuse (analogous to comparing no drug use to drug use), the focus now is shifting to chronic administration and the acute and long-term neuroadaptive changes in the brain that result in relapse. The purpose of current neurobiological drug abuse research is to understand the genetic/epigenetic, cellular, and molecular mechanisms that mediate the transition from occasional, controlled drug use to the loss of behavioral control over drug-seeking and drug-taking and to chronic relapse even after protracted abstinence which is a hallmark of addiction.
A psychiatric-motivational framework that provides sources of both positive and negative reinforcement for drug-taking is the conceptualization that drug addiction has aspects of both impulse control disorders and compulsive disorders (Table 1). Impulse control disorders are characterized by an increasing sense of tension or arousal before committing an impulsive act and pleasure, gratification, or relief at the time of committing the act. Impulse control disorders are largely associated with positive reinforcement mechanisms (American Psychiatric Association, 1994). In contrast, compulsive disorders are characterized by anxiety and stress before committing a compulsive repetitive behavior and relief from the stress by performing the compulsive behavior. Compulsive disorders are largely associated with negative reinforcement mechanisms and automaticity.
Collapsing the cycles of impulsivity and compulsivity yields a composite addiction cycle composed of three stages—binge/intoxication, withdrawal/negative affect, preoccupation/anticipation—in which impulsivity often dominates at the early stages and impulsivity combined with compulsivity dominates at the later stages. As an individual moves from impulsivity to compulsivity, a shift occurs from positive reinforcement driving the motivated behavior to negative reinforcement and automaticity driving the motivated behavior (Koob, 2004; Table 1). These three stages are conceptualized as interacting with each other, becoming more intense, and ultimately leading to the pathological state known as addiction (Koob and Le Moal, 1997; Table 2). The transition from occasional drug use to addiction involves neuroplasticity in all of these elements and may begin with initial drug use in vulnerable individuals or individuals at particularly vulnerable developmental periods (eg, adolescence; Koob et al, 2008b). The present review focuses on the brain neurocircuitry that is engaged at each stage of the addiction cycle, how it changes with increasing engagement with drugs of abuse, and how it interacts to produce the pathological state known as addiction.
Sources of Reinforcement: Motivation, Opponent Process, Incentive Salience
Changes in the motivation for drugs and natural rewards are a key component of addiction (Table 1). Early work by Wikler (1952) stressed the function of changes in drive states associated with dependence (herein referred to as addiction. Subjects described withdrawal changes as a ‘hunger’ or primary need and the effects of morphine on such a state as ‘satiation’ or gratification of the primary need (Wikler, 1952). Although Wikler argued that positive reinforcement was retained even in heavily dependent subjects (eg, thrill of the intravenous opioid injection), addiction produced a new source of gratification, that of negative reinforcement (Table 1).
The concept of motivation was linked inextricably with hedonic, affective, or emotional states in the transition to addiction by Solomon's opponent process theory of motivation. Solomon and Corbit (1974) postulated that hedonic, affective, or emotional states, once initiated, are automatically modulated by the central nervous system with mechanisms that reduce the intensity of hedonic feelings. Positive hedonic responses in drug use occur shortly after presentation of a stimulus, correlate closely with the intensity, quality, and duration of the reinforcer, and show tolerance and affective or hedonic withdrawal (abstinence). In contrast, negative hedonic responses follow the positive hedonic responses, are sluggish in onset, slow to build up to an asymptote, slow to decay, and get larger with repeated exposure. The role of opponent processes begins early in drug-taking, reflects changes in the brain reward and stress systems, and later forms one of the major motivations for compulsivity in drug-taking in the form of a motivational withdrawal syndrome.
In this formulation, manifestation of a withdrawal syndrome after removal of chronic drug administration, either acute or protracted, is defined in terms of motivational aspects of dependence such as the emergence of a negative emotional state (eg, dysphoria, anxiety, irritability) when access to the drug is prevented (Koob and Le Moal, 2001), rather than on the physical signs of dependence, which tend to be of short duration. Indeed, some have argued that the development of such a negative affective state can define dependence relative to addiction (Russell, 1976; Baker et al, 1987) and that such a negative affective state contributes to compulsivity through negative reinforcement mechanisms (Koob and Le Moal, 2005).
Another conceptualization of the motivational changes associated with addiction is derived from early work on conditioned reinforcement, incentive motivation, behavioral sensitization, and maladaptive stimulus–response learning, all of which are subsumed under the motivational conceptualization of incentive salience. Drugs are hypothesized to usurp systems in the brain that are put in place to direct animals to stimuli with salience for preservation of the species. The incentive salience hypothesis has significant heuristic value as a common element of drug addiction because it narrows the focus to drug-seeking at the expense of natural rewards. The clinical observation that individuals with substance use disorders have an unusual focus on drug-seeking to the exclusion of natural rewards fits the incentive salience view.
The increase in incentive salience produced by psychostimulant drugs has early roots in the facilitation of conditioned reinforcement and drug-seeking (Robbins, 1976; Hill, 1970). Here, drug-seeking is controlled by a succession of drug-associated discriminative stimuli that can also function as conditioned reinforcers when presented as a consequence of instrumental responses (Everitt et al, 2008). Many have argued that by means of associative learning, the enhanced incentive salience state becomes oriented specifically toward drug-related stimuli, leading to escalating compulsion for seeking and taking drugs (Hyman et al, 2006; Kalivas and Volkow, 2005). The underlying activation of neural structures involved in maintaining the incentive salience state persists, making addicts vulnerable to long-term relapse.
Another view of incentive salience involved behavioral sensitization, usually measured as increased locomotor responses to repeated administration of a drug. The behavioral sensitization paradigm has provided a major impetus to exploring not only the neurocircuitry of addiction but also a model of the neuroplasticity that may occur during the transition from drug use to addiction. Here, a shift in an incentive salience state, described as ‘wanting’ linked to compulsive use, as opposed to ‘liking’ linked to hedonic responses, was hypothesized to be progressively increased by repeated exposure to drugs of abuse (Robinson and Berridge, 1993).
Transition to Addiction: Patterns of Drug-Taking, Animal Models
Different drugs produce different patterns of neuroadaptations with chronic drug exposures. For example, opioid-addicted subjects meet most of the DSM criteria for addiction, including dramatic tolerance and withdrawal (classic symptoms associated with physical dependence) and most of the symptoms associated with motivational withdrawal. A pattern of intravenous or smoked drug-taking evolves, including intoxication, tolerance, escalation in intake, and profound dysphoria, physical discomfort, and somatic withdrawal signs during abstinence. Intense preoccupation with obtaining opioids (craving) develops that often precedes the somatic signs of withdrawal and is linked not only to stimuli associated with obtaining the drug but also to stimuli associated with withdrawal and the aversive motivational state. A pattern develops in which the drug must be obtained to avoid the severe dysphoria and discomfort of abstinence. Other drugs of abuse follow a similar pattern but may involve more the binge/intoxication stage (psychostimulants) or less binge/intoxication and more withdrawal/negative affect and preoccupation/anticipation stages (nicotine and cannabinoids).
Much of the recent progress in understanding the neurobiology of addiction has derived from the study of animal models of addiction to specific drugs such as stimulants, opioids, alcohol, nicotine, and Δ9-tetrahydrocannabinol (Δ9-THC). Although no animal model of addiction fully emulates the human condition, animal models do permit investigation of specific elements of the process of drug addiction. Such elements can be defined by models of different stages of the addiction cycle (see above; Table 2).
A progressive increase in the frequency and intensity of drug use is one of the major behavioral phenomena characterizing the development of addiction and has face validity with the DSM criteria: ‘The substance is often taken in larger amounts and over a longer period than was intended’ (American Psychiatric Association, 1994). Two animal models, one involving experimenter-administered drug, and the other involving self-administered drug, have been used to explore the effects of repeated drug administration on neuroplasticity in the neurocircuits identified above. Behavioral sensitization typically involved repeated administration by the experimenter of a drug, usually a psychostimulant, in a specific environmental context and the dependent measure was usually locomotor activity. Here, animals that received drug showed a much more dramatic increase in locomotor activity to a challenge dose of drug (sensitization) than controls that had received only repeated measures of vehicle injections.
A framework, perhaps with more face validity with which to model the transition from drug use to drug addiction, can be found in animal models of prolonged access to self- administration of drugs. Here, using intravenous drug self-administration, extended access to drugs is associated with an escalation in intake over days (Koob, 2009a). Such increased self-administration also has been observed with alcohol in which rats drink excessively during acute and protracted withdrawal from dependence induction using either chronic liquid diet or chronic vapor exposure (Gilpin and Koob, 2008). Animals made dependent on alcohol reliably obtain blood alcohol levels in the 100–150 mg% range, which are equivalent to the levels abused by moderate to heavy alcohol abusers. Changes in the reinforcing and incentive effects of the drug have been observed following extended access and induction of dependence and include increased progressive-ratio responding (Koob, 2009a), increased drug-induced reinstatement after extinction, decreased latency to goal time in a runway model for drug reward (Deroche-Gamonet et al, 2004), and increased resistance to punishment in which the animal will sustain higher aversive punishment to obtain drug (Vanderschuren and Everitt, 2004). Whether the enhanced drug-taking with extended access reflects a sensitization of reward (or of incentive motivation) or a reward-deficit state, or both, remains under discussion (Vezina, 2004).
NEUROCIRCUITRY OF ADDICTION: NEUROPSYCHOPHARMACOLOGICAL EVIDENCE FROM ANIMAL STUDIES
Binge/Intoxication Stage
Our understanding of the neurobiological substrates for the reinforcing effects of drugs of abuse can be traced to early work on the identification of a reward system in the brain with the discovery of electrical brain stimulation reward or intracranial self-stimulation by Olds and Milner (1954). Brain stimulation reward involves widespread neurocircuitry in the brain, but the most sensitive sites defined by the lowest thresholds involve the trajectory of the medial forebrain bundle that connects the ventral tegmental area (VTA) to the basal forebrain (Olds and Milner, 1954). All drugs of abuse, when administered acutely, decrease brain stimulation reward thresholds (ie, increased reward; Kornetsky and Esposito, 1979) and when administered chronically increase reward thresholds during withdrawal (ie, decreased reward; see below). Although much emphasis was focused initially on the role of the ascending monoamine systems in the medial forebrain bundle in reward, first norepinephrine (Stein, 1962) and then dopamine (Crow, 1973; Wise, 1978), other nondopaminergic systems in the medial forebrain bundle clearly have a key role in mediating brain stimulation reward (Hernandez et al, 2006). Indeed, much work suggests that activation of the midbrain dopamine system has multiple roles to give incentive salience to stimuli in the environment (Robinson and Berridge, 1993) to promote performance of goal-directed behavior (Salamone et al, 2007) or activation in general (Le Moal and Simon, 1991). More recently, the hypothesis has been raised that the time course of dopamine signaling is a key factor, with the fastest time course predominantly having a preferential role in reward and valuation of predicted outcomes of behavior and steady activation of dopamine release having a preferential role in providing an enabling effect on specific behavior-related systems (Schultz, 2007). Work in the domain of the acute reinforcing effects of drugs of abuse supports this hypothesis in which the mesolimbic dopamine system is critical for the acute rewarding effects of psychostimulant drugs but has a more enabling function for all drugs of abuse.
The acute rewarding properties of psychostimulant drugs have long been known to depend on activation of the mesolimbic dopamine system, but activation of this system is not necessarily critical for the acute reinforcing effects of other drugs of abuse (Koob, 1992; Nestler, 2005; Hnasko et al, 2005). Neurotoxin-selective lesions of the mesocorticolimbic dopamine system block the reinforcing effects of cocaine and D-amphetamine (McGregor and Roberts, 1993). In contrast, neurochemically specific lesions of dopamine in the nucleus accumbens with 6-hydroxydopamine failed to block heroin or ethanol self-administration, supporting this hypothesis (Koob and Le Moal, 2006).
Using the technique of intracranial self-administration (Table 1) and intracranial place conditioning (Table 1), opioids and alcohol have been shown to be directly self-administered into the VTA. Opioids also produce conditioned place preference when injected into the VTA. Opioids, phencyclidine, and psychostimulants are directly self-administered into the nucleus accumbens, and psychostimulants produce a conditioned place preference when injected into the nucleus accumbens. Cocaine and phencyclidine are directly self-administered into the frontal cortex (McBride et al, 1999). The mesolimbic dopamine system is activated by acute administration of opioids, ethanol, nicotine, and Δ9-THC (Di Chiara and Imperato, 1988).
Intravenous nicotine self-administration is blocked by neurotoxin-specific lesions of the mesocorticolimbic dopamine system, and the neuropharmacological action has been hypothesized to be through nicotinic receptor activation of release of dopamine primarily in the VTA and also presynaptically in the nucleus accumbens (Watkins et al, 2000). However, nicotine reward measured by conditioned place preference appears to be independent of the mesocorticolimbic dopamine system (Laviolette et al, 2002). Other substrates implicated in nicotine reward include cholinergic inputs to the pedunculopontine nucleus (Yeomans and Baptista, 1997). In the VTA, activation of the β2 subunit of nicotinic receptors appears to be critical for nicotine activation of dopamine neurons (Mameli-Engvall et al, 2006). Neuropharmacological studies on cannabinoids have implicated both cannabinoid and opioid mechanisms. Opioid and cannabinoid CB1 antagonists block intravenous self-administration of Δ9-THC in squirrel monkeys (Justinova et al, 2003). Similar to other drugs of abuse, Δ9-THC administration activates dopamine release in the nucleus accumbens shell (Tanda et al, 1997).
Thus, all drugs of abuse activate the mesolimbic dopamine system, but much evidence suggests that dopamine-independent reinforcement occurs at the level of the nucleus accumbens, suggesting multiple inputs to the activation of critical reinforcement circuitry in these brain regions (Koob, 1992; Nestler, 2005).
The central nucleus of the amygdala (CeA) also has a key function in the acute reinforcing actions of drugs of abuse. Microinjections of dopamine D1 receptor antagonists into the CeA block cocaine self-administration (Caine et al, 1995; McGregor and Roberts, 1993). The most sensitive site for γ-aminobutyric acid (GABA) and opioid antagonism of oral alcohol self-administration in nondependent rats was the CeA (Hyytia and Koob, 1995; Heyser et al, 1999). Lesions of the CeA block oral self-administration of alcohol (Moller et al, 1997). Serotonin-3 antagonists injected into the CeA block oral ethanol self-administration in nondependent rats, an effect hypothesized to possibly involve the ability of serotonin-3 receptor antagonists to block drug-induced dopamine release (Dyr and Kostowski, 1995).
A major output from the nucleus accumbens is to the ventral pallidum/substantia innominata. Consistent with the nucleus accumbens as a key substrate for drug reward, lesions of the ventral pallidum are particularly effective in blocking the motivation to work for intravenous cocaine and intravenous heroin (Hubner and Koob, 1990; Robledo and Koob, 1993). In addition, blockade of dopamine and GABAA receptors in the ventral pallidum blocks the reinforcing effects of alcohol (Melendez et al, 2004; June et al, 2003). Thus, elements of the ventral pallidum may not only be critical for further processing of the drug reward signal but may also be directly modulated by drugs of abuse.
The dorsal striatum does not appear to have a major role in the acute reinforcing effects of drugs abuse but appears to be recruited during the development of compulsive drug-seeking (Everitt et al, 2008). 6-Hydroxydopamine lesions of the dorsal striatum do not block cocaine-induced locomotor activity or cocaine self-administration (Roberts, 1992) but do block amphetamine-induced stereotyped behavior (Kelly and Iversen, 1976; Creese and Iversen, 1974). Using a second-order schedule (Table 1), lesions of the nucleus accumbens and basolateral amygdala blocked the acquisition of cocaine-seeking (Whitelaw et al, 1996). Similarly, when the nucleus accumbens core was selectively lesioned on one side of the brain and combined with dopamine receptor blockade in the contralateral dorsal striatum, no effect was observed in animals immediately after acquisition, but greatly decreased cocaine-seeking was observed in rats with stable responding on a second-order schedule (Belin and Everitt, 2008). These results suggest that the dorsal striatum may have a minor role in the acute reinforcing effects of psychostimulant drugs but a key role in the transition to compulsive use (Everitt et al, 2008).
Data with knockout mice also provide key insights into the role of dopamine in the rewarding effects of drugs of abuse. Genetically altered mice homozygous with a lack of the dopamine D1 receptor do not self-administer cocaine (Caine et al, 2007). Although the initial report that dopamine transporter (DAT) knockout mice continued to self-administer cocaine (Rocha et al, 1998) questioned the function of the DAT in cocaine's reinforcing effects, a recent study showed that transgenic animals that expressed DAT that did not bind cocaine but that was functional as a dopamine reuptake carrier did not show cocaine reward measured by conditioned place preference (Chen et al, 2006a). These results support the hypothesis of a crucial role of the DAT in cocaine's reinforcing effects.
On the basis of this synthesis, an early neurobiological circuit for drug reward was proposed (Koob, 1992) that has been elaborated and expanded (Koob and Nestler, 1997; Figure 1). The starting point for the reward circuit was the medial forebrain bundle, composed of myelinated fibers connecting bidirectionally the olfactory tubercle and nucleus accumbens with the hypothalamus and VTA (Nauta and Haymaker, 1969) and including the ascending monoamine pathways such as the mesocorticolimbic dopamine system.
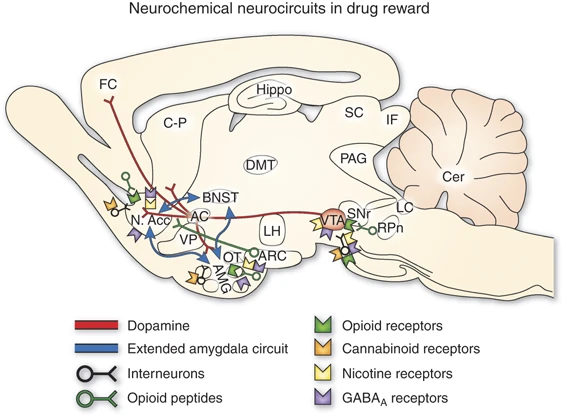
Sagittal section through a representative rodent brain illustrating the pathways and receptor systems implicated in the acute reinforcing actions of drugs of abuse. Cocaine and amphetamines activate the release of dopamine in the nucleus accumbens and amygdala through direct actions on dopamine terminals. Opioids activate opioid receptors in the VTA, nucleus accumbens, and amygdala through direct or indirect actions via interneurons. Opioids facilitate the release of dopamine in the nucleus accumbens by an action either in the VTA or the nucleus accumbens, but also are hypothesized to activate elements independent of the dopamine system. Alcohol activates γ-aminobutyric acid-A (GABAA) receptors or GABA release in the VTA, nucleus accumbens, and amygdala by either direct actions at the GABAA receptor or through indirect release of GABA. Alcohol is hypothesized to facilitate the release of opioid peptides in the VTA, nucleus accumbens, and central nucleus of the amygdala. Alcohol facilitates the release of dopamine in the nucleus accumbens through an action either in the VTA or the nucleus accumbens. Nicotine activates nicotinic acetylcholine receptors in the VTA, nucleus accumbens, and amygdala, either directly or indirectly, through actions on interneurons. Cannabinoids activate cannabinoid CB1 receptors in the VTA, nucleus accumbens, and amygdala. Cannabinoids facilitate the release of dopamine in the nucleus accumbens through an unknown mechanism either in the VTA or the nucleus accumbens. The blue arrows represent the interactions within the extended amygdala system hypothesized to have a key function in drug reinforcement. The medial forebrain bundle represents ascending and descending projections between the ventral forebrain (nucleus accumbens, olfactory tubercle, septal area) and the ventral midbrain (VTA) (not shown in figure for clarity). AC, anterior commissure; AMG, amygdala; ARC, arcuate nucleus; BNST, bed nucleus of the stria terminalis; Cer, cerebellum; C-P, caudate-putamen; DMT, dorsomedial thalamus; FC, frontal cortex; Hippo, hippocampus; IF, inferior colliculus; LC, locus coeruleus; LH, lateral hypothalamus; N Acc., nucleus accumbens; OT, olfactory tract; PAG, periaqueductal gray; RPn, reticular pontine nucleus; SC, superior colliculus; SNr, substantia nigra pars reticulata; VP, ventral pallidum; VTA, ventral tegmental area (taken with permission from Koob, 2005).
The initial action of drug reward was hypothesized to depend on dopamine release in the nucleus accumbens for cocaine, amphetamine, and nicotine; opioid peptide receptor activation in the VTA (dopamine activation) and nucleus accumbens (independent of dopamine activation) for opiates; and GABAA systems in the nucleus accumbens and amygdala for alcohol. The nucleus accumbens is situated strategically to receive important limbic information from the amygdala, frontal cortex, and hippocampus that could be converted to motivational action through its connections with the extrapyramidal motor system. Thus, an early critical role for the nucleus accumbens was established for the acute reinforcing effects of drugs, with a supporting role for the CeA and ventral pallidum (Figures 1 and 2a).
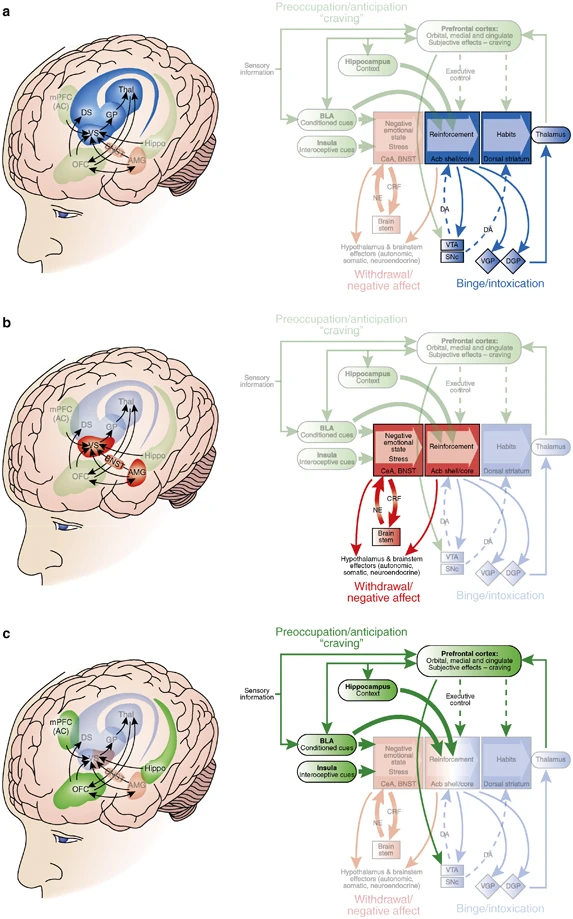
Neural circuitry associated with the three stages of the addiction cycle. (a) Binge/intoxication stage. Reinforcing effects of drugs may engage reward neurotransmitters and associative mechanisms in the nucleus accumbens shell and core and then engage stimulus–response habits that depend on the dorsal striatum. Two major neurotransmitters mediating the rewarding effects of drugs of abuse are dopamine and opioid peptides. (b) Withdrawal/negative affect stage. The negative emotional state of withdrawal may engage the activation of the extended amygdala. The extended amygdala is composed of several basal forebrain structures, including the bed nucleus of the stria terminalis, central nucleus of the amygdala, and possibly a transition zone in the medial portion (or shell) of the nucleus accumbens. Major neurotransmitters in the extended amygdala hypothesized to have a function in negative reinforcement are corticotropin-releasing factor, norepinephrine, and dynorphin. Major projections of the extended amygdala are to the hypothalamus and brainstem. (c) Preoccupation/anticipation (craving) stage. This stage involves the processing of conditioned reinforcement in the BLA and the processing of contextual information by the hippocampus. Executive control depends on the prefrontal cortex and includes representation of contingencies, representation of outcomes, and their value and subjective states (ie, craving and, presumably, feelings) associated with drugs. The subjective effects termed drug craving in humans involve activation in functional imaging studies of the orbital and anterior cingulate cortices and temporal lobe, including the amygdala. A major neurotransmitter involved in the craving stage is glutamate localized in pathways from frontal regions and the BLA that project to the ventral striatum. Green/blue arrows, glutamatergic projections; orange arrows, dopaminergic projections; pink arrows, GABAergic projections; Acb, nucleus accumbens; BLA, basolateral amygdala; VTA, ventral tegmental area; SNc, substantia nigra pars compacta; VGP, ventral globus pallidus; DGP, dorsal globus pallidus; BNST, bed nucleus of the stria terminalis; CeA, central nucleus of the amygdala; NE, norepinephrine; CRF, corticotropin-releasing factor; PIT, Pavlovian instrumental transfer (modified with permission from Koob et al, 2008a).
Withdrawal/Negative Affect Stage
The neuroanatomical entity termed the extended amygdala (Heimer and Alheid, 1991) may represent a common anatomical substrate integrating brain arousal–stress systems with hedonic processing systems to produce the negative emotional states that promote negative reinforcement mechanisms associated with the development of addiction. The extended amygdala is composed of the CeA, bed nucleus of the stria terminalis (BNST), and a transition zone in the medial (shell) subregion of the nucleus accumbens (Figure 2b). Each of these regions has cytoarchitectural and circuitry similarities (Heimer and Alheid, 1991). The extended amygdala receives numerous afferents from limbic structures such as the basolateral amygdala and hippocampus and sends efferents to the medial part of the ventral pallidum and a large projection to the lateral hypothalamus, thus further defining the specific brain areas that interface classical limbic (emotional) structures with the output of extrapyramidal motor system (Alheid et al, 1995). The extended amygdala has long been hypothesized to have a key role not only in fear conditioning (Le Doux, 2000) but also in the emotional component of pain processing (Neugebauer et al, 2004).
Within-system neuroadaptations to chronic drug exposure include decreases in function of the neurotransmitter systems in the neurocircuits implicated in the acute reinforcing effects of drug of abuse. One prominent hypothesis is that dopamine systems are compromised in crucial phases of the addiction cycle, such as withdrawal, and lead to decreased motivation for nondrug-related stimuli and increased sensitivity to the abused drug (Melis et al, 2005; see brain imaging studies below). Psychostimulant withdrawal in humans is associated with fatigue, decreased mood and psychomotor retardation, and in animals is associated with decreased motivation to work for natural rewards (Barr and Phillips, 1999) and decreased locomotor activity (Pulvirenti and Koob, 1993), behavioral effects that may involve decreased dopaminergic function. Animals during amphetamine withdrawal show decreased responding on a progressive-ratio schedule for a sweet solution, and this decreased responding was reversed by the dopamine partial agonist terguride (Orsini et al, 2001), suggesting that low dopamine tone contributes to the motivational deficits associated with psychostimulant withdrawal. Decreases in activity of the mesolimbic dopamine system and decreases in serotonergic neurotransmission in the nucleus accumbens occur during acute drug withdrawal from all major drugs of abuse in animal studies (Rossetti et al, 1992; Weiss et al, 1992, 1996).
A second component of the withdrawal/negative affect stage is a between-system neuroadaptation in which different neurochemical systems involved in stress modulation also may be engaged within the neurocircuitry of the brain stress and aversive systems in an attempt to overcome the chronic presence of the perturbing drug to restore normal function despite the presence of drug. Both the hypothalamic–pituitary–adrenal axis and the brain stress/aversive system mediated by corticotropin-releasing factor (CRF) are activated during withdrawal from chronic administration of all major drugs with abuse potential, with a common response of elevated adrenocorticotropic hormone, corticosterone, and amygdala CRF during acute withdrawal (Koob, 2008; Koob and Kreek, 2007). Acute withdrawal from all drugs of abuse also produces an aversive or anxiety-like state in which CRF and other stress-related systems (including noradrenergic pathways) have key roles.
The aversive stimulus effects of drug withdrawal can be measured using place aversion (Hand et al, 1988), and the opioid partial agonist buprenorphine dose dependently decreased the place aversion produced by precipitated opioid withdrawal. Systemic administration of a CRF1 receptor antagonist and direct intracerebral administration of a peptide CRF1/CRF2 antagonist also decreased opioid withdrawal-induced place aversions (Stinus et al, 2005; Heinrichs et al, 1995). Functional noradrenergic antagonists administered directly into the BNST blocked opioid withdrawal-induced place aversion, implicating the importance of noradrenergic stimulation in the stress responses that follow acute drug withdrawal (Delfs et al, 2000). Indeed, classical medications used to treat physical withdrawal in heroin abusers and alcoholics include α-adrenergic drugs (eg, clonidine) that inhibit noradrenergic release and decrease some symptoms of alcohol and heroin withdrawal.
Another candidate for the aversive effects of drug withdrawal is dynorphin. Much evidence shows that dynorphin is increased in the nucleus accumbens in response to dopaminergic activation and, in turn, that overactivity of the dynorphin systems can decrease dopaminergic function. κ-Opioid agonists are aversive, and cocaine, opioid, and ethanol withdrawal is associated with increased dynorphin in the nucleus accumbens and/or amygdala (Koob, 2008). An exception is salvidorin A, which is a κ-agonist abused by humans, but this may reflect its hallucinogenic effects rather than any pleasurable properties (Gonzalez et al, 2006).
Another common between-system response to acute withdrawal and protracted abstinence from all major drugs of abuse is the manifestation of anxiety-like responses. For example, withdrawal from repeated administration of cocaine produces an anxiogenic-like response in the elevated plus maze and defensive burying test, both of which are reversed by CRF antagonists. Similarly, ethanol withdrawal produces anxiety-like behavior that is reversed by intracerebroventricular administration of CRF1/CRF2 peptidergic antagonists, systemic administration of a small molecule CRF1 antagonist, and microinjection of a peptidergic CRF1/CRF2 antagonist into the amygdala (Funk et al, 2006; Koob, 2008). CRF antagonists injected intracerebroventricularly or systemically also block the potentiated anxiety-like responses to stressors observed during protracted abstinence from chronic ethanol, and the effects of CRF antagonists have been localized to the CeA (Koob, 2008). Precipitated withdrawal from nicotine produces anxiety-like responses that are also reversed by CRF antagonists (Tucci et al, 2003; George et al, 2007).
Thus, acute withdrawal is associated with within-system changes reflected in a decrease in dopaminergic activity in the mesolimbic dopamine system and with between-system recruitment of neurotransmitter systems that convey stress and anxiety-like effects such as CRF and dynorphin. Other neurotransmitter systems known to be involved in emotional dysregulation of the motivational effects of drug withdrawal include norepinephrine, substance P, vasopressin, neuropeptide Y (NPY), endocannabinoids, and nociceptin (Koob, 2008).
Preoccupation/Anticipation (Craving) Stage
The preoccupation/anticipation or craving stage of the addiction cycle has long been hypothesized to be a key element of relapse in humans and defines addiction as a chronic relapsing disorder. Although often linked to the construct of craving, craving per se has been difficult to measure clinically (Tiffany et al, 2000) and often does not correlate well with relapse. Nevertheless, the stage of the addiction cycle in which the individual reinstates drug-seeking behavior after abstinence remains a challenging focus for neurobiological mechanisms and medications development for treatment. Animal models of craving can be divided into two domains: drug-seeking induced by drug or stimuli paired with drug-taking, and drug-seeking induced by an acute stressor or a residual negative emotional state, often a state of stress, termed protracted abstinence (see Transition to addiction: patterns of drug-taking, animal models section).
Much evidence from animal studies suggests that drug-induced reinstatement is localized to the medial prefrontal cortex/nucleus accumbens/ventral pallidum circuit mediated by the neurotransmitter glutamate (McFarland and Kalivas, 2001). In contrast, cue-induced reinstatement appears to involve the basolateral amygdala as a critical substrate with a possible feed-forward mechanism through the prefrontal cortex system involved in drug-induced reinstatement (Everitt and Wolf, 2002; Weiss et al, 2001). The association of previously neutral stimuli paired with precipitated opioid withdrawal (conditioned withdrawal) also depends critically on the basolateral amygdala (Schulteis et al, 2000), and such stimuli may have motivational significance (Kenny et al, 2006). Neurocircuitry changes associated with drug- and cue-induced reinstatement after extinction have been linked to a glutamatergic pathway from the prefrontal cortex to the nucleus accumbens core, the dopamine projection from the VTA to the medial prefrontal cortex, and the GABA projection from the nucleus accumbens to the ventral pallidum (Kalivas and O’Brien, 2008).
In contrast, stress-induced reinstatement of drug-related responding in animal models appears to depend on the activation of both CRF and norepinephrine in elements of the extended amygdala (both the CeA and BNST; for reviews, see Shaham et al, 2003; Shalev et al, 2002). Protracted abstinence, largely described in alcohol dependence models, appears to involve overactive glutamatergic and CRF systems, presumably in the extended amygdala, although to a large extent this remains to be explored (de Witte et al, 2005; Valdez et al, 2002).
Human subjects with cocaine addiction show impaired performance in tasks involving attention, cognitive flexibility, and delayed reward discounting that are mediated by the medial and orbital prefrontal cortices, as well as spatial, verbal, and recognition memory impairments that are mediated by the hippocampus, and these deficits can predict poor treatment outcomes (Aharonovich et al, 2006; Bolla et al, 2003). Parallel animal studies of the orbitofrontal, prefrontal cortex, and hippocampus in addiction using animal models have begun to show some of the deficits reflected in human studies. Experimenter-administered cocaine produced impairments in reversal learning (an orbital frontal task) in rats and monkeys (Jentsch et al, 2002; Schoenbaum et al, 2004; Calu et al, 2007). Perhaps even more compelling, animals allowed extended access, but not limited access, to cocaine showed deficits in working memory (a prefrontal-cortex-dependent task), sustained attention task (a prefrontal-cortex-dependent task), and an object recognition task (a hippocampus-dependent task; Briand et al, 2008a, 2008b; George et al, 2008). In one study (Briand et al, 2008a), these deficits were associated with a significant decrease in dopamine D2 receptor mRNA in the medial and orbital prefrontal cortices, an observation also consistent with human imaging studies. Thus, animal studies using models of compulsive stimulant administration are beginning to show deficits associated with human cocaine addiction (see Human studies: imaging and neuropsychopharmacology).
HUMAN STUDIES: IMAGING AND NEUROPSYCHOPHARMACOLOGY
As noted above, evidence from preclinical and clinical studies suggests that addiction represents sequential neuroadaptations. As a result, an initial impulsive action turns compulsive and becomes (eventually) chronic and relapsing. Work from imaging studies has provided evidence that this transition involves reprogramming of neuronal circuits that process (1) reward and motivation; (2) memory, conditioning, and habituation; (3) executive function and inhibitory control; (4) interoception and self-awareness; and (5) stress reactivity. This transition is heavily influenced by genetic, developmental, and environmental factors and their dynamic interactions, which will determine the course and severity of the addiction.
Similar to preclinical investigations, distinguishing the three stages in the recurring course of addiction in humans (intoxication, withdrawal, and craving/relapse) has been useful. The following sections describe these stages and some of the relevant neuronal circuits that underlie them.
Binge/Intoxication Stage
Most cases of addiction are initiated by the abuse of substances that are sought because of their hedonic properties. However, drug experimentation also results from the reinforcing effects of conforming to social groups (peer pressure) with the eventual subsequent transfer of motivation to taking the drug for its reinforcing effects. Infrequently, the first use of a drug may be related to its therapeutic properties (such as opiate analgesics for pain or stimulants for attention-deficit hyperactivity disorder). As shown by preclinical studies, a key element of the reinforcing effects of drugs is broadly accepted to involve their ability to trigger large increases in extracellular dopamine in limbic regions (including the nucleus accumbens). Although acute drug self-administration is a good animal model for drug intoxication, using animal models to assess the subjective correlates of drug-induced dopamine increases is difficult. Brain imaging studies in humans have been instrumental in showing that drug-induced increases in dopamine in the striatum (including the ventral striatum where the nucleus accumbens is located) are associated with subjective descriptors of reward (eg, pleasure, high, euphoria; Volkow et al, 1996b). Moreover, these studies have shown that fast dopamine changes are associated with the subjective perception of reward, whereas slow and stable dopamine increases do not induce these subjective responses (Grace, 2000; Volkow and Swanson, 2003).
The pharmacokinetic properties of drugs, which influence the speed of delivery into the brain as well as the duration of their actions, are key elements of their addiction potential. Pharmacokinetic properties determine the doses, routes of administration, and frequency of drug use within a given binge episode. For example, comparison of the brain pharmacokinetics of cocaine and of methamphetamine reveals that both reach the brain very rapidly (although cocaine is somewhat faster than methamphetamine) but that cocaine clears out of the brain much faster than methamphetamine (Figure 3). This difference helps explain why cocaine is taken every 30–60 min during a binge, whereas methamphetamine is taken every couple of hours (Fowler et al, 2008). The importance of pharmacokinetics also helps explain why most abused drugs (with the exception of alcohol) are injected, smoked, or snorted. These routes allow for a much faster delivery of the drug to the brain than when taken orally (Volkow et al, 2000). Pharmacokinetics also help explain why stimulant drugs such as methylphenidate or amphetamine, which also increase dopamine, are not typically perceived as reinforcing when taken orally as prescribed therapeutically (Chait, 1994; Volkow et al, 2001b).
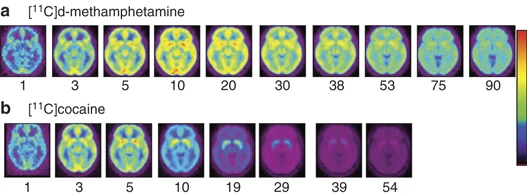
Brain images obtained at different times after administration for [11C]D-methamphetamine and for [11C]cocaine (n=19 for each drug) showing axial planes at a level that transects the basal ganglia. Note the fast uptake of both drugs in the brain and the much slower clearance for [11C]D-methamphetamine than for [11C]cocaine (taken with permission from Fowler et al, 2008).
Clinical studies have also shown that the expectation of the drug's effects significantly influences the rewarding responses to drugs, such that the behavioral as well as regional brain activation response of the brain to the drug tends to be more intense when a rewarding drug is expected compared with when the same drug is received unexpectedly (Volkow et al, 2003). The dependency of the drug's rewarding effects on context and expectation suggests the importance of other neurotransmitters such as glutamate, which modulates the reactivity of dopamine cells and dopamine release in the nucleus accumbens, in the rewarding effects of drugs of abuse (Kalivas and Volkow, 2005).
Withdrawal/Negative Affect Stage
The response that follows the stage of drug intoxication differs markedly across drugs and is influenced by the chronicity and frequency of its abuse. For some drugs such as opiates, alcohol, and sedative hypnotics, drug discontinuation in chronic drug users can trigger an intense, acute physical withdrawal syndrome that, if not properly managed and when severe, can sometimes be fatal. All drugs of abuse are associated with a motivational withdrawal syndrome characterized by dysphoria, irritability, emotional distress, and sleep disturbances that persist even after protracted withdrawal. The neurobiology of acute withdrawal is distinct from protracted or motivational withdrawal, and both contribute to relapse. Few imaging studies have been carried out during acute withdrawal. One such study that measured changes in dopamine during heroin withdrawal failed to document the dopamine decreases in the nucleus accumbens that had previously been reported with microdialysis in the rodent brain (Wang et al, 1997). From this study, it is unclear whether the results reflect the lack of involvement of striatal dopamine during acute withdrawal in heroin abusers or the limited sensitivity of the positron emission tomography (PET) technology.
The mechanisms underlying acute withdrawal are likely to be drug specific and reflect adaptations in the molecular targets of these drugs. For example, during the first few days of cocaine withdrawal, enhanced sensitivity of the brain to the effects of GABA-enhancing drugs occurs that may reflect the downregulation of this neurotransmitter with chronic cocaine use (Volkow et al, 1998). Similarly, brain imaging studies have also revealed decreases in endogenous opioids during cocaine withdrawal, which may contribute to the irritability, malaise, and dysphoria that occur during this phase of motivational withdrawal (Zubieta et al, 1996).
During protracted withdrawal, once the signs and symptoms of acute withdrawal have subsided, imaging studies have documented hypofunction in dopamine pathways, demonstrated by decreases in D2 receptor expression and decreases in dopamine release, which may contribute to the anhedonia (ie, decreased sensitivity to rewarding stimuli) and amotivation reported by drug-addicted subjects during protracted withdrawal (Volkow et al, 1997b, 2007; Martinez et al, 2004, 2005). The decreased reactivity of dopamine to reinforcing stimuli is also present after protracted withdrawal from alcohol when acute physical withdrawal has subsided. In contrast to the decreased sensitivity to rewards (including drug rewards), imaging studies have reported that during detoxification, enhanced sensitivity to conditioned cues also occurs. Abstinence from smoking, for example, can dramatically potentiate neural responses to smoking-related cues (McClernon et al, 2009). These conditioned responses sustain the cycle of abstinence and relapse that characterizes substance use disorders (Childress et al, 1988).
In addition, imaging studies evaluating markers of brain function have shown that drug abusers tested during protracted detoxification show evidence of disrupted activity of frontal regions, including dorsolateral prefrontal regions, cingulate gyrus, and orbitofrontal cortex, which is hypothesized to underlie their impaired inhibitory control and impulsivity and contribute to relapse (see the following section for discussion).
Preoccupation/Anticipation (Craving) Stage
The enhanced sensitivity to conditioned cues, which include emotional states, triggers the latent preoccupation/anticipation (craving) stage, which is characterized by an increase in drug craving. Indeed, stress is a powerful trigger of relapse to drug-taking behaviors through the activation of brain circuits involved in reward processing and in the attentional and mnemonic bias for drug use reminders (Duncan et al, 2007). This chronic relapse phenomenon is broadly recognized as one of the most challenging problems in fighting drug addiction. Addicted subjects are liable to return to compulsive drug-taking long after experiencing acute withdrawal symptoms (Langleben et al, 2008). The gradual reorganization of reward and memory circuits, brought about by chronic drug abuse, is hypothesized to be crucial to the mounting of these responses. Both dopamine and glutamate have been identified in preclinical studies as contributing to the neuroplastic changes associated with conditioned responses. Moreover, plastic changes in CRF and glucocorticoid receptors likely participate in the enhanced sensitivity to stressors. In humans, the lack of suitable radiotracers to assess glutamate neurotransmission and the lack of ligands for CRF or glucocorticoid receptors have limited the studies of craving mostly to the dopamine system.
NEUROCIRCUITRY DYNAMICS IN THE TRANSITION TO ADDICTION
The neurocircuitry outlined above forms the basis for the neuroplasticity associated with the development of addiction. Summarized below are neuroadaptive changes engaged within the circuits that represent the stages of the addiction cycle outlined above. Five circuits are hypothesized to be engaged in succession, including (1) mesolimbic dopamine system, (2) ventral striatum, (3) ventral striatum/dorsal striatum/thalamus circuits, (4) dorsolateral frontal cortex/inferior frontal cortex/hippocampus circuits, and (5) extended amygdala (Figure 4). The relative weighting and direction of these neuroadaptive changes is illustrated in the circuit diagram of the addicted state (Figure 5).
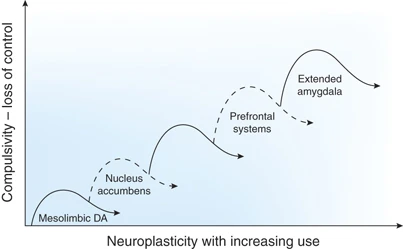
Schematic drawing describing the sequential and cumulative effects of neuroadaptive changes hypothesized to contribute to the neuroplasticity that promotes compulsive drug-seeking. An early neuroadaptation, common to all drugs of abuse and observed after a single injection of cocaine, is increased excitability of the mesolimbic dopamine system reflected in long-term potentiation dependent on changes in glutamate activity. Subsequently, the activation of dopamine contributes to increased excitability of the ventral striatum with decreased glutamatergic activity during withdrawal and increased glutamatergic activity during drug-primed and cue-induced drug-seeking. The engagement of ventral striatal-pallidal-thalamic loops is hypothesized to translate to the dorsal striatum to contribute to engagement of habits and automaticity that resemble compulsive-like behavior. As compulsivity evolves into full-blown addiction, loss of function occurs in the frontal cortex systems that control executive function, contributing to poor decision-making and gain of function in the brain stress systems but contributing to incentive salience for drugs over natural reinforcers.
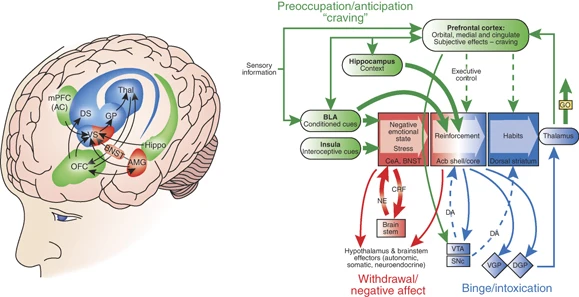
Neurocircuitry schematic illustrating the combination of neuroadaptations in the brain circuitry for the three stages of the addiction cycle that promote drug-seeking behavior in the addicted state. Note the activation of the ventral striatum/dorsal striatum/extended amygdala driven by cues through the hippocampus and basolateral amygdala and stress through the insula. The frontal cortex system is compromised, producing deficits in executive function and contributing to the incentive salience of drugs compared to natural reinforcers. Dopamine systems are compromised, and brain stress systems such as CRF are activated to reset further the salience of drugs and drug-related stimuli in the context of an aversive dysphoric state (modified with permission from Koob et al, 2008a).
Mesolimbic Dopamine System: Incentive Salience Pathways, Salience Attribution
One major hypothesis guiding the neuroplasticity associated with addiction is focused on the mesolimbic dopamine system. The hypothesis is that drugs of abuse, particularly cocaine and amphetamine, increase dopamine release in a more prolonged and unregulated manner than natural stimuli, resulting in changes in synaptic plasticity both within the dopamine system and in dopamine-receptive neurons (Wolf, 2002). These changes ultimately usurp normal learning mechanisms to shift neurocircuitry to associations or a form of habit-learning that persists in the face of significant adverse consequences (a component of compulsivity; Everitt and Wolf, 2002; Hyman et al, 2006).
Animal models of behavioral sensitization have focused largely on the increased locomotor-activating effects of psychomotor stimulant drugs in animals with a history of stimulant exposure. Such studies have revealed a rich neuroplasticity associated with mesolimbic dopamine systems and its terminal projection to the ventral striatum (where the nucleus accumbens is located). Drugs of abuse induce short- and long-term modifications of firing of dopamine neurons in the VTA (Bonci et al, 2003). Studies have shown that burst firing of dopamine neurons in the VTA appears to be correlated with an orienting response to a sensory stimulus (Freeman et al, 1985). A single in vivo exposure to cocaine or amphetamine induces long-term potentiation (LTP) of AMPA-mediated excitatory neurotransmission in dopamine neurons (Ungless et al, 2001). The potentiation of synaptic AMPA responses has been hypothesized to increase the incidence of burst firing (Jones and Bonci, 2005). Persistent LTP lasting for 3 months of abstinence was induced in the VTA in rats that actively self-administered cocaine but not in passively injected rats (Chen et al, 2008). Similar effects of induction of LTP of glutamate transmission on dopamine neurons have been observed with morphine and nicotine (Saal et al, 2003).
However, more chronic repeated administration of psychostimulants failed to produce sensitization of mesolimbic dopamine activity as measured by in vivo microdialysis (Maisonneuve et al, 1995). In addition, extended access to cocaine fails to produce locomotor sensitization (Ben-Shahar et al, 2004) but does produce a sensitized stereotyped behavior response (Ferrario et al, 2005). Moreover, human cocaine abusers showed attenuated dopamine responses when challenged with a stimulant drug, which is opposite to that predicted by the enhanced sensitization of mesolimbic dopamine activity (Volkow et al, 1997b; Martinez et al, 2007).
Ventral Striatum: Incentive Salience Pathways, Salience Attribution
Another plasticity associated with behavioral sensitization is a persistent potentiation of nucleus accumbens excitatory synapses that is observed after repeated drug exposure followed by an extended drug-free period (Kourrich et al, 2007). Repeated cocaine administration increases glutamate neurotransmission only in rats that showed behavioral sensitization (Pierce et al, 1996). In addition, cocaine-sensitized mice showed an enhancement of LTP in nucleus accumbens slices during withdrawal, presumably reflecting increased activity of glutamatergic activity (Yao et al, 2004). An increased surface-to-intracellular ratio of glutamate-1 receptors (GluR1) has been observed 21 days after the last injection of cocaine, suggesting a slowly developing redistribution of AMPA receptors to the surface of nucleus accumbens neurons, particularly in those lacking GluR2 (Boudreau and Wolf, 2005; Conrad et al, 2008). The increases in cell-surface AMPA receptors depends on activation of dopamine D1 receptors and subsequent protein kinase A signaling (Chao et al, 2002). Functionally, overexpression of GluR1 in the nucleus accumbens facilitated extinction of cocaine-seeking responses (Sutton et al, 2003) and increased brain stimulation reward thresholds, reflecting decreased reward and possibly decreased motivated behavior (Todtenkopf et al, 2006). However, a single reexposure to cocaine during extended withdrawal produced synaptic depression, which may reflect the enhanced glutamate release during cocaine reexposure (Kourrich et al, 2007). Curiously, the increase in AMPA receptor expression observed with cocaine does not occur in amphetamine-sensitized rats, leading to the hypothesis of different functional effects of glutamate projections to the nucleus accumbens during cocaine vs amphetamine withdrawal (Nelson et al, 2009).
Consistent with the results of altered glutamate neurotransmission in cocaine-sensitized rats, microdialysis and microinjection studies have shown that following chronic cocaine, decreased basal release of glutamate occurs but sensitized synaptic glutamate release during reinstatement of extinguished drug-seeking in rats (Kalivas and O’Brien, 2008; McFarland et al, 2003). This glutamate dysregulation has been hypothesized to be caused by decreased function of the cystine–glutamate exchanger (Baker et al, 2003) and desensitization of the metabotropic glutamate mGlu2/3 receptor. Lower basal levels of glutamate, combined with increased release of synaptic glutamate from activation of prefrontal cortex afferents to the nucleus accumbens, are hypothesized to result in a drive to engage in drug-seeking (Kalivas, 2004).
These long-lasting synaptic effects produce both a decrease in glutamate neurotransmission during chronic administration of the drug and a persistent increase in the efficacy of glutamatergic synaptic neurotransmission during reinstatement following withdrawal. These dynamic changes may promote cellular excitation, which has been hypothesized to be an important substrate for sensitization and drug-related learning in the addictive state (Kauer and Malenka, 2007; Wolf et al, 2004).
As previously suggested by animal models, the magnitude of striatal dopamine release (particularly in its ventral aspect) in humans correlates positively with the hedonic response to most drugs of abuse, including amphetamine (Drevets et al, 2001), cocaine (Volkow et al, 1997a), methylphenidate (Volkow et al, 2002), and nicotine (Sharma and Brody, 2009). The drug-dependent, fast, and supraphysiological increases in dopamine are likely to mimic the dopamine changes induced by the phasic dopamine cell firing that occurs in response to salient stimuli, thus categorizing the drug experience as one that is highly salient, an experiential outcome that commands attention and promotes arousal, conditioned learning, and motivation (Volkow et al, 2004b). On the basis of findings in laboratory animals, the frequent exposure to these drug responses in drug abusers is postulated to result in the recalibration of dopamine-activating (reward) thresholds for natural reinforcers.
Thus, one can envision the development of a change in firing in mesolimbic dopamine neurons that begins with one administration of the drug, develops into LTP first in the VTA then nucleus accumbens, and via feedback loops subsequently engages the dorsal striatum. Moreover, long-term changes in the CeA and medial prefrontal cortex may follow, and combined with dysregulation of the brain stress systems (see below) may provide a powerful drive for drug-seeking behavior even months after drug withdrawal (Figure 4 and 5).
Ventral Striatum/Dorsal Striatum/Thalamus: Voluntary to Habitual Drug-Seeking
The hypothesis that dorsal striatal circuitry has a key role in the development of habitual compulsive cocaine use is supported by data showing the importance for the dorsal striatum in stimulus–response habit learning (Yin et al, 2005) and microdialysis studies showing that prolonged cocaine-seeking increased dopamine release in the dorsal striatum but not ventral striatum (Ito et al, 2002). Disconnection of the ventral striatum from the dorsal striatum in rats self-administering cocaine on a second-order schedule only showed a deficit in animals with well-established ‘compulsive’ intake but not in animals that recently acquired the second-order schedule (Belin and Everitt, 2008). Thus, the hypothesis is that drug addiction represents changes in associative structures to become automatic or habitual and involves a gradual engagement of dorsal striatal mechanisms.
Animal studies have strongly suggested that with repeated drug exposure neutral stimuli that are associated with the drug can eventually acquire the ability to increase dopamine by themselves. Brain imaging studies confirmed this in addicted humans (Volkow et al, 2008a; Heinz et al, 2004). These studies showed that drug-associated cues induced dopamine increases in the dorsal striatum (caudate and putamen), an effect that correlated with self-reports of craving. The fact that the magnitude of the dopamine increases triggered by the cues was associated with the degree of addiction severity highlights the importance of these conditioned dopamine responses in the process of drug addiction in humans.
Clinical studies have also shown that the striatal slow dopamine increases induced by acute administration of oral methylphenidate do not elicit craving in cocaine abusers unless they are coupled to drug-associated cues (Volkow et al, 2008a). This most likely reflects the fact that the craving results from fast dopamine increases achieved with phasic dopamine firing, as opposed to slow dopamine increases achieved with tonic dopamine firing and in the experiment with oral methylphenidate. In fact, intravenous administration of methylphenidate, which results in fast dopamine increases, induces intense craving.
Brain imaging studies have also shown that, in drug-addicted subjects, these processes involve the orbitofrontal cortex, a brain region implicated in salience attribution and motivation, disruption of which results in compulsivity, and is a brain region with heavy projections to the dorsal striatum. The cingulate gyrus is also involved and is a brain region implicated in inhibitory control and conflict resolution, disruption of which results in impulsivity (Volkow et al, 2004b). Moreover, in cocaine-addicted, but not nonaddicted, subjects, the intravenous administration of methylphenidate, which cocaine abusers report has effects similar to those of cocaine, activated the orbital and medial prefrontal cortices, and this activation was associated with cocaine craving (Volkow et al, 2005). Similarly, in marijuana-addicted subjects, but not in nonaddicted individuals, acute administration of Δ9-THC activated the obitofrontal cortex (Volkow et al, 1996a). Activation of the obitofrontal cortex and cingulate gyrus is also triggered by conditioned cues that predict reward and trigger craving (McClernon et al, 2009). Interestingly, these are regions that regulate dopamine cell firing and release, which have been postulated to be necessary for the enhanced incentive motivational values of drugs in addicted individuals (mirroring a hypothesis based on animal studies; Volkow et al, 1999). When combined, these observations strongly suggest that the dopamine increases associated with conditioned cues are not primary responses, but rather the result of feedback stimulation of dopamine cells, most likely glutamatergic afferents from the prefrontal cortex and/or amygdala. On the basis of these findings, the activation of the obitofrontal cortex, with concomitant increases in dopamine produced by the drug, has been hypothesized to contribute to the compulsive drug consumption that characterizes drug bingeing in addicted individuals (Volkow et al, 2007).
Indeed, human neuroimaging studies show that the prefrontal cortex (orbitofrontal, medial prefrontal, prelimbic/cingulate) and the basolateral amygdala are critical in drug- and cue-induced craving in humans (Franklin et al, 2007). In prefrontal regions (eg, cingulate gyrus and obitofrontal cortex), these changes have been associated with a reduction in striatal dopamine D2 receptor availability observed in addicted subjects (Heinz et al, 2004; Volkow et al, 1993, 2001a, 2007). These associations could either reflect a disruption of frontal brain regions secondary to changes in striatal dopamine activity, or alternatively they could reflect a primary disruption of frontal regions that regulate dopamine cell activity. Indeed, a recent PET study provided evidence that prefrontal brain regions regulate the value of rewards by modulating dopamine increases in the ventral striatum, a regulatory mechanism that becomes dysfunctional in addicted individuals (Volkow et al, 2007).
Thus, concomitant dopamine and glutamate neurotransmission in the dorsal striatum, a region implicated in habit learning and action initiation, is involved in cue/context-dependent craving. As such, the dorsal striatum may be a fundamental component of addiction (Volkow et al, 2006). Research on novel strategies to inhibit cue-conditioned dopamine and glutamate responses is a major focus of current medications development efforts.
The thalamus has not been studied as extensively in the context of addiction. However, because of its integrative function in the regulation of arousal and attentional modulation, this region has been increasingly implicated in the addiction process. For example, intravenous administration of a stimulant drug in cocaine abusers, but not in controls, increased dopamine neurotransmission in the thalamus, an effect associated with craving (Volkow et al, 1997a). In contrast, compared with controls, cocaine abusers show hypoactivation of the thalamus, possibly reflecting noradrenergic and/or dopaminergic deficits, when performing a cognitive task (Tomasi et al, 2007b). Similarly, the thalamus was reported to show attenuated activation while performing a visual cognitive task in smokers exposed to nicotine (Sharma and Brody, 2009). These results suggest that thalamic abnormalities in cocaine abusers may contribute not only to impairments in sensory processing and attention but also to craving. Interestingly, changes in dopamine transmission in the thalamus and striatum appear to be involved in the deterioration of cognitive performance (eg, visual attention and working memory) that inexorably follows a period of sleep deprivation (Volkow et al, 2008b). Thus, more research that builds upon the available preliminary data is warranted.
Dorsolateral Frontal Cortex, Inferior Frontal Cortex, Hippocampus: Cognitive Control, Delayed Gratification, and Memory
Addiction also entails perturbations in cortically regulated cognitive and emotional processes, which cause the overvaluing of drug reinforcers at the expense of the undervaluing of natural reinforcers, and deficits in inhibitory control of drug responses (Goldstein and Volkow, 2002). As a result, an underperforming prefrontal system is widely believed to be crucial to the addiction process.
One of the components in such a system is impulse control, which is among the most robust cognitive risk factors for substance use disorders. Cocaine appears to have a direct effect on the neurobiology underlying impulse control. After an intravenous injection of cocaine, cocaine users actually showed an improvement in a motor response inhibition task and concomitant increased activation in their right dorsolateral and inferior frontal cortices (Garavan et al, 2008). Because these areas are considered to be important in impulse control, this observation suggests that some of the acute effects of cocaine could in fact mediate a transient reversal of the chronic hypofunction in impulse control circuitry.
Another important function that resides in frontocortical areas is the ability to choose between small and immediate rewards compared to large but deferred rewards, which can be measured using a delayed discounting task. A recent study found that both the dorsolateral and inferolateral frontal cortex gray matter volumes inversely correlated with preference for immediate gratification during decision-making (Bjork et al, 2009). This finding suggests that abnormalities in frontocortical regions may underlie the inability to delay gratification, a trait that is characteristic of addiction and other psychiatric disorders.
The neural substrates of memory and conditioned learning are among the major circuits undergoing aberrant neuroadaptations in response to chronic drug exposure (Volkow et al, 2004a). Different memory systems have been proposed to be involved in drug addiction, including conditioned-incentive learning (via the nucleus accumbens and amygdala), habit learning (via the caudate and putamen), and declarative memory (via the hippocampus; White, 1996), which is the focus of this section.
Over the past decade, many provocative animal studies have suggested that addictive drugs can disrupt neurogenesis in the adult hippocampus (Canales, 2007). Damage to the ventral subiculum of the hippocampus was shown to affect cocaine self-administration in rats (Caine et al, 2001). Such observations have provided insights into the possible involvement of a dysregulated hippocampus in human addiction. This hypothesis is an extension of current knowledge because the hippocampus is broadly viewed as important in contextual conditioning, namely in the processing of contextual cues by which memories can be accessed and retrieved. In fact, declarative memory has been long recognized to be involved in learning and the linking of affective conditions or circumstances with drug-taking experiences. Studies with PET and functional magnetic resonance imaging have shown that cue-elicited craving, as well as acute intoxication, activates the hippocampus and amygdala (Volkow et al, 2004a). For example, the craving that cocaine users experience while exposed to drug-related stimuli is accompanied by blood flow increases in a distributed region implicated in several forms of memory, including the amygdala (Childress et al, 1999; Grant et al, 1996; Kilts et al, 2001) and hippocampus (Kilts et al, 2001).
Therefore, new approaches to disrupt memory reconsolidation may help erode the strong associations between context and drug (Lee, 2008; Lee et al, 2005). Interestingly, β-blockers have already shown a promising capacity to inhibit conditioned responses to both natural reinforcers and aversive stimuli (Miranda et al, 2003). Moreover, results from a more recent study suggest that drug-induced conditioned responses may also be sensitive to β-blockade treatment (Milton et al, 2008). Similarly, further research on GABA-enhancing drugs also seems warranted. GABAergic stimulation, which can attenuate Pavlovian conditioning, appears to disrupt the response to drugs of abuse in animals (Volkow et al, 2004a) and may be a useful strategy to treat addiction in humans (Dewey et al, 1998).
Extended Amygdala: Negative Reinforcement Pathways
Compulsive drug use defined by increased intake of drug with extended access is accompanied by a chronic perturbation in brain reward homeostasis using measures of brain stimulation reward thresholds. The differential exposure to drug self-administration has dramatic effects on reward thresholds that progressively increase (ie, decreased reward) in extended-access, but not in limited-access, rats across successive self-administration sessions (Ahmed et al, 2002; Kenny et al, 2006; Wee et al, unpublished results). Animals with extended access to cocaine are more sensitive to the blockade of self-administration by dopamine antagonists and partial agonists (Ahmed and Koob, 2004; Wee et al, 2007), and the opioid partial agonist buprenorphine dose dependently decreased heroin self-administration in extended-access, opioid-dependent rats (Chen et al, 2006b), suggesting that reversal of reward deficits can blunt the motivational drives of drug addiction. This mechanism could underlie the benefit of methadone and buprenorphine treatment in heroin addiction.
As noted above, CRF antagonists blocked the anxiogenic- and aversive-like effects of drug withdrawal, and withdrawal from all drugs of abuse activated CRF in the CeA. These observations led to the hypothesis that activation of CRF, specifically extrahypothalamic CRF in the CeA, contributed to the motivational state driving compulsivity from the negative reinforcement perspective (Koob and Le Moal, 2008). Thus, one would predict that blockade of the brain stress systems in animal models of extended access to drugs may block the motivation for excessive drug intake. CRF antagonists selectively blocked the increased self-administration of drugs associated with extended access to intravenous self-administration of cocaine, nicotine (Koob, 2008), heroin (Greenwell et al, 2009), and alcohol (Koob, 2008). A particularly dramatic example of the motivational effects of CRF in the extended amygdala in dependence can be observed in animal models of ethanol self-administration in dependent animals in which a CRF1/2 peptide antagonist injected into the amygdala blocked the increase in ethanol self-administration during withdrawal (Funk et al, 2006; Koob, 2008).
Although less well developed, evidence suggests involvement of norepinephrine systems in the extended amygdala in the negative motivational state and increased self-administration associated with dependence (Koob, 2009b). Consistent with the role of the dynorphin-κ opioid system in the aversive effects of drug withdrawal, a κ-opioid antagonist blocked the excessive drinking associated with ethanol withdrawal in dependent rats and selectively blocked the increased progressive-ratio performance in rats with extended access to cocaine (Koob, 2009b; Wee et al, 2009).
Neuropeptide Y has dramatic anxiolytic-like properties localized to the amygdala and has been hypothesized to have effects opposite to CRF in the negative motivational state of withdrawal from drugs of abuse (Heilig et al, 1994; Heilig and Koob, 2007). NPY administered intracerebroventricularly blocked the increased drug intake associated with ethanol dependence (Thorsell et al, 2005a, 2005b). Injection of NPY into the CeA (Gilpin et al, 2008) and viral vector-enhanced expression of NPY in the CeA also blocked the increased drug intake associated with ethanol dependence (Thorsell et al, 2007).
Thus, the CRF increases in the CeA that occur with acute withdrawal from drugs have motivational significance not only for the anxiety/aversive-like effects of acute withdrawal but also for the increased drug intake associated with dependence. Acute withdrawal also may increase the release of norepinephrine in the BNST and dynorphin in the nucleus accumbens, both of which may contribute to the negative emotional state associated with dependence. Decreased activity of NPY in the CeA also may contribute to the anxiety-like state associated with ethanol dependence. Activation of brain stress systems (CRF, norepinephrine, dynorphin), combined with inactivation of brain antistress systems (NPY) in the extended amygdala may elicit powerful emotional dysregulation with motivational significance to addiction. A number of other neurotransmitter systems have been hypothesized to modulate the extended amygdala both from the stress-induction domain (vasopressin, substance P, orexin) and the antistress domain (nociceptin, endocannabinoids; for review, see Koob, 2008). Such dysregulation may be a significant contribution to the between-system opponent processes that help maintain dependence and also sets the stage for more prolonged state changes in emotionality such as protracted abstinence.
Research on negative reinforcement mechanisms in human addiction has been very limited. With cocaine, for example, the amygdala and lateral orbitofrontal cortex were shown to be activated by unexpected but not expected cocaine infusions in active cocaine abusers (Kufahl et al, 2008), but cocaine abstinence was associated with large reductions in the activity of dopamine projection regions, including the amygdala (Tomasi et al, 2007a). In apparent contrast, smoking abstinence was associated with increased cerebral blood flow in the extended amygdala, among other regions (Wang et al, 2007), whereas a nasal nicotine spray reduced regional cerebral blood flow in the right amygdala and left anterior temporal cortex of habitual smokers subjected to 12 h of smoking deprivation (Zubieta et al, 2001).
The amygdala may be equally important for processing positive reward (Murray, 2007) and reward expectancy (Holland and Gallagher, 2004), similar to processing negative reward. Particularly interesting in the context of brain imaging research will be to understand the function of the amygdala in generating the anxiety and negative emotion frequently seen during abstinence.
A recent report highlighted the importance in addiction of the interoceptive circuit that most likely interfaces with the extended amygdala and ventral striatum. The study showed that smokers with damage to their insula (but not smokers with extrainsular lesions) were able to stop smoking easily and without experiencing either cravings or relapse (Naqvi et al, 2007). The insula, particularly its more anterior regions, is reciprocally connected to several limbic regions (eg, ventromedial prefrontal cortex, amygdala, and ventral striatum) and appears to have an interoceptive function, integrating the autonomic and visceral information with emotion and motivation and providing conscious awareness of these urges (Naqvi and Bechara, 2009). Indeed, brain lesion studies suggest that the ventromedial prefrontal cortex and insula are necessary components of the distributed circuits that support emotional decision-making (Clark et al, 2008). Consistent with this hypothesis, many imaging studies show differential activation in the insula during craving (Naqvi and Bechara, 2009). The reactivity of this brain region has been suggested to serve as a biomarker to help predict relapse.
MOLECULAR TARGETS FOR NEUROPLASTICITY: BINGE/INTOXICATION, WITHDRAWAL/NEGATIVE AFFECT, AND PREOCCUPATION/ANTICIPATION (CRAVING)
The focus of the present review is on the neurocircuitry of addiction. However, parallel to the neuroplasticity of the neurocircuitry are the molecular changes that occur in these same structures. Chronic exposure to opiates and cocaine leads to activation of cyclic adenosine monophosphate response-element binding protein (CREB) in the nucleus accumbens and CeA (Shaw-Lutchman et al, 2002; Edwards et al, 2007). CREB can be phosphorylated by protein kinase A and by protein kinase regulated by growth factors, putting it at a point of convergence for several intracellular messenger pathways that can regulate gene expression. Activation of CREB in the nucleus accumbens with psychostimulant drugs is linked to the motivational symptoms of psychostimulant withdrawal, such as dysphoria, possibly through induction of the opioid peptide dynorphin, which binds to κ-opioid receptors and has been hypothesized to represent a mechanism of motivational tolerance and dependence (Nestler, 2005). Repeated CREB activation promotes dynorphin expression in the nucleus accumbens, which in turn decreases dopaminergic activity, both of which can contribute to negative emotional states. Extracellular signal-regulated kinase is another key element of intracellular signaling considered a key component in the plasticity associated with repeated administration of cocaine, specifically behavioral sensitization, cocaine reward, and time-dependent increases in cocaine-seeking after withdrawal (ie, incubation effect; Lu et al, 2006; Li et al, 2008).
Another molecular target for regulating the plasticity that leads to addiction is dysregulation of cystine–glutamate exchange, which is hypothesized to promote pathological glutamate signaling related to several components of the addiction cycle. Here, repeated administration of cocaine blunts cystine–glutamate exchange, leading to reduced basal and increased cocaine-induced glutamate in the nucleus accumbens that persists for at least 3 weeks after the last cocaine treatment (Baker et al, 2003). Most compelling is the observation that treatment with N-acetylcysteine, by activating cystine–glutamate exchange, prevented cocaine-induced escalation and behavioral sensitization, restored the ability to induce LTP and long-term depression in the nucleus accumbens, and blunted reinstatement in animals and conditioned reactivity to drug cues in humans (Moussawi et al, 2009; LaRowe et al, 2007; Madayag et al, 2007).
CREB and other intracellular messengers can activate transcription factors, which can change gene expression and produce long-term changes in protein expression, and, as a result, neuronal function. Although acute administration of drugs of abuse can cause a rapid (within hours) activation of members of the Fos protein family, such as c-fos, FosB, Fra-1, and Fra-2 in the nucleus accumbens, other transcription factors, isoforms of ΔFosB, a highly stable form of FosB, have been shown to accumulate over longer periods of time (days) with repeated drug administration (Nestler, 2005). Animals with activated ΔFosB have exaggerated sensitivity to the rewarding effects of drugs of abuse, and ΔFosB may be a sustained molecular ‘switch’ that helps to initiate and maintain a state of addiction (McClung et al, 2004). Whether (and how) such transcription factors influence the function of the brain stress systems, such as CRF and those described above, remains to be determined.
SUMMARY AND CONCLUSIONS
In summary, multiple brain regions and circuits are disrupted in drug addiction and are likely to contribute differentially to the complex phenotype observed in addicted individuals (Figure 5). Although some of these functional abnormalities may be present to a greater or lesser extent across all classes of drug addictions, some of the changes may be specific to certain types of drugs. For example, long-lasting decrements in the DAT in the striatum are observed in methamphetamine but not in alcohol or cocaine addictions. Conversely, decrements in dopamine D2 receptors in the striatum are observed in subjects addicted to all of the drugs of abuse that have been investigated, and increased activation of brain stress systems such as CRF has been observed in animal models during acute withdrawal for all types of drugs. Importantly, the neuronal abnormalities that become manifest in an addicted individual and that can be uncovered by imaging and/or neuropsychopharmacological studies are a reflection of not only a given chronic drug exposure trajectory, but also an individual's specific constellations of genetic, developmental, and environmental characteristics.
FUTURE RESEARCH DIRECTIONS
The advances outlined above point the way to future directions for research in the neurocircuitry of addiction in the same conceptual framework of binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation. The rich resources of modern neurosciences applied to the neurobiology of addiction offer an opportunity to not only understand the neurocircuitry of the addiction process but also to provide the keys to understanding vulnerability and providing treatment for this devastating disease.
In the binge/intoxication stage of the addiction cycle, how neuroplasticity that begins with a change in firing in mesolimbic dopamine neurons during initial drug exposure is translated to engagement of the dorsal striatum, disruption of frontal system function, and recruitment of brain stress systems and results in a residual powerful drive for drug-seeking behavior even months after withdrawal remains to be determined. For example, what is the relationship between vulnerability to impulsivity and subsequent compulsivity in the neuroplasticity of the circuits described above? Such future studies may involve molecular genetic approaches that range from selective breeding to upregulation or knockdown of molecular mechanisms within specific brain circuits using short-hairpin RNA technology.
In the withdrawal/negative affect stage, engagement of the brain stress systems, such as CRF, in animal models needs to be extended to other interactive brain stress systems and explored in human studies. Numerous other neurotransmitter systems that interact with the brain stress system are only now being explored, such as dynorphin, NPY, substance P, nociceptin, and orexin. Virtually unexplored at this stage are human imaging studies of this component of the addiction cycle and human imaging of brain neurotransmitter systems implicated in motivational aspects of drug withdrawal. The development of novel radioactive ligands for human imaging studies that bind to the receptors of the above neurotransmitter systems would be a great boost to the field.
In the preoccupation/anticipation stage, human neuroimaging studies show that the prefrontal cortex (orbitofrontal, medial prefrontal, prelimbic/cingulate) and the basolateral amygdala are critical in drug- and cue-induced craving. Whether such associations reflect a disruption of frontal brain regions secondary to changes in striatal dopamine activity, or alternatively reflect a primary disruption of frontal regions that regulate dopamine cell activity, remains to be determined. New approaches to the study of memory reconsolidation may help elucidate the strong associations between context and drug. The importance in addiction of the interoceptive circuit involving the insula and other regions that most likely interface with the extended amygdala and ventral striatum remains to be determined. The reactivity of these brain circuits may serve as a biomarker to help predict relapse and help predict treatment efficacy. Human post-mortem studies, human laboratory studies, and neurocircuitry studies in parallel animal models will likely yield promising results in this domain.
Finally, molecular and genetic changes that convey the changes in activity of the neurocircuits in all three stages of the addiction cycle described above are only now being elucidated. Changes in transmitter regulatory systems, transcription factors, and even gene regulation at the epigenetic level may explain how circuits are dysregulated, stay dysregulated, and provide vulnerability to dysregulation initially or long into abstinence. Ultimately, neurobiological targets elucidated through the framework of the neurocircuitry of addiction will provide targets for identifying genetic vulnerability in the human population, and genetic vulnerability in the human studies may identify novel targets to be explored at the mechanistic level in animal studies.
References
Aharonovich E, Hasin DS, Brooks AC, Liu X, Bisaga A, Nunes EV (2006). Cognitive deficits predict low treatment retention in cocaine dependent patients. Drug Alcohol Depend 81: 313–322.
Ahmed SH, Kenny PJ, Koob GF, Markou A (2002). Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat Neurosci 5: 625–626.
Ahmed SH, Koob GF (1998). Transition from moderate to excessive drug intake: change in hedonic set point. Science 282: 298–300. This study showed that rats given extended access to cocaine escalate intake and show behavior consistent with an increase in hedonic set point (lower reward) for the drug.
Ahmed SH, Koob GF (2004). Changes in response to a dopamine antagonist in rats with escalating cocaine intake. Psychopharmacology 172: 450–454.
Ahmed SH, Walker JR, Koob GF (2000). Persistent increase in the motivation to take heroin in rats with a history of drug escalation. Neuropsychopharmacology 22: 413–421.
Alheid GF, De Olmos JS, Beltramino CA (1995). Amygdala and extended amygdala. In: Paxinos G (ed). The Rat Nervous System. Academic Press: San Diego. pp 495–578.
Allen TJ, Moeller FG, Rhoades HM, Cherek DR (1998). Impulsivity and history of drug dependence. Drug Alcohol Depend 50: 137–145.
American Psychiatric Association (1994). Diagnostic and Statistical Manual of Mental Disorders 4th edn. American Psychiatric Press: Washington, DC.
American Psychiatric Association (2000). Diagnostic and Statistical Manual of Mental Disorders 4th edn, text revision American Psychiatric Press: Washington, DC.
Arroyo M, Markou A, Robbins TW, Everitt BJ (1998). Acquisition, maintenance and reinstatement of intravenous cocaine self-administration under a second-order schedule of reinforcement in rats: effects of conditioned cues and continuous access to cocaine. Psychopharmacology 140: 331–344.
Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S et al (2003). Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6: 743–749.
Baker TB, Morse E, Sherman JE (1987). The motivation to use drugs: a psychobiological analysis of urges. In: River PC (ed). Alcohol and Addictive Behavior (series title: Nebraska Symposium on Motivation, vol 34). University of Nebraska Press: Lincoln, NE. pp 257–323.
Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT (1991). CRF antagonist reverses the ‘anxiogenic’ response to ethanol withdrawal in the rat. Psychopharmacology 103: 227–232.
Barr AM, Phillips AG (1999). Withdrawal following repeated exposure to d-amphetamine decreases responding for a sucrose solution as measured by a progressive ratio schedule of reinforcement. Psychopharmacology 141: 99–106.
Belin D, Everitt BJ (2008). Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron 57: 432–441. This study showed that the interactions between the ventral and dorsal striatum are critical for the development of compulsive-like cocaine-seeking behavior.
Ben-Shahar O, Ahmed SH, Koob GF, Ettenberg A (2004). The transition from controlled to compulsive drug use is associated with a loss of sensitization. Brain Res 995: 46–54.
Bjork JM, Momenan R, Hommer DW (2009). Delay discounting correlates with proportional lateral frontal cortex volumes. Biol Psychiatry 65: 710–713.
Bolla KI, Eldreth DA, London ED, Kiehl KA, Mouratidis M, Contoreggi C et al (2003). Orbitofrontal cortex dysfunction in abstinent cocaine abusers performing a decision-making task. Neuroimage 19: 1085–1094.
Bonci A, Bernardi G, Grillner P, Mercuri NB (2003). The dopamine-containing neuron: maestro or simple musician in the orchestra of addiction? Trends Pharmacol Sci 24: 172–177.
Boudreau AC, Wolf ME (2005). Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci 25: 9144–9151.
Briand LA, Flagel SB, Garcia-Fuster MJ, Watson SJ, Akil H, Sarter M et al (2008a). Persistent alterations in cognitive function and prefrontal dopamine D2 receptors following extended, but not limited, access to self-administered cocaine. Neuropsychopharmacology 33: 2969–2980.
Briand LA, Gross JP, Robinson TE (2008b). Impaired object recognition following prolonged withdrawal from extended-access cocaine self-administration. Neuroscience 155: 1–6.
Caine SB, Heinrichs SC, Coffin VL, Koob GF (1995). Effects of the dopamine D-1 antagonist SCH 23390 microinjected into the accumbens, amygdala or striatum on cocaine self-administration in the rat. Brain Res 692: 47–56.
Caine SB, Humby T, Robbins TW, Everitt BJ (2001). Behavioral effects of psychomotor stimulants in rats with dorsal or ventral subiculum lesions: locomotion, cocaine self-administration, and prepulse inhibition of startle. Behav Neurosci 115: 880–894.
Caine SB, Thomsen M, Gabriel KI, Berkowitz JS, Gold LH, Koob GF et al (2007). Lack of self-administration of cocaine in dopamine D1 receptor knock-out mice. J Neurosci 27: 13140–13150.
Calu DJ, Stalnaker TA, Franz TM, Singh T, Shaham Y, Schoenbaum G (2007). Withdrawal from cocaine self-administration produces long-lasting deficits in orbitofrontal-dependent reversal learning in rats. Learn Mem 14: 325–328.
Canales JJ (2007). Adult neurogenesis and the memories of drug addiction. Eur Arch Psychiatry Clin Neurosci 257: 261–270.
Chait LD (1994). Reinforcing and subjective effects of methylphenidate in humans. Behav Pharmacol 5: 281–288.
Chao SZ, Ariano MA, Peterson DA, Wolf ME (2002). D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J Neurochem 83: 704–712.
Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM et al (2008). Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59: 288–297.
Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S et al (2006a). Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci USA 103: 9333–9338.
Chen SA, O’Dell L, Hoefer M, Greenwell TN, Zorrilla EP, Koob GF (2006b). Unlimited access to heroin self-administration: independent motivational markers of opiate dependence. Neuropsychopharmacology 31: 2692–2707 (corrigendum: 31: 2802).
Childress AR, McLellan AT, Ehrman R, O’Brien CP (1988). Classically conditioned responses in opioid and cocaine dependence: a role in relapse?. In: Ray BA (ed). Learning Factors in Substance Abuse (series title: NIDA Research Monograph, vol 84). National Institute on Drug Abuse: Rockville, MD. pp 25–43.
Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP (1999). Limbic activation during cue-induced cocaine craving. Am J Psychiatry 156: 11–18.
Clark L, Bechara A, Damasio H, Aitken MR, Sahakian BJ, Robbins TW (2008). Differential effects of insular and ventromedial prefrontal cortex lesions on risky decision-making. Brain 131: 1311–1322.
Collins RJ, Weeks JR, Cooper MM, Good PI, Russell RR (1984). Prediction of abuse liability of drugs using IV self-administration by rats. Psychopharmacology 82: 6–13.
Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y et al (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454: 118–121.
Creese I, Iversen SD (1974). The role of forebrain dopamine systems in amphetamine-induced stereotyped behavior in the rat. Psychopharmacology 39: 345–357.
Crow TJ (1973). Catecholamine-containing neurones and electrical self-stimulation: 2. A theoretical interpretation and some psychiatric implications. Psychol Med 3: 66–73.
de Witte P, Littleton J, Parot P, Koob G (2005). Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs 19: 517–537.
Delfs JM, Zhu Y, Druhan JP, Aston-Jones G (2000). Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature 403: 430–434.
Deroche-Gamonet V, Belin D, Piazza PV (2004). Evidence for addiction-like behavior in the rat. Science 305: 1014–1017.
Dewey SL, Morgan AE, Ashby Jr CR, Horan B, Kushner SA, Logan J et al (1998). A novel strategy for the treatment of cocaine addiction. Synapse 30: 119–129.
Di Chiara G, Imperato A (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85: 5274–5278.
Drevets WC, Gautier C, Price JC, Kupfer DJ, Kinahan PE, Grace AA et al (2001). Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol Psychiatry 49: 81–96.
Duncan E, Boshoven W, Harenski K, Fiallos A, Tracy H, Jovanovic T et al (2007). An fMRI study of the interaction of stress and cocaine cues on cocaine craving in cocaine-dependent men. Am J Addict 16: 174–182.
Dyr W, Kostowski W (1995). Evidence that the amygdala is involved in the inhibitory effects of 5-HT3 receptor antagonists on alcohol drinking in rats. Alcohol 12: 387–391.
Edwards S, Graham DL, Bachtell RK, Self DW (2007). Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur J Neurosci 25: 2201–2213.
Everitt BJ, Belin D, Economidou D, Pelloux Y, Dalley JW, Robbins TW (2008). Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Phil Trans Royal Soc London B Biol Sci 363: 3125–3135.
Everitt BJ, Robbins TW (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8: 1481–1489 (erratum: 9(7): 979).
Everitt BJ, Wolf ME (2002). Psychomotor stimulant addiction: a neural systems perspective. J Neurosci 22: 3312–3320 (erratum: 22(16): 1a).
Ferrario CR, Gorny G, Crombag HS, Li Y, Kolb B, Robinson TE (2005). Neural and behavioral plasticity associated with the transition from controlled to escalated cocaine use. Biol Psychiatry 58: 751–759.
Fowler JS, Volkow ND, Logan J, Alexoff D, Telang F, Wang GJ et al (2008). Fast uptake and long-lasting binding of methamphetamine in the human brain: comparison with cocaine. Neuroimage 43: 756–763.
Franklin TR, Wang Z, Wang J, Sciortino N, Harper D, Li Y et al (2007). Limbic activation to cigarette smoking cues independent of nicotine withdrawal: a perfusion fMRI study. Neuropsychopharmacology 32: 2301–2309.
Freeman AS, Meltzer LT, Bunney BS (1985). Firing properties of substantia nigra dopaminergic neurons in freely moving rats. Life Sci 36: 1983–1994.
Funk CK, O’Dell LE, Crawford EF, Koob GF (2006). Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci 26: 11324–11332. This study showed that blockade of CRF receptors in the area of the central nucleus of the amygdala blocks the increased alcohol intake associated with dependence but not alcohol intake in nondependent animals.
Garavan H, Kaufman JN, Hester R (2008). Acute effects of cocaine on the neurobiology of cognitive control. Phil Trans Royal Soc London B Biol Sci 363: 3267–3276.
George O, Ghozland S, Azar MR, Cottone P, Zorrilla EP, Parsons LH et al (2007). CRF-CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc Natl Acad Sci USA 104: 17198–17203.
George O, Mandyam CD, Wee S, Koob GF (2008). Extended access to cocaine self-administration produces long-lasting prefrontal cortex-dependent working memory impairments. Neuropsychopharmacology 33: 2474–2482.
Gilpin NW, Koob GF (2008). Overview: neurobiology of alcohol dependence with a focus on motivational mechanisms. Alcohol Res Health 31: 185–195.
Gilpin NW, Misra K, Koob GF (2008). Neuropeptide Y in the central nucleus of the amygdala suppresses dependence-induced increases in alcohol drinking. Pharmacol Biochem Behav 90: 475–480.
Goldstein RZ, Volkow ND (2002). Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 159: 1642–1652.
Gonzalez D, Riba J, Bouso JC, Gomez-Jarabo G, Barbanoj MJ (2006). Pattern of use and subjective effects of Salvia divinorum among recreational users. Drug Alcohol Depend 85: 157–162.
Grace AA (2000). The tonic/phasic model of dopamine system regulation and its implications for understanding alcohol and psychostimulant craving. Addiction 95 (Suppl 2): S119–S128.
Grant BF, Dawson DA (1998). Age of onset of drug use and its association with DSM-IV drug abuse and dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. J Subst Abuse 10: 163–173.
Grant BF, Dawson DA, Stinson FS, Chou SP, Dufour MC, Pickering RP (2004). The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend 74: 223–234.
Grant S, London ED, Newlin DB, Villemagne VL, Liu X, Contoreggi C et al (1996). Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci USA 93: 12040–12045.
Greenwell TN, Funk CK, Cottone P, Richardson HN, Chen SA, Rice K et al (2009). Corticotropin-releasing factor-1 receptor antagonists decrease heroin self-administration in long-, but not short-access rats. Addict Biol 14: 130–143.
Hand TH, Koob GF, Stinus L, Le Moal M (1988). Aversive properties of opiate receptor blockade: evidence for exclusively central mediation in naive and morphine-dependent rats. Brain Res 474: 364–368.
Hebb DO (1972). Textbook of Psychology 3rd edn. WB Saunders: Philadelphia.
Heilig M, Koob GF (2007). A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci 30: 399–406.
Heilig M, Koob GF, Ekman R, Britton KT (1994). Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci 17: 80–85.
Heimer L, Alheid G (1991). Piecing together the puzzle of basal forebrain anatomy. In: Napier TC, Kalivas PW, Hanin I (eds). The Basal Forebrain: Anatomy to Function (series title: Advances in Experimental Medicine and Biology, vol 295). Plenum Press: New York. pp 1–42.
Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L (1995). Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav Pharmacol 6: 74–80.
Heinz A, Siessmeier T, Wrase J, Hermann D, Klein S, Grusser SM et al (2004). Correlation between dopamine D(2) receptors in the ventral striatum and central processing of alcohol cues and craving. Am J Psychiatry 161: 1783–1789 (erratum: 161: 2344).
Hernandez G, Hamdani S, Rajabi H, Conover K, Stewart J, Arvanitogiannis A et al (2006). Prolonged rewarding stimulation of the rat medial forebrain bundle: neurochemical and behavioral consequences. Behav Neurosci 120: 888–904.
Heyser CJ, Roberts AJ, Schulteis G, Koob GF (1999). Central administration of an opiate antagonist decreases oral ethanol self-administration in rats. Alcohol Clin Exp Res 23: 1468–1476.
Hill RT (1970). Facilitation of conditioned reinforcement as a mechanism of psychomotor stimulation. In: Cost E, Garattini S (eds). Amphetamines and Related Compounds. Raven Press: New York. pp 781–795.
Hnasko TS, Sotak BN, Palmiter RD (2005). Morphine reward in dopamine-deficient mice. Nature 438: 854–857.
Holland PC, Gallagher M (2004). Amygdala–frontal interactions and reward expectancy. Curr Opin Neurobiol 14: 148–155.
Hubner CB, Koob GF (1990). The ventral pallidum plays a role in mediating cocaine and heroin self-administration in the rat. Brain Res 508: 20–29.
Hyman SE, Malenka RC, Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598.
Hyytia P, Koob GF (1995). GABA-A receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol 283: 151–159.
Ito R, Dalley JW, Robbins TW, Everitt BJ (2002). Dopamine release in the dorsal striatum during cocaine-seeking behavior under the control of a drug-associated cue. J Neurosci 22: 6247–6253.
Jentsch JD, Olausson P, de la Garza II R, Taylor JR (2002). Impairments of reversal learning and response perseveration after repeated, intermittent cocaine administrations to monkeys. Neuropsychopharmacology 26: 183–190.
Jones S, Bonci A (2005). Synaptic plasticity and drug addiction. Curr Opin Pharmacol 5: 20–25.
June HL, Foster KL, McKay PF, Seyoum R, Woods JE, Harvey SC et al (2003). The reinforcing properties of alcohol are mediated by GABA(A1) receptors in the ventral pallidum. Neuropsychopharmacology 28: 2124–2137.
Justinova Z, Tanda G, Redhi GH, Goldberg SR (2003). Self-administration of delta9-tetrahydrocannabinol (THC) by drug naive squirrel monkeys. Psychopharmacology 169: 135–140.
Kalivas PW (2004). Glutamate systems in cocaine addiction. Curr Opin Pharmacol 4: 23–29.
Kalivas PW, O’Brien C (2008). Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology 33: 166–180.
Kalivas PW, Volkow ND (2005). The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry 162: 1403–1413.
Kauer JA, Malenka RC (2007). Synaptic plasticity and addiction. Nat Rev Neurosci 8: 844–858.
Kelly PH, Iversen SD (1976). Selective 6-OHDA-induced destruction of mesolimbic dopamine neurons: abolition of psychostimulant-induced locomotor activity in rats. Eur J Pharmacol 40: 45–56.
Kenny PJ, Chen SA, Kitamura O, Markou A, Koob GF (2006). Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J Neurosci 26: 5894–5900.
Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Faber TL, Muhammad F et al (2001). Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry 58: 334–341.
Kitamura O, Wee S, Specio SE, Koob GF, Pulvirenti L (2006). Escalation of methamphetamine self-administration in rats: a dose–effect function. Psychopharmacology 186: 48–53.
Koob GF (1992). Drugs of abuse: anatomy, pharmacology, and function of reward pathways. Trends Pharmacol Sci 13: 177–184.
Koob GF (2004). Allostatic view of motivation: implications for psychopathology. In: Bevins RA, Bardo MT (eds). Motivational Factors in the Etiology of Drug Abuse (series title: Nebraska Symposium on Motivation, vol 50). University of Nebraska Press: Lincoln, NE. pp 1–18.
Koob GF (2005). The neurocircuitry of addiction: implications for treatment. Clin Neurosci Res 5: 89–101.
Koob GF (2008). A role for brain stress systems in addiction. Neuron 59: 11–34.
Koob GF (2009a). Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology 56 (Suppl 1): 18–31.
Koob GF (2009b). Brain stress systems in the amygdala and addiction. Brain Res (in press).
Koob GF, Everitt BJ, Robbins TW (2008a). Reward, motivation, and addiction. In: Squire LG, Berg D, Bloom FE, Du Lac S, Ghosh A, Spitzer N (eds). Fundamental Neuroscience 3rd edn. Academic Press: Amsterdam. pp 987–1016.
Koob GF, Kandel D, Volkow ND (2008b). Pathophysiology of addiction. In: Tasman A, Kay J, Lieberman JA, First MB, Maj M (eds). Psychiatry 3rd edn, vol 1 Wiley: Chichester. pp 354–378.
Koob GF, Kreek MJ (2007). Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry 164: 1149–1159.
Koob GF, Le Moal M (1997). Drug abuse: hedonic homeostatic dysregulation. Science 278: 52–58. This theoretical review argued that drug addiction involves decreased hedonic homeostatic dysregulation (dysregulation of reward function) driven by both decreased activity in reward pathways and recruitment of brain stress systems.
Koob GF, Le Moal M (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24: 97–129.
Koob GF, Le Moal M (2005). Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci 8: 1442–1444.
Koob GF, Le Moal M (2006). Neurobiology of Addiction. Academic Press: London.
Koob GF, Le Moal M (2008). Addiction and the brain antireward system. Annu Rev Psychol 59: 29–53.
Koob GF, Lloyd GK, Mason BJ (2009). Development of pharmacotherapies for drug addiction: a Rosetta Stone approach. Nat Rev Drug Discov 8: 500–515.
Koob GF, Nestler EJ (1997). The neurobiology of drug addiction. J Neuropsychiatry Clin Neurosci 9: 482–497.
Kornetsky C, Bain G (1990). Brain-stimulation reward: a model for drug induced euphoria. In: Adler MW, Cowan A (eds). Testing and Evaluation of Drugs of Abuse (series title: Modern Methods in Pharmacology, vol 6). Wiley-Liss: New York. pp 211–231.
Kornetsky C, Esposito RU (1979). Euphorigenic drugs: effects on the reward pathways of the brain. Fed Proc 38: 2473–2476.
Kourrich S, Rothwell PE, Klug JR, Thomas MJ (2007). Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci 27: 7921–7928.
Kufahl P, Li Z, Risinger R, Rainey C, Piacentine L, Wu G et al (2008). Expectation modulates human brain responses to acute cocaine: a functional magnetic resonance imaging study. Biol Psychiatry 63: 222–230.
Langleben DD, Ruparel K, Elman I, Busch-Winokur S, Pratiwadi R, Loughead J et al (2008). Acute effect of methadone maintenance dose on brain FMRI response to heroin-related cues. Am J Psychiatry 165: 390–394.
LaRowe SD, Myrick H, Hedden S, Mardikian P, Saladin M, McRae A et al (2007). Is cocaine desire reduced by N-acetylcysteine? Am J Psychiatry 164: 1115–1117.
Laviolette SR, Alexson TO, van der Kooy D (2002). Lesions of the tegmental pedunculopontine nucleus block the rewarding effects and reveal the aversive effects of nicotine in the ventral tegmental area. J Neurosci 22: 8653–8660.
Le Doux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184.
Le Moal M, Simon H (1991). Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev 71: 155–234.
Lee JL (2008). Memory reconsolidation mediates the strengthening of memories by additional learning. Nat Neurosci 11: 1264–1266.
Lee JL, Di Ciano P, Thomas KL, Everitt BJ (2005). Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron 47: 795–801.
Li YQ, Li FQ, Wang XY, Wu P, Zhao M, Xu CM et al (2008). Central amygdala extracellular signal-regulated kinase signaling pathway is critical to incubation of opiate craving. J Neurosci 28: 13248–13257.
Logan GD, Schachar RJ, Tannock R (1997). Impulsivity and inhibitory control. Psychol Sci 8: 60–64.
Lu L, Koya E, Zhai H, Hope BT, Shaham Y (2006). Role of ERK in cocaine addiction. Trends Neurosci 29: 695–703.
Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M et al (2007). Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci 27: 13968–13976.
Maisonneuve IM, Ho A, Kreek MJ (1995). Chronic administration of a cocaine ‘binge’ alters basal extracellular levels in male rats: an in vivo microdialysis study. J Pharmacol Exp Ther 272: 652–657.
Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP et al (2006). Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron 50: 911–921.
Markou A, Kosten TR, Koob GF (1998). Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology 18: 135–174.
Martinez D, Broft A, Foltin RW, Slifstein M, Hwang DR, Huang Y et al (2004). Cocaine dependence and d2 receptor availability in the functional subdivisions of the striatum: relationship with cocaine-seeking behavior. Neuropsychopharmacology 29: 1190–1202 (erratum: 29: 1763).
Martinez D, Gil R, Slifstein M, Hwang DR, Huang Y, Perez A et al (2005). Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biol Psychiatry 58: 779–786.
Martinez D, Narendran R, Foltin RW, Slifstein M, Hwang DR, Broft A et al (2007). Amphetamine-induced dopamine release: markedly blunted in cocaine dependence and predictive of the choice to self-administer cocaine. Am J Psychiatry 164: 622–629.
McBride WJ, Murphy JM, Ikemoto S (1999). Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res 101: 129–152.
McClernon FJ, Kozink RV, Lutz AM, Rose JE (2009). 24-h smoking abstinence potentiates fMRI-BOLD activation to smoking cues in cerebral cortex and dorsal striatum. Psychopharmacology 204: 25–35.
McClung CA, Ulery PG, Perrotti LI, Zachariou V, Berton O, Nestler EJ (2004). DeltaFosB: a molecular switch for long-term adaptation in the brain. Mol Brain Res 132: 146–154.
McFarland K, Kalivas PW (2001). The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 21: 8655–8663. This study established a key role of the dorsal frontal cortex-nucleus accumbens-ventral pallidal circuit in cocaine-induced reinstatement.
McFarland K, Lapish CC, Kalivas PW (2003). Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531–3537.
McGregor A, Roberts DCS (1993). Dopaminergic antagonism within the nucleus accumbens or the amygdala produces differential effects on intravenous cocaine self-administration under fixed and progressive ratio schedules of reinforcement. Brain Res 624: 245–252.
Melendez RI, Rodd ZA, McBride WJ, Murphy JM (2004). Involvement of the mesopallidal dopamine system in ethanol reinforcement. Alcohol 32: 137–144.
Melis M, Spiga S, Diana M (2005). The dopamine hypothesis of drug addiction: hypodopaminergic state. Int Rev Neurobiol 63: 101–154.
Milton AL, Lee JL, Everitt BJ (2008). Reconsolidation of appetitive memories for both natural and drug reinforcement is dependent on β-adrenergic receptors. Learn Mem 15: 88–92.
Miranda MI, LaLumiere RT, Buen TV, Bermudez-Rattoni F, McGaugh JL (2003). Blockade of noradrenergic receptors in the basolateral amygdala impairs taste memory. Eur J Neurosci 18: 2605–2610.
Moeller FG, Barratt ES, Dougherty DM, Schmitz JM, Swann AC (2001). Psychiatric aspects of impulsivity. Am J Psychiatry 158: 1783–1793.
Moller C, Wiklund L, Sommer W, Thorsell A, Heilig M (1997). Decreased experimental anxiety and voluntary ethanol consumption in rats following central but not basolateral amygdala lesions. Brain Res 760: 94–101.
Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A et al (2009). N-acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci 12: 182–189.
Murray EA (2007). The amygdala, reward and emotion. Trends Cogn Sci 11: 489–497.
Naqvi NH, Bechara A (2009). The hidden island of addiction: the insula. Trends Neurosci 32: 56–67.
Naqvi NH, Rudrauf D, Damasio H, Bechara A (2007). Damage to the insula disrupts addiction to cigarette smoking. Science 315: 531–534. This study showed that damage to the insula in human smokers was associated with cessation of smoking, establishing a link between the insula and nicotine addiction.
Nauta JH, Haymaker W (1969). Hypothalamic nuclei and fiber connections. In: Haymaker W, Anderson E, Nauta WJH (eds). The Hypothalamus. Charles C Thomas: Springfield, IL. pp 136–209.
Nelson CL, Milovanovic M, Wetter JB, Ford KA, Wolf ME (2009). Behavioral sensitization to amphetamine is not accompanied by changes in glutamate receptor surface expression in the rat nucleus accumbens. J Neurochem 109: 35–51.
Nestler EJ (2005). Is there a common molecular pathway for addiction? Nat Neurosci 8: 1445–1449. This review summarizes a body of work characterizing the role of molecular changes mediating the transition from drug taking to addiction with a special emphasis on the accumulation of the transcription factor ΔFosB in the nucleus accumbens following chronic drug exposure.
Neugebauer V, Li W, Bird GC, Han JS (2004). The amygdala and persistent pain. Neuroscientist 10: 221–234.
O’Dell LE, Koob GF (2007). Nicotine deprivation effect in rats with intermittent 23-h access to intravenous nicotine self-administration. Pharmacol Biochem Behav 86: 346–353.
Olds J, Milner P (1954). Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47: 419–427.
Orsini C, Koob GF, Pulvirenti L (2001). Dopamine partial agonist reverses amphetamine withdrawal in rats. Neuropsychopharmacology 25: 789–792.
Pierce RC, Bell K, Duffy P, Kalivas PW (1996). Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16: 1550–1560.
Pulvirenti L, Koob GF (1993). Lisuride reduces psychomotor retardation during withdrawal from chronic intravenous amphetamine self-administration in rats. Neuropsychopharmacology 8: 213–218.
Rachlin H, Green L (1972). Commitment, choice and self-control. J Exp Anal Behav 17: 15–22.
Robbins TW (1976). Relationship between reward-enhancing and stereotypical effects of psychomotor stimulant drugs. Nature 264: 57–59.
Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF (2000). Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology 22: 581–594.
Roberts DCS (1992). Neural substrates mediating cocaine reinforcement: the role of monoamine systems. In: Lakoski JM, Galloway MP, White FJ (eds). Cocaine: Pharmacology, Physiology and Clinical Strategies. CRC Press: Boca Raton, FL. pp 73–90.
Robinson TE, Berridge KC (1993). The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev 18: 247–291.
Robledo P, Koob GF (1993). Two discrete nucleus accumbens projection areas differentially mediate cocaine self-administration in the rat. Behav Brain Res 55: 159–166.
Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B et al (1998). Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci 1: 132–137.
Rossetti ZL, Hmaidan Y, Gessa GL (1992). Marked inhibition of mesolimbic dopamine release: a common feature of ethanol, morphine, cocaine and amphetamine abstinence in rats. Eur J Pharmacol 221: 227–234.
Russell MAH (1976). What is dependence?. In: Edwards G (ed). Drugs and Drug Dependence. Lexington Books: Lexington, MA. pp 182–187.
Saal D, Dong Y, Bonci A, Malenka RC (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582 (erratum: 38: 359).
Salamone JD, Correa M, Farrar A, Mingote SM (2007). Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology 191: 461–482.
Sanchis-Segura C, Spanagel R (2006). Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol 11: 2–38.
Sarnyai Z, Biro E, Gardi J, Vecsernyes M, Julesz J, Telegdy G (1995). Brain corticotropin-releasing factor mediates ‘anxiety-like’ behavior induced by cocaine withdrawal in rats. Brain Res 675: 89–97.
Schoenbaum G, Saddoris MP, Ramus SJ, Shaham Y, Setlow B (2004). Cocaine-experienced rats exhibit learning deficits in a task sensitive to orbitofrontal cortex lesions. Eur J Neurosci 19: 1997–2002.
Schulteis G, Ahmed SH, Morse AC, Koob GF, Everitt BJ (2000). Conditioning and opiate withdrawal: the amygdala links neutral stimuli with the agony of overcoming drug addiction. Nature 405: 1013–1014.
Schulteis G, Stinus L, Risbrough VB, Koob GF (1998). Clonidine blocks acquisition but not expression of conditioned opiate withdrawal in rats. Neuropsychopharmacology 19: 406–416.
Schultz W (2007). Multiple dopamine functions at different time courses. Annu Rev Neurosci 30: 259–288.
Shaham Y, Shalev U, Lu L, de Wit H, Stewart J (2003). The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology 168: 3–20.
Shalev U, Grimm JW, Shaham Y (2002). Neurobiology of relapse to heroin and cocaine seeking: a review. Pharmacol Rev 54: 1–42. This review summarizes the neurocircuitry associated with drug-, cue-, and stress-induced relapse determined from extensive work with animal models.
Sharma A, Brody AL (2009). In vivo brain imaging of human exposure to nicotine and tobacco. Handb Exp Pharmacol 192: 145–171.
Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S et al (2002). Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci 22: 3663–3672.
Solomon RL, Corbit JD (1974). An opponent-process theory of motivation: 1. Temporal dynamics of affect. Psychol Rev 81: 119–145.
Stein L (1962). Effects and interactions of imipramine, chlorpromazine, reserpine, and amphetamine on self-stimulation: possible neurophysiological basis of depression. Recent Adv Biol Psychiatry 4: 288–309.
Stinus L, Cador M, Zorrilla EP, Koob GF (2005). Buprenorphine and a CRF1 antagonist block the acquisition of opiate withdrawal-induced conditioned place aversion in rats. Neuropsychopharmacology 30: 90–98.
Substance Abuse and Mental Health Services Administration (2008). Results from the 2007 National Survey on Drug Use and Health: National Findings (Office of Applied Statistics, NSDUH Series H-34, DHHS Publication No. SMA 08-4343). Rockville, MD.
Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D et al (2003). Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature 421: 70–75.
Tanda G, Pontieri FE, Di Chiara G (1997). Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common μ1 opioid receptor mechanism. Science 276: 2048–2050.
Thorsell A, Rapunte-Canonigo V, O’Dell L, Chen SA, King A, Lekic D et al (2007). Viral vector-induced amygdala NPY overexpression reverses increased alcohol intake caused by repeated deprivations in Wistar rats. Brain 130: 1330–1337.
Thorsell A, Slawecki CJ, Ehlers CL (2005a). Effects of neuropeptide Y and corticotropin-releasing factor on ethanol intake in Wistar rats: interaction with chronic ethanol exposure. Behav Brain Res 161: 133–140.
Thorsell A, Slawecki CJ, Ehlers CL (2005b). Effects of neuropeptide Y on appetitive and consummatory behaviors associated with alcohol drinking in Wistar rats with a history of ethanol exposure. Alcohol Clin Exp Res 29: 584–590.
Tiffany ST, Carter BL, Singleton EG (2000). Challenges in the manipulation, assessment and interpretation of craving relevant variables. Addiction 95 (Suppl 2): s177–s187.
Todtenkopf MS, Parsegian A, Naydenov A, Neve RL, Konradi C, Carlezon Jr WA (2006). Brain reward regulated by AMPA receptor subunits in nucleus accumbens shell. J Neurosci 26: 11665–11669.
Tomasi D, Goldstein RZ, Telang F, Maloney T, Alia-Klein N, Caparelli EC et al (2007a). Widespread disruption in brain activation patterns to a working memory task during cocaine abstinence. Brain Res 1171: 83–92.
Tomasi D, Goldstein RZ, Telang F, Maloney T, Alia-Klein N, Caparelli EC et al (2007b). Thalamo-cortical dysfunction in cocaine abusers: implications in attention and perception. Psychiatry Res 155: 189–201.
Tornatzky W, Miczek KA (2000). Cocaine self-administration ‘binges’: transition from behavioral and autonomic regulation toward homeostatic dysregulation in rats. Psychopharmacology 148: 289–298.
Tucci S, Cheeta S, Seth P, File SE (2003). Corticotropin releasing factor antagonist, α-helical CRF9−41, reverses nicotine-induced conditioned, but not unconditioned, anxiety. Psychopharmacology 167: 251–256.
Tzschentke TM (1998). Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 56: 613–672.
Ungless MA, Whistler JL, Malenka RC, Bonci A (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583–587.
Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP et al (2002). Increased ethanol self-administration and anxiety-like behavior during acute withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res 26: 1494–1501.
Vanderschuren LJ, Everitt BJ (2004). Drug seeking becomes compulsive after prolonged cocaine self-administration. Science 305: 1017–1019.
Vezina P (2004). Sensitization of midbrain dopamine neuron reactivity and the self-administration of psychomotor stimulant drugs. Neurosci Biobehav Rev 27: 827–839.
Volkow ND, Chang L, Wang GJ, Fowler JS, Ding YS, Sedler M et al (2001a). Low level of brain dopamine D2 receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. Am J Psychiatry 158: 2015–2021. This study shows an association between the decreases in dopamine function in addiction and decreased function of the orbitofrontal cortex, establishing a key link between compromised striatal activity and orbitofrontal dysfunction in addiction.
Volkow ND, Fowler JS, Wang GJ, Ding YS, Gatley SJ (2002). Role of dopamine in the therapeutic and reinforcing effects of methylphenidate in humans: results from imaging studies. Eur Neuropsychopharmacol 12: 557–566.
Volkow ND, Fowler JS, Wang GJ, Hitzemann R, Logan J, Schlyer DJ et al (1993). Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 14: 169–177.
Volkow ND, Fowler JS, Wang GJ (2004a). The addicted human brain viewed in the light of imaging studies: brain circuits and treatment strategies. Neuropharmacology 47 (Suppl 1): 3–13.
Volkow ND, Fowler JS, Wang GJ, Swanson JM (2004b). Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry 9: 557–569.
Volkow ND, Gillespie H, Mullani N, Tancredi L, Grant C, Valentine A et al (1996a). Brain glucose metabolism in chronic marijuana users at baseline and during marijuana intoxication. Psychiatry Res 67: 29–38.
Volkow ND, Swanson JM (2003). Variables that affect the clinical use and abuse of methylphenidate in the treatment of ADHD. Am J Psychiatry 160: 1909–1918.
Volkow ND, Wang G, Fowler JS, Logan J, Gerasimov M, Maynard L et al (2001b). Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J Neurosci 21: RC121.
Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN et al (1997a). Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature 386: 827–830.
Volkow ND, Wang GJ, Fowler JS, Franceschi D, Thanos PK, Wong C et al (2000). Cocaine abusers show a blunted response to alcohol intoxication in limbic brain regions. Life Sci 66: PL161–PL167.
Volkow ND, Wang GJ, Fowler JS, Gatley SJ, Ding YS, Logan J et al (1996b). Relationship between psychostimulant-induced ‘high’ and dopamine transporter occupancy. Proc Natl Acad Sci USA 93: 10388–10392.
Volkow ND, Wang GJ, Fowler JS, Hitzemann R, Angrist B, Gatley SJ et al (1999). Association of methylphenidate-induced craving with changes in right striato-orbitofrontal metabolism in cocaine abusers: implications in addiction. Am J Psychiatry 156: 19–26.
Volkow ND, Wang GJ, Fowler JS, Hitzemann R, Gatley SJ, Dewey SS et al (1998). Enhanced sensitivity to benzodiazepines in active cocaine-abusing subjects: a PET study. Am J Psychiatry 155: 200–206.
Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R et al (1997b). Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature 386: 830–833. This study using PET showed decreased release of dopamine in the striatum and a decreased ‘high’ produced by methylphenidate, suggesting a compromised striatal dopamine system in addiction.
Volkow ND, Wang GJ, Ma Y, Fowler JS, Wong C, Ding YS et al (2005). Activation of orbital and medial prefrontal cortex by methylphenidate in cocaine-addicted subjects but not in controls: relevance to addiction. J Neurosci 25: 3932–3939.
Volkow ND, Wang GJ, Ma Y, Fowler JS, Zhu W, Maynard L et al (2003). Expectation enhances the regional brain metabolic and the reinforcing effects of stimulants in cocaine abusers. J Neurosci 23: 11461–11468.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR et al (2006). Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. J Neurosci 26: 6583–6588.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR et al (2008a). Dopamine increases in striatum do not elicit craving in cocaine abusers unless they are coupled with cocaine cues. Neuroimage 39: 1266–1273.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Jayne M et al (2007). Profound decreases in dopamine release in striatum in detoxified alcoholics: possible orbitofrontal involvement. J Neurosci 27: 12700–12706.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Wong C et al (2008b). Sleep deprivation decreases binding of [11C]raclopride to dopamine D2/D3 receptors in the human brain. J Neurosci 28: 8454–8461.
Wang GJ, Volkow ND, Fowler JS, Logan J, Abumrad NN, Hitzemann RJ et al (1997). Dopamine D2 receptor availability in opiate-dependent subjects before and after naloxone-precipitated withdrawal. Neuropsychopharmacology 16: 174–182.
Wang Z, Faith M, Patterson F, Tang K, Kerrin K, Wileyto EP et al (2007). Neural substrates of abstinence-induced cigarette cravings in chronic smokers. J Neurosci 27: 14035–14040.
Watkins SS, Stinus L, Koob GF, Markou A (2000). Reward and somatic changes during precipitated nicotine withdrawal in rats: centrally and peripherally mediated effects. J Pharmacol Exp Ther 292: 1053–1064.
Wee S, Wang Z, Woolverton WL, Pulvirenti L, Koob GF (2007). Effect of aripiprazole, a partial D2 receptor agonist, on increased rate of methamphetamine self-administration in rats with prolonged access. Neuropsychopharmacology 32: 2238–2247.
Wee S, Orio L, Ghirmai S, Cashman J, Koob GF (2009). Inhibition of kappa opioid receptors attenuates the increased motivation for cocaine in rats with extended access to cocaine. Psychopharmacology (in press).
Weiss F, Ciccocioppo R, Parsons LH, Katner S, Liu X, Zorrilla EP et al (2001). Compulsive drug-seeking behavior and relapse: neuroadaptation, stress, and conditioning factors. In: Quinones-Jenab V (ed). The Biological Basis of Cocaine Addiction (series title: Annals of the New York Academy of Sciences, vol 937) New York Academy of Sciences: New York. pp 1–26.
Weiss F, Markou A, Lorang MT, Koob GF (1992). Basal extracellular dopamine levels in the nucleus accumbens are decreased during cocaine withdrawal after unlimited-access self-administration. Brain Res 593: 314–318.
Weiss F, Parsons LH, Schulteis G, Hyytia P, Lorang MT, Bloom FE et al (1996). Ethanol self-administration restores withdrawal-associated deficiencies in accumbal dopamine and 5-hydroxytryptamine release in dependent rats. J Neurosci 16: 3474–3485.
White NM (1996). Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction 91: 921–949.
Whitelaw RB, Markou A, Robbins TW, Everitt BJ (1996). Excitotoxic lesions of the basolateral amygdala impair the acquisition of cocaine-seeking behaviour under a second-order schedule of reinforcement. Psychopharmacology 127: 213–224.
Wikler A (1952). A psychodynamic study of a patient during experimental self-regulated re-addiction to morphine. Psychiatr Q 26: 270–293.
Wise RA (1978). Catecholamine theories of reward: a critical review. Brain Res 152: 215–247.
Wolf ME (2002). Addiction: making the connection between behavioral changes and neuronal plasticity in specific pathways. Mol Intervent 2: 146–157.
Wolf ME, Sun X, Mangiavacchi S, Chao SZ (2004). Psychomotor stimulants and neuronal plasticity. Neuropharmacology 47 (Suppl 1): 61–79.
Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM et al (2004). Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41: 625–638.
Yeomans J, Baptista M (1997). Both nicotinic and muscarinic receptors in ventral tegmental area contribute to brain-stimulation reward. Pharmacol Biochem Behav 57: 915–921.
Yin HH, Ostlund SB, Knowlton BJ, Balleine BW (2005). The role of the dorsomedial striatum in instrumental conditioning. Eur J Neurosci 22: 513–523.
Zubieta JK, Gorelick DA, Stauffer R, Ravert HT, Dannals RF, Frost JJ (1996). Increased mu opioid receptor binding detected by PET in cocaine-dependent men is associated with cocaine craving. Nat Med 2: 1225–1229.
Zubieta J, Lombardi U, Minoshima S, Guthrie S, Ni L, Ohl LE et al (2001). Regional cerebral blood flow effects of nicotine in overnight abstinent smokers. Biol Psychiatry 49: 906–913.
Acknowledgements
This is publication number 20084 from The Scripps Research Institute. Preparation of this work was supported by the Pearson Center for Alcoholism and Addiction Research and National Institutes of Health grants AA12602, AA08459, and AA06420 from the National Institute on Alcohol Abuse and Alcoholism; DA04043, DA04398, and DA10072 from the National Institute on Drug Abuse; DK26741 from the National Institute of Diabetes and Digestive and Kidney Diseases; and 17RT-0095 from the Tobacco-Related Disease Research Program from the State of California. We thank Michael Arends and Ruben Baler for their assistance with paper preparation.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE
The authors declare no conflicts of interest.
Rights and permissions
About this article
Cite this article
Koob, G., Volkow, N. Neurocircuitry of Addiction. Neuropsychopharmacol 35, 217–238 (2010). https://doi.org/10.1038/npp.2009.110
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2009.110
This article is cited by
-
Addiction biotypes: a paradigm shift for future treatment strategies?
Molecular Psychiatry (2024)
-
Dorsal hippocampus to nucleus accumbens projections drive reinforcement via activation of accumbal dynorphin neurons
Nature Communications (2024)
-
Effect of prenatal and early post-natal oxycodone exposure on the reinforcing and antinociceptive effects of oxycodone in adult C57BL/6 J mice
Psychopharmacology (2024)
-
Changes in distinct brain systems identified with fMRI during smoking cessation treatment with varenicline: a review
Psychopharmacology (2024)
-
The novel psychoactive substance 25E-NBOMe induces reward-related behaviors via dopamine D1 receptor signaling in male rodents
Archives of Pharmacal Research (2024)
