
Abstract
Experiential factors, such as stress, are major determinants of vulnerability to drug addiction and relapse. The behavioral controllability of the stressor is a major determinant of how exposure to a stressor impacts addictive processes. Recent evidence suggests that controllable stressors, such as escapable shock (ES), activate ventral regions of the medial prefrontal cortex (mPFCv), whereas physically identical, but uncontrollable stress (inescapable shock, IS) does not. This activation is critical to the blunting effect that control has on neurochemical and behavioral sequelae of stress. Our laboratory has previously shown that IS, but not ES, potentiates morphine-conditioned place preference (CPP). However, the role of the mPFCv in this phenomenon is unknown. The present experiments investigated the role of the mPFCv during ES and IS in determining the effects of the stressor on subsequent morphine-CPP. Intra-mPFCv microinjection of the GABAA agonist muscimol 1 h before ES led ES to potentiate morphine-CPP, as does IS. Conversely, the potentiation of morphine-CPP normally observed in IS rats was blocked by intra-mPFCv microinjection of the GABAA antagonist picrotoxin 1 h before IS. These results suggest that during stress, activation of the mPFCv prevents subsequent potentiation of morphine-CPP, whereas inactivation of the mPFCv during stress does not. Thus, activation of the mPFCv during a stress experience is both necessary and sufficient to block the impact of stress on morphine-CPP, and control over stress blunts stress-induced potentiation of morphine effects by activating the mPFCv.
Similar content being viewed by others


INTRODUCTION
Environmental and experiential factors are critical determinants of an individual's vulnerability to drug addiction and relapse (Harrison et al, 1997; Koob and Le Moal, 1997; Sinha et al, 1999). However, not all individuals exposed to stress develop drug dependence (DuMont et al, 2007), and the mechanisms determining vulnerability remain largely unknown. Some of these determinants may relate to the type of stressor to which the individual is exposed.
Acute uncontrollable, but not behaviorally controllable, stress has persistent behavioral effects that occur outside the original stressor environment. These behavioral outcomes, which are dependent upon stressor controllability, have been called ‘learned helplessness’ (Maier and Seligman, 1976) or ‘behavioral depression’ (Weiss et al, 1981). Uncontrollable stress (inescapable shock, IS), but not controllable stress (escapable shock, ES) intensely activates and sensitizes serotonin (5HT) cells in the dorsal raphe nucleus (DRN) (Amat et al, 1998a, 1998b; Grahn et al, 1999). Furthermore, these alterations in DRN 5HT activity are both necessary and sufficient to produce ‘learned helplessness’ (Maier et al, 1995a, 1995b, 1994).
Rats exposed to IS, but not ES, exhibit potentiated morphine reward in a long-term, trans-situational manner as measured by conditioned place preference (CPP) (Will et al, 1998), a behavioral paradigm used to assess reward (Tzschentke, 2007). As with all consequences of exposure to IS, the potentiation of morphine-CPP by IS is mediated by IS-induced sensitization of DRN serotonergic neurons (Will et al, 2004). Consistent with potentiated morphine-CPP following IS, Bland et al (2004) reported potentiated dopamine (DA) efflux in the nucleus accumbens (NAc) shell following acute morphine in IS, but not ES rats. This morphine-induced potentiation of DA efflux by IS is also dependent on IS sensitization of DRN 5HT neurons (Bland et al, 2003).
Although the DRN sends serotonergic projections to the mesolimbic pathway (Van Bockstaele et al, 1993) and activation of the DRN during IS is critical in the potentiation of morphine-CPP, the DRN may be a proximate mediator in a more extended neural circuit regulating this phenomenon. Glutamatergic neurons from the ventral regions of the medial prefrontal cortex (mPFCv) project to the DRN (Peyron et al, 1998; Vertes, 2004), and synapse preferentially on GABAergic neurons within the DRN (Varga et al, 2001). Consistent with this anatomy, stimulation of the mPFCv inhibits DRN 5HT neuronal firing (Celada et al, 2001; Hajos et al, 1998). Interestingly, the mPFC is implicated in decision making, rule learning, appreciation of event significance, and goal-directed behaviors (Miller and Cohen, 2001). The mPFCv is also associated with affective- (Bremner et al, 2007; Killgore and Yurgelun-Todd, 2006; van Reekum et al, 2007) and addiction-related (Ballesteros-Yanez et al, 2007; Volkow et al, 2005) disorders.
Given the anatomy reviewed above and the proximal role of DRN 5HT in IS-induced behaviors, Amat et al (2005) investigated the role of the mPFCv in mediating the DRN changes that are produced by ES and IS. ES, but not IS, appeared to activate mPFCv output to the DRN, thereby reducing DRN 5HT activation. Thus, inactivation of mPFCv output by microinjection of the GABAA receptor agonist muscimol during ES led ES to produce the level of DRN 5HT activation and the behavioral deficits normally produced by IS. That is, having control did not reduce the DRN-activating effects of stress when the mPFCv could not be activated. Indeed, the mPFCv has also been implicated in stressor controllability studies examining fear conditioning (Baratta et al, 2007) and behavioral immunization (Amat et al, 2006).
Whether the mPFCv is a critical mediator of the effects of stressor controllability on morphine-CPP is unknown. Thus, in the present experiments the mPFCv was inactivated by microinjection of the GABAA receptor agonist muscimol. This procedure has been used in other experiments that have examined the role of the mPFCv in mediating the impact of stressor controllability on later escape learning and fear conditioning (Amat et al, 2005). The present study also employed a new approach. If inactivation of the mPFCv eliminates the protection afforded by behavioral control, then pharmacological activation of the mPFCv during uncontrollable stressor exposure might be expected to provide such protection. That is, IS should now no longer potentiate later morphine-CPP. The GABAA receptor antagonist picrotoxin has been used before to activate the mPFCv (Berretta et al, 2005), and so picrotoxin was used here.
MATERIALS AND METHODS
Subjects
Adult, male Sprague–Dawley rats (Harlan Inc., Indianapolis, IN) weighing 275–375 g were housed in pairs in Plexiglas cages with food and water available ad libitum. Rats were maintained in a climate-controlled colony room at 21°C on a 12 h light–dark cycle (lights on at 0700 hours), and all experiments were conducted during the light phase. Rats were allowed, at minimum, 1 week of acclimation prior to any procedures. All animal care and experimental procedures were in accord with protocols approved by the University of Colorado Institutional Animal Care and Use Committee.
Surgery and Cannulation
Surgery was performed under halothane (Halocarbon Laboratories, River Edge, NJ) or isoflurane (Webster Veterinary, Sterling, MA) anesthesia. Because of the shortage of halothane, rats in experiments 2 and 3 were anesthetized with isoflurane. All rats were implanted with a 26-gauge dual guide cannula (Plastics One, Roanoke, VA), 1 mm center-to-center distance. For the majority of the rats, the tips of the cannulae were aimed at 1 mm above the infralimbic/prelimbic (IL/PL) regions of the mPFCv: 2.7 mm rostral to bregma, 3.3 mm ventral from the dura mater, and 0.5 mm relative to midline (Paxinos, 1998). Rats used in site-specificity studies were implanted with cannulae aimed at the ventromedial orbital cortex (vmOC): 4.2 mm rostral to bregma, 3.0 mm ventral from the dura mater, and 0.5 mm relative to midline (Paxinos, 1998). Rats were allowed to recover 1–2 weeks before any experimentation.
Cannula Verification
At 24 h following an experiment rats were overdosed with sodium pentobarbital and brains were removed and rapidly frozen in chilled isopentane. A cryostat was used to section 40-μm brain slices, which were mounted on gelatin-treated slides and stained with Cresyl Violet for cannula placement verification. Data were only analyzed from animals whose cannulae were verified to terminate within the IL/PL (mPFCv placements) or vmOC (site-specificity controls). Four rats were eliminated due to cannula misplacement.
Drugs
All drugs were dissolved in 0.9% sterile saline according to required dose. Rats were microinjected with a volume of 0.5 μl of either saline, 100 ng of picrotoxin (Sigma Co., St Louis, MO), or 50 ng of muscimol (Sigma). Morphine sulfate (NIDA) was injected subcutaneously at a dose of 3.0 mg/kg for all CPP experiments at a volume of 1 ml/kg of body weight. This dose was used, as in prior studies (Will et al, 1998), to produce only minimal CPP in controls, thereby allowing facilitation to be observed.
Stressor Controllability
In a separate and distinctly different environment than the CPP environment, rats received tailshock in clear Plexiglas boxes measuring 14 cm × 11 cm × 17 cm and containing a wheel mounted on the front wall. Each rat's tail extended from the rear of the box and was taped to a Plexiglas rod. Two copper electrodes coated with electrode paste were affixed to the mid-section of the tail and shocks were delivered to yoked pairs (ES, IS) of rats using a Precision-Regulated Animal Shocker with Graphic State 3.0 software (Coulbourn Instruments, Allentown, PA). Stress sessions consisted of 80 trials of tailshock with an average intertrial interval of 60 s. This session was divided into 30 trials at 1.0 mA intensity, 30 trials at 1.3 mA, and 20 trials at 1.6 mA. This escalating shock intensity procedure maintains escape behavior in ES subjects. Tailshock was terminated for both ES and IS rats after the ES rat performed the required escape response. The initial response requirement involved a ¼ turn of the wheel. The response requirement increased a ½ turn when three consecutive trials were terminated in less than 5 s. Each subsequent response performed in less than 5 s resulted in a 50% increase in the response requirement until the maximum response requirement of four full wheel turns was achieved. If the operant response was not performed within 10 s at any time during the stress session, the response requirement was incrementally decreased. If the correct response was not performed within 30 s, shock was terminated and the response requirement returned to a ¼ turn. Prior studies indicate that restraint in the wheel-turn box does not potentiate morphine-CPP (Will et al, 1998), for this reason non-stressed homecage control (HC) rats remained undisturbed in the colony.
Microinjections
Drugs for microinjection were delivered via two 10 μl Hamilton microsyringes attached to Kopf Instruments (Tujunga, CA) Model 5000 microinjector. Polyethylene 50 tubing connected the microinjector apparatus to a dual 33-gauge microinjector (Plastics One), which was inserted into the cannula guides, extending 1 mm beyond the tip of the cannula. Injections were made by continuous infusion over a 1-min period. After infusion, the injector remained in place for 2 min. Microinjections were considered successful if upon removal of the microinjector from the guide cannula fluid was dispensable from the microinjector tip.
Conditioned Place Preference
The Plexiglas place preference apparatus measured 72 cm × 30 cm × 30 cm (length, width, and height) and consisted of three distinct environments, two conditioning, and a neutral area. Each conditioning environment measured 30 cm × 30 cm × 30 cm. The environments differed from each other both visually and tactilely. One conditioning environment was striped horizontally with alternating 2 cm black and white electrical tape on the walls, while the other conditioning environment was striped vertically in the same manner. The floor was black sanded Plexiglas with a 2 cm wire grid on the horizontal side and a 3 mm wire mesh on the vertical side. The neutral area measured 12 cm × 30 cm × 30 cm, was painted gray with no texturing. During the conditioning phase, vertically and horizontally striped Plexiglas partitions were inserted on the respective sides of the neutral area to restrict the rats to their conditioning environment. Rat activity was monitored by a Philips TC352A video camera (Lancaster, PA) mounted 1.5 m above the center of the CPP apparatus. The camera relayed information of the rat's location to the Chromotrack Version 4.02 tracking software (Prototype Systems Ltd., Boulder, CO), run on a PC-compatible computer in a separate room. A SA-3 tracker (San Diego Instruments, San Diego, CA) simultaneously measured the time spent by each rat within the three compartments, the distance traveled, and the number of crossings between the environments.
Prior to the experiment, subjects were handled and fitted with a plastic rat collar that fit loosely around the neck (BAS, West Lafayette, IN). A 1 cm × 2 cm piece of reflective tape affixed to the collar was used to track the subjects while they were in the apparatus. On day 1, between 1200 and 1330 hours, all subjects were initially placed in the neutral area and the time spent in each environment of the apparatus was recorded for 20 min. Day 1 served as an assessment of individual subject bias for a given environment, any subject spending less than 20% of total time in either conditioning environment was eliminated from the experiment. Thirty-one rats were eliminated due to bias. On day 2, animals were randomly assigned to receive either ES, IS, or HC control treatment. One hour prior to ES, IS, or HC control, subjects received either an intra-mPFCv or intra-vmOC microinjection of picrotoxin, muscimol, or saline. On day 3, rats were weighed in the morning and, using a counterbalanced conditioning procedure, were randomly assigned to conditioning environments. Conditioning occurred at 1030 and 1430 hours, lasting 45 min per session. All rats received morphine and saline during this day; half were administered s.c. morphine at 1030 hours, while the other half received equivolume of s.c. saline. All rats were injected within 3 min and then placed in the appropriate conditioning environment. At 1430 hours, the injections were alternated such that a rat that had received morphine at 1030 hours, received saline at 1430 hours. Day 4 conditioning was similar to day 3 conditioning, except the order in which the subject was presented morphine and saline on day 3 was reversed. Day 5 was the test of preference. In a drug-free state, subjects were placed in the neutral area and allowed to explore the entire CPP apparatus for 20 min. The length of the pre-exposure and conditioning phases of this CPP protocol were chosen because under these parameters and this dose of morphine, CPP is potentiated in IS, but not ES, rats (Will et al, 1998). The dependent variable for measuring preference for each subject was the difference in time spent in the drug-paired environment before drug conditioning sessions (day 1) and after drug conditioning (day 5). Thus, a positive score indicates a shift in preference for the drug-paired compartment. Locomotor and neutral area crossing data are expressed as a difference after and before conditioning; therefore, a positive number reflects the amount by which the measure was reduced.
Statistical Analysis
All data were expressed as mean±SEM. Data were analyzed by ANOVA to determine differences between groups. All statistically significant main effects and interactions were followed with a Student–Newman–Keuls post hoc test (α=0.05).
RESULTS
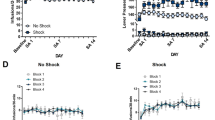
Figure 1 shows the cannula placements for both mPFCv and site-specificity control microinjections across experiments.

Placements of microinjection cannula. Numerals indicate distance from bregma (mm). Summary of microinjection cannula placements in ventral regions of the medial prefrontal cortex and ventromedial orbital cortex for all experiments. Ventromedial orbital cortex located at +4.2 mm from bregma. Medial prefrontal cortex located from +3.2 to +2.2 mm from bregma. Not all cannulae are shown due to overlapping placements. Medial orbital cortex (MO), ventral orbital cortex (VO), prelimbic cortex (PrL), and infralimbic cortex (IL).
Experiment 1: Intra-mPFCv Muscimol Administered during Stress Selectively Potentiates Morphine-CPP in Rats that Previously Experienced ES
Rats (n=7–8 per group) received an intra-mPFCv microinjection of either muscimol or saline 1 h prior to ES, IS, or HC and subsequently underwent morphine conditioning 24 h later. Importantly, intra-mPFCv microinjection of muscimol at this dose does not affect the learning of the escape response during stress exposure (Amat et al, 2005). The CPP data are presented in Figure 2a. As is typical, prior IS potentiated morphine-CPP and ES did not. The important new finding is that intra-mPFCv muscimol microinjection before stressor exposure did not alter the effects of IS, but now ES also potentiated morphine-CPP. A 3 × 2 ANOVA revealed a significant main effect of stress [F(2,41)=7.890, p<0.01], but no significant effect of microinjection or interaction between stressor treatment and microinjection. This failure to obtain a significant interaction is attributable to the fact that muscimol had no effect in either IS or HC subjects. Student–Newman–Keuls post hoc tests revealed that morphine-CPP in saline-microinjected groups did not differ between ES and HC subjects, but morphine-CPP was significantly elevated in rats that received IS. Post hoc tests also revealed that intra-mPFCv muscimol significantly potentiated morphine-CPP in ES rats, relative to saline ES and muscimol HC, but not muscimol IS rats. Lastly, post hoc tests revealed no effect of intra-mPFCv microinjection in IS and HC groups. The differences in locomotor activity after and before conditioning were calculated and are presented in Figure 3a. All groups showed a reduction in locomotor activity, but a 3 × 2 ANOVA revealed no significant interaction of stressor treatment and microinjection. The differences in the number of neutral area crossings after and before conditioning were calculated and are presented in Figure 4a. Again, all groups showed a reduction in neutral area crossings and a 3 × 2 ANOVA revealed no significant interaction.

Morphine-conditioned place preference (CPP) following escapable stress (ES), inescapable stress (IS), or homecage control (HC) treatments. Data are expressed as mean±SEM difference in time (s) spent on the drug-paired side before and after conditioning. Positive scores indicate a shift in preference for the drug-paired compartment. (a) Intra-mPFCv microinjection of the GABAA receptor agonist muscimol 1 h before stress potentiates morphine-CPP in rats that received ES (n=7–8 rats per group). *Different from muscimol-microinjected HC group (p<0.05). **Different from saline-microinjected ES and HC groups (p<0.05). #Different from saline-microinjected ES group (p<0.05). (b) Intra-mPFCv microinjection of the GABAA receptor antagonist picrotoxin 1 h before stress attenuates IS-induced potentiation of morphine-CPP (n=7–11 rats per group). *Different from all groups (p<0.05). (c) Intra-vmOC microinjection of the GABAA receptor agonist muscimol 1 h before stress did not potentiate morphine-CPP in rats that received ES (n=6–8 rats per group). *Different from rats that received intra-mPFCv muscimol 1 h prior to ES (p<0.05). (d) Intra-vmOC microinjection of the GABAA receptor antagonist picrotoxin 1 h before stress potentiated morphine-CPP in rats that received IS (n=7–9 rats per group). *Different from rats that received intra-mPFCv picrotoxin 1 h prior to IS (p<0.05).

Difference in locomotor behavior following escapable stress (ES), inescapable stress (IS), or homecage control (HC) treatments. Data are expressed as mean±SEM difference in distance traveled (cm) after and before conditioning. Positive scores represent the decrease in locomotor activity following stressor and conditioning phases. (a) Locomotor behavior is unaltered by intra-mPFCv muscimol 1 h before ES, IS, or HC treatments (n=7–8 rats per group). (b) Locomotor behavior is unaltered by intra-mPFCv picrotoxin 1 h before ES, IS, or HC treatments (n=7–11 rats per group). (c) Locomotor behavior is unaltered by intra-mPFCv or intra-vmOC muscimol 1 h before ES (n=6–8 rats per group). (d) Locomotor behavior is unaltered by intra-mPFCv or intra-vmOC picrotoxin 1 h before IS (n=7–9 rats per group).

Difference in the number of neutral zone crossings following escapable stress (ES), inescapable stress (IS), or homecage control (HC) treatments. Data are expressed as mean±SEM difference in neutral zone crossings after and before conditioning. Positive scores represent a decrease in the number of neutral zone crossings following stressor and conditioning phases. (a) The number of neutral zone crossings is unaltered by intra-mPFCv muscimol 1 h before ES, IS, or HC treatments (n=7–8 rats per group). (b) The number of neutral zone crossings is unaltered by intra-mPFCv picrotoxin 1 h before ES, IS, or HC treatments (n=7–11 rats per group). (c) The number of neutral zone crossings is unaltered by intra-mPFCv or intra-vmOC muscimol 1 h before ES (n=6–8 rats per group). (d) The number of neutral zone crossings is unaltered by intra-mPFCv or intra-vmOC picrotoxin 1 h before IS (n=7–9 rats per group).
Experiment 2: Intra-mPFCv Picrotoxin Administered during Stress Selectively Blunts Morphine-CPP in Rats that Previously Experienced IS
Rats (n=7–11 per group) received an intra-mPFCv microinjection of either picrotoxin or saline 1 h prior to ES, IS, or HC and subsequently underwent morphine conditioning 24 h later. The results are shown in Figure 2b. As above, IS but not ES potentiated subsequent morphine-CPP. Picrotoxin did not alter the protective effects of ES, but now IS no longer potentiated morphine-CPP. A 3 × 2 ANOVA revealed a significant main effect of stress [F(2,48)=4.383, p<0.05], but no significant effect of microinjection or interaction between stressor treatment and microinjection. As in experiment 1, Student–Newman–Keuls post hoc tests revealed that within saline-microinjected groups there was no CPP difference between ES and HC subjects, but a significant potentiation of morphine-CPP in IS rats. Post hoc tests revealed morphine-CPP in picrotoxin-microinjected IS rats was significantly reduced relative to saline-microinjected IS rats, but there was no effect of microinjection in ES or HC rats. No significant differences of morphine-CPP was found between ES, IS, and HC rats receiving intra-mPFCv picrotoxin. As shown in Figure 3b, locomotor activity after and before conditioning was reduced but a 3 × 2 ANOVA revealed no significant interaction of stressor treatment and microinjection. The differences in the number of neutral area crossings after and before conditioning were calculated and are shown in Figure 4b. Again, all groups showed a reduction in neutral area crossings and a 3 × 2 ANOVA revealed no significant interaction.
Experiment 3: Site-Specificity Control Microinjections do not Alter Morphine-CPP
Two site-specificity control studies were conducted. Rats were implanted with bilateral cannulae directed at the vmOC (see Materials and methods) and given the CPP protocol. ES rats received microinjections of muscimol (n=6–8 per group) and IS rats were microinjected with picrotoxin (n=7–9 per group) 1 h before stress. Only ES rats were tested with muscimol and IS rats with picrotoxin because mPFCv muscimol only altered the effects of ES and picrotoxin only altered the effects of IS. This strategy saved a large number of subjects. As shown in Figure 2c, rats receiving an intra-vmOC microinjection of muscimol demonstrated significantly less morphine-CPP compared to the intra-mPFCv-microinjected ES rats in experiment 1. That is, muscimol microinjected at the control site did not duplicate the effects of intra-mPFCv muscimol. A one-way ANOVA revealed a significant effect of microinjection location [F(1,12)=5.703, p<0.05). As shown in Figure 3c, locomotor activity was reduced in both groups and a one-way ANOVA revealed no significant difference. The number of neutral area crossings are shown in Figure 4c, both groups showed a reduction and a one-way ANOVA revealed no difference. As shown in Figure 2d, rats receiving an intra-vmOC microinjection of picrotoxin showed potentiated morphine-CPP compared to the intra-mPFCv-microinjected IS rats in Experiment 2. Again, picrotoxin microinjected at the control site did not duplicate the effects of intra-mPFCv picrotoxin. A one-way ANOVA revealed a significant effect of microinjection location [F(1,14)=6.139, p<0.05]. Changes in locomotor behavior and neutral area crossings are shown in Figures 3d and 4d, respectively. Both groups showed a reduction in distance traveled and neutral area crossings, but ANOVA tests revealed no significant differences.
DISCUSSION
The research presented here is the first to investigate the role of the mPFCv in mediating the effects of controllable and uncontrollable stress on subsequent morphine reward. The present study replicates the previous finding that a brief exposure to uncontrollable, but not controllable, tailshock potentiates morphine-CPP (Will et al, 1998). The results of these experiments demonstrate that regardless of stressor controllability, activation of the mPFCv during stress is both necessary and sufficient in blocking stress-induced potentiation of morphine-CPP. Indeed, the degree of activation of the mPFCv during stress exposure is a critical determinant of subsequent morphine-CPP. The microinjection of the GABAA receptor agonist muscimol into the mPFCv during stress led to potentiated morphine-CPP in ES subjects, whereas morphine-CPP in saline-microinjected ES subjects remained at the level of non-stressed controls. Interestingly, HC control subjects that received microinjections of either muscimol or saline exhibited similar morphine-CPP. This finding suggests that the stress-induced changes in the expression of morphine-CPP are indeed dependent upon the co-occurrence of stress and the activation state of the mPFCv. Conversely, pharmacological activation of the mPFCv during IS with the GABAA receptor antagonist picrotoxin significantly blunted potentiation of morphine-CPP, relative to IS subjects microinjected with saline. Additionally, microinjection of muscimol into the vmOC did not potentiate morphine-CPP in ES rats, suggesting that the mediation of the protective effects of ES against facilitated drug reward is specific to the mPFCv. Picrotoxin site-specificity controls also demonstrated that simple neural activation during stress was not sufficient for protection, but that activation had to be within the mPFCv. Furthermore, the stress-induced changes in morphine-CPP were not attributable to reductions in locomotor activity. Lastly, although pharmacological manipulations during IS and ES altered the CPP normally observed, we cannot conclude from these studies that the pharmacological manipulations used exactly mimic mPFCv inactivation/activation during the stress experience.
The present study utilized CPP, a widely used paradigm for measuring reward (Tzschentke, 2007), to assess differences in morphine reward due to stressor controllability. However, a number of interpretations for the IS-induced potentiation of morphine-CPP are possible. One interpretation is that morphine's rewarding properties have increased due to neuronal alterations following IS, but not ES and HC control, treatments. This interpretation is bolstered by neurochemical studies examining NAc DA following morphine injection in ES, IS, and HC rats (Bland et al, 2004). However, since CPP is a learning paradigm, another possibility is that IS rats simply condition better than do ES and HC controls, that is, the associative process itself is facilitated. This interpretation would posit that IS rats would also exhibit potentiated CPP to other drugs of abuse; however, IS-induced potentiation of CPP is opioid specific (Der-Avakian et al, 2007; Will et al, 1998). Lastly, another possible interpretation is that increases in CPP reflect novelty seeking rather than reward. The novelty-seeking confound can be assessed by using a CPP apparatus with three conditioning environments (Bardo and Bevins, 2000). Although the present study used a CPP apparatus with two conditioning environments, morphine-CPP using three conditioning environments has revealed that increased time spent in the drug-paired environment reflects reward rather than novelty seeking (Mucha and Iversen, 1984).
Previously, our laboratory has demonstrated that activation of the mPFCv during ES inhibits the stress-induced serotonergic response of the DRN (Amat et al, 2005). That is, aversive experiences activate DRN 5HT release, but when the mPFCv detects the presence of behavioral control over the aversive event, the DRN and other stress-sensitive brain structures are actively inhibited. Indeed, DRN 5HT efflux during IS, but not ES, is elevated almost 500% (Maswood et al, 1998). This serotonergic response characteristic of IS causes a downregulation of inhibitory 5HT1A autoreceptor mRNA in the DRN (Greenwood et al, 2003), thereby sensitizing DRN 5HT cells. Thus, activation of the mPFCv during stress either by the presence of behavioral control or by pharmacological manipulation was expected to eliminate sensitization of DRN serotonergic neurons.
Muscimol, a GABAA receptor agonist known to facilitate neuronal hyperpolarization in the rat brain (Andrews and Johnston, 1979; Edeline et al, 2002), has been previously used to inhibit the mPFCv during stress (Amat et al, 2005). Using the same dose of muscimol as in the present study, Amat et al (2005) demonstrated that intra-mPFCv muscimol potentiates DRN 5HT efflux in ES rats to the levels of that observed in IS rats that received only vehicle microinjections. Muscimol is a potent agonist of the GABAA receptor and can have long-lasting effects (Martin and Ghez, 1999). Nevertheless, the potentiation of morphine-CPP following intra-mPFCv muscimol in ES rats is unlikely due to a long-lasting presence of muscimol during CPP conditioning because muscimol-microinjected HC controls did not exhibit potentiated morphine-CPP. Picrotoxin, a GABAA receptor antagonist, has been shown to effectively suppress cortical inhibitory postsynaptic potentials (Connors et al, 1988; Yoon et al, 1993) and in turn inhibits subcortical structures (Berretta et al, 2005). Although picrotoxin can elevate cortical spiking in the rat brain for 12–24 h (Turski et al, 1985), this is not problematic because morphine conditioning in the present study occurred 24 h following stress. Additionally, our laboratory has investigated DRN 5HT efflux following intra-mPFCv picrotoxin during ES, IS, and HC control (Amat et al, 2008). At the same dose of picrotoxin used in this study, Amat et al (2008) found no potentiation of DRN 5HT efflux in rats that received intra-mPFCv picrotoxin during IS compared to vehicle microinjection during ES. However, the possibility of a long-term rebound effect, that is, increased GABAB receptor activation due to blockade of GABAA receptors (Bal et al, 1995), must be noted and cannot be ruled out in the present study. Lastly, the binding of muscimol and picrotoxin to other substrates besides the GABAA receptor has been documented (Das et al, 2003; Etter et al, 1999; Shen and Johnson, 2001); therefore, interpretive cautions regarding GABAA receptors in the mPFCv and behavioral outcomes of stressor controllability should be exercised.
Anatomical studies indicate that the mPFCv can influence the mesolimbic pathway. Glutamatergic efferents originating in the IL (Hurley et al, 1991) and PL (Vertes, 2004) cortices project to the NAc and the ventral tegmental area (VTA). Pyramidal cells from the mPFCv to the NAc appear to synapse on medium spiny GABA neurons (Carr and Sesack, 2000), with these GABAergic neurons then projecting to DA cells in the VTA (Sesack and Pickel, 1992). Projections from the mPFCv target GABA interneurons within the VTA, which project to both VTA DA cells and DA terminals in the NAc (Carr and Sesack, 2000) and also synapse directly on VTA DA cells (Omelchenko and Sesack, 2007). Additionally, the mPFCv can indirectly influence the VTA via ventral pallidal relays (Groenewegen et al, 1993; Heimer et al, 1991).
Although the anatomy described above is complex, studies using electrical and pharmacological stimulation provide a clearer account. Initial studies found that stimulation of the mPFC increases NAc DA (Karreman and Moghaddam, 1996; Taber and Fibiger, 1995; You et al, 1998). However, later experiments using more physiological levels of stimulation in the mPFC revealed a decrease in release of DA in the NAc (Jackson et al, 2001). Moreover, this circuitry appears to be tonically active as blockade of AMPA/kainate receptors in the VTA causes increases in NAc DA efflux (Takahata and Moghaddam, 2000). The implication of the present data is that morphine-induced inhibition of the mPFCv causes disinhibition of NAc DA; however, the pathway by which the mPFCv exerts this effect is unknown.
While the present study emphasizes inhibition of the mPFCv leading to potentiated morphine-CPP, inactivation of the dorsal mPFC has been implicated in prevention of both footshock-induced (McFarland et al, 2004) as well as cue-induced reinstatement of cocaine self-administration (McLaughlin and See, 2003). These data suggest that the modulatory role of the mPFC in drug-related paradigms is subregion and drug specific, with dorsal and ventral mPFC serving quite different functions. For example, morphine-CPP is attenuated by IL, but not PL or anterior cingulate, lesions and cocaine-CPP is blocked by PL, but not IL or anterior cingulate, lesions (Tzschentke and Schmidt, 1999); moreover, inactivation of the mPFCv, but not the dorsal mPFC, enhances spontaneous recovery of cocaine seeking (Peters et al, 2007). Although in the present study inactivation of the mPFCv is hypothesized to potentiate morphine-CPP due to changes induced by IS, the present experiments only address the role of mPFCv during the stress experience.
The results of the present experiments are consistent with studies examining the prefrontal cortex and addiction-related processes. Inhibition/hypoactivity of the mPFC has been linked to other psychiatric disorders (eg PTSD), resulting in reduced inhibitory control over limbic structures (Bremner, 2006). One proposed consequence of reduced mPFCv activity is a disinhibition of structures in the ventral striatum (Ernst et al, 2006). Reductions of prefrontal cortical activity have also been correlated with impulsive, perseverative, and deleterious behaviors (Dalley et al, 2004; Passetti et al, 2002; Soloff et al, 2003; Tanabe et al, 2007), which can predispose an individual to enter an addictive state (Jentsch and Taylor, 1999; Mitchell, 1999; Perry et al, 2005; Poulos et al, 1995). Similarly, as in the present experiments, experiential factors that behaviorally (or pharmacologically) inhibit the mPFCv following opioid administration result in a potentiation of approach behavior to previously neutral stimuli paired with morphine.
The present experiments demonstrate that activation of the mPFCv during the stress experience is a critical determinant in whether stress will produce long-term trans-situational changes in morphine reward. Activation of the mPFCv appears to be both necessary and sufficient to attenuate stress-induced potentiation of morphine-CPP. Our findings further suggest a role of the mPFCv in the complex interaction between stress and drugs of abuse. It is possible that an activated mPFCv is responsible for both the detection of stressor controllability and the subsequent dampening of stress-activated neural structures as well as maintaining tonic inhibition of the mesolimbic pathway.
References
Amat J, Baratta MV, Paul E, Bland ST, Watkins LR, Maier SF (2005). Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat Neurosci 8: 365–371.
Amat J, Matus-Amat P, Watkins LR, Maier SF (1998a). Escapable and inescapable stress differentially alter extracellular levels of 5-HT in the basolateral amygdala of the rat. Brain Res 812: 113–120.
Amat J, Matus-Amat P, Watkins LR, Maier SF (1998b). Escapable and inescapable stress differentially and selectively alter extracellular levels of 5-HT in the ventral hippocampus and dorsal periaqueductal gray of the rat. Brain Res 797: 12–22.
Amat J, Paul E, Watkins LR, Maier SF (2008). Activation of the ventral medial prefrontal cortex during an uncontrollable stressor reproduces both the immediate and long-term protective effects of behavioral control (submitted).
Amat J, Paul E, Zarza C, Watkins LR, Maier SF (2006). Previous experience with behavioral control over stress blocks the behavioral and dorsal raphe nucleus activating effects of later uncontrollable stress: role of the ventral medial prefrontal cortex. J Neurosci 26: 13264–13272.
Andrews PR, Johnston GA (1979). GABA agonists and antagonists. Biochem Pharmacol 28: 2697–2702.
Bal T, von Krosigk M, McCormick DA (1995). Role of the perigeniculate nucleus in the generation of synchronized oscillations in vitro. J Physiol 483: 665–685.
Ballesteros-Yanez I, Ambrosio E, Benavides-Piccione R, Perez J, Torres I, Miguens M et al (2007). The effects of morphine self-administration on cortical pyramidal cell structure in addiction-prone Lewis rats. Cereb Cortex 17: 238–249.
Baratta MV, Christianson JP, Gomez DM, Zarza CM, Amat J, Masini CV et al (2007). Controllable versus uncontrollable stressor bi-directionally conditioned but not innate fear. Neuroscience 146: 1495–1503.
Bardo MT, Bevins RA (2000). Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl) 153: 31–43.
Berretta S, Pantazopoulos H, Caldera M, Pantazopoulos P, Pare D (2005). Infralimbic cortex activation increases c-Fos expression in intercalated neurons of the amygdala. Neuroscience 132: 943–953.
Bland ST, Hargrave D, Pepin JL, Amat J, Watkins LR, Maier SF (2003). Stressor controllability modulates stress-induced dopamine and serotonin efflux and morphine-induced serotonin efflux in the medial prefrontal cortex. Neuropsychopharmacology 28: 1589–1596.
Bland ST, Twining C, Schmid MJ, Der-Avakian A, Watkins LR, Maier SF (2004). Stress potentiation of morphine-induced dopamine efflux in the nucleus accumbens shell is dependent upon stressor uncontrollability and is mediated by the dorsal raphe nucleus. Neuroscience 126: 705–715.
Bremner JD (2006). Traumatic stress: effects on the brain. Dialogues Clin Neurosci 8: 445–461.
Bremner JD, Elzinga B, Schmahl C, Vermetten E (2007). Structural and functional plasticity of the human brain in posttraumatic stress disorder. Prog Brain Res 167: 171–186.
Carr DB, Sesack SR (2000). Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci 20: 3864–3873.
Celada P, Puig MV, Casanovas JM, Guillazo G, Artigas F (2001). Control of dorsal raphe serotonergic neurons by the medial prefrontal cortex: involvement of serotonin-1A, GABA(A), and glutamate receptors. J Neurosci 21: 9917–9929.
Connors BW, Malenka RC, Silva LR (1988). Two inhibitory postsynaptic potentials, and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. J Physiol 406: 443–468.
Dalley JW, Cardinal RN, Robbins TW (2004). Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev 28: 771–784.
Das P, Bell-Horner CL, Machu TK, Dillon GH (2003). The GABAA receptor antagonist picrotoxin inhibits 5-hydroxytryptamine type 3A receptors. Neuropharmacology 44: 431–438.
Der-Avakian A, Bland ST, Rozeske RR, Tamblyn JP, Hutchinson MR, Watkins LR et al (2007). The effects of a single exposure to uncontrollable stress on the subsequent conditioned place preference responses to oxycodone, cocaine, and ethanol in rats. Psychopharmacology (Berl) 191: 909–917.
DuMont KA, Widom CS, Czaja SJ (2007). Predictors of resilience in abused and neglected children grown-up: the role of individual and neighborhood characteristics. Child Abuse Negl 31: 255–274.
Edeline JM, Hars B, Hennevin E, Cotillon N (2002). Muscimol diffusion after intracerebral microinjections: a reevaluation based on electrophysiological and autoradiographic quantifications. Neurobiol Learn Mem 78: 100–124.
Ernst M, Pine DS, Hardin M (2006). Triadic model of the neurobiology of motivated behavior in adolescence. Psychol Med 36: 299–312.
Etter A, Cully DF, Liu KK, Reiss B, Vassilatis DB, Schaeffer JM et al (1999). Picrotoxin blockade of invertebrate glutamate gated chloride channels: subunit dependence and evidence for binding within the pore. J Neurochem 72: 318–326.
Grahn RE, Will MJ, Hammack SE, Maswood S, McQueen MB, Watkins LR et al (1999). Activation of serotonin-immunoreactive cells in the dorsal raphe nucleus in rats exposed to an uncontrollable stressor. Brain Res 826: 35–43.
Greenwood BN, Foley TE, Day HE, Campisi J, Hammack SH, Campeau S et al (2003). Freewheel running prevents learned helplessness/behavioral depression: role of dorsal raphe serotonergic neurons. J Neurosci 23: 2889–2898.
Groenewegen HJ, Berendse HW, Haber SN (1993). Organization of the output of the ventral striatopallidal system in the rat: ventral pallidal efferents. Neuroscience 57: 113–142.
Hajos M, Richards CD, Szekely AD, Sharp T (1998). An electrophysiological and neuroanatomical study of the medial prefrontal cortical projection to the midbrain raphe nuclei in the rat. Neuroscience 87: 95–108.
Harrison PA, Fulkerson JA, Beebe TJ (1997). Multiple substance use among adolescent physical and sexual abuse victims. Child Abuse Negl 21: 529–539.
Heimer L, Zahm DS, Churchill L, Kalivas PW, Wohltmann C (1991). Specificity in the projection patterns of accumbal core and shell in the rat. Neuroscience 41: 89–125.
Hurley KM, Herbert H, Moga MM, Saper CB (1991). Efferent projections of the infralimbic cortex of the rat. J Comp Neurol 308: 249–276.
Jackson ME, Frost AS, Moghaddam B (2001). Stimulation of prefrontal cortex at physiologically relevant frequencies inhibits dopamine release in the nucleus accumbens. J Neurochem 78: 920–923.
Jentsch JD, Taylor JR (1999). Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology (Berl) 146: 373–390.
Karreman M, Moghaddam B (1996). The prefrontal cortex regulates the basal release of dopamine in the limbic striatum: an effect mediated by ventral tegmental area. J Neurochem 66: 589–598.
Killgore WD, Yurgelun-Todd DA (2006). Ventromedial prefrontal activity correlates with depressed mood in adolescent children. Neuroreport 17: 167–171.
Koob GF, Le Moal M (1997). Drug abuse: hedonic homeostatic dysregulation. Science 278: 52–58.
Maier SF, Busch CR, Maswood S, Grahn RE, Watkins LR (1995a). The dorsal raphe nucleus is a site of action mediating the behavioral effects of the benzodiazepine receptor inverse agonist DMCM. Behav Neurosci 109: 759–766.
Maier SF, Grahn RE, Watkins LR (1995b). 8-OH-DPAT microinjected in the region of the dorsal raphe nucleus blocks and reverses the enhancement of fear conditioning and interference with escape produced by exposure to inescapable shock. Behav Neurosci 109: 404–412.
Maier SF, Kalman BA, Grahn RE (1994). Chlordiazepoxide microinjected into the region of the dorsal raphe nucleus eliminates the interference with escape responding produced by inescapable shock whether administered before inescapable shock or escape testing. Behav Neurosci 108: 121–130.
Maier SF, Seligman MEP (1976). Learned helplessness: theory and evidence. J Exp Psychol Gen 105: 3–46.
Martin JH, Ghez C (1999). Pharmacological inactivation in the analysis of the central control of movement. J Neurosci Methods 86: 145–159.
Maswood S, Barter JE, Watkins LR, Maier SF (1998). Exposure to inescapable but not escapable shock increases extracellular levels of 5-HT in the dorsal raphe nucleus of the rat. Brain Res 783: 115–120.
McFarland K, Davidge SB, Lapish CC, Kalivas PW (2004). Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci 24: 1551–1560.
McLaughlin J, See RE (2003). Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 168: 57–65.
Miller EK, Cohen JD (2001). An integrative theory of prefrontal cortex function. Annu Rev Neurosci 24: 167–202.
Mitchell SH (1999). Measures of impulsivity in cigarette smokers and non-smokers. Psychopharmacology (Berl) 146: 455–464.
Mucha RF, Iversen SD (1984). Reinforcing properties of morphine and naloxone revealed by conditioned place preference: a procedural examination. Psychopharmacology (Berl) 82: 241–247.
Omelchenko N, Sesack SR (2007). Glutamate synaptic inputs to ventral tegmental area neurons in the rat derive primarily from subcortical sources. Neuroscience 146: 1259–1274.
Passetti F, Chudasama Y, Robbins TW (2002). The frontal cortex of the rat and visual attentional performance: dissociable functions of distinct medial prefrontal subregions. Cereb Cortex 12: 1254–1268.
Paxinos GWC (1998). The Rat Brain in Stereotaxic Coordinates, 4th edn. Academic Press: San Diego.
Perry JL, Larson EB, German JP, Madden GJ, Carroll ME (2005). Impulsivity (delay discounting) as a predictor of acquisition of IV cocaine self-administration in female rats. Psychopharmacology (Berl) 178: 193–201.
Peters J, Vallone J, Laurendi K, Kalivas PW (2007). Opposing roles for the ventral prefrontal cortex and the basolateral amygdala on the spontaneous recovery of cocaine-seeking in rats. Psychopharmacology (Berl) E-pub ahead of print.
Peyron C, Petit JM, Rampon C, Jouvet M, Luppi PH (1998). Forebrain afferents to the rat dorsal raphe nucleus demonstrated by retrograde and anterograde tracing methods. Neuroscience 82: 443–468.
Poulos CX, Le AD, Parker JL (1995). Impulsivity predicts individual susceptibility to high levels of alcohol self-administration. Behav Pharmacol 6: 810–814.
Sesack SR, Pickel VM (1992). Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol 320: 145–160.
Shen KZ, Johnson SW (2001). Potentiation of GABA(A) receptor agonists by GABA uptake inhibitors in the rat ventral midbrain. Eur J Pharmacol 428: 1–7.
Sinha R, Catapano D, O’Malley S (1999). Stress-induced craving and stress response in cocaine dependent individuals. Psychopharmacology (Berl) 142: 343–351.
Soloff PH, Meltzer CC, Becker C, Greer PJ, Kelly TM, Constantine D (2003). Impulsivity and prefrontal hypometabolism in borderline personality disorder. Psychiatry Res 123: 153–163.
Taber MT, Fibiger HC (1995). Electrical stimulation of the prefrontal cortex increases dopamine release in the nucleus accumbens of the rat: modulation by metabotropic glutamate receptors. J Neurosci 15 (5 Part 2): 3896–3904.
Takahata R, Moghaddam B (2000). Target-specific glutamatergic regulation of dopamine neurons in the ventral tegmental area. J Neurochem 75: 1775–1778.
Tanabe J, Thompson L, Claus E, Dalwani M, Hutchison K, Banich MT (2007). Prefrontal cortex activity is reduced in gambling and nongambling substance users during decision-making. Hum Brain Mapp 28: 1276–1286.
Turski WA, Cavalheiro EA, Calderazzo-Filho LS, Kleinrok Z, Czuczwar SJ, Turski L (1985). Injections of picrotoxin and bicuculline into the amygdaloid complex of the rat: an electroencephalographic, behavioural and morphological analysis. Neuroscience 14: 37–53.
Tzschentke TM (2007). Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol 12: 227–462.
Tzschentke TM, Schmidt WJ (1999). Functional heterogeneity of the rat medial prefrontal cortex: effects of discrete subarea-specific lesions on drug-induced conditioned place preference and behavioural sensitization. Eur J Neurosci 11: 4099–4109.
Van Bockstaele EJ, Biswas A, Pickel VM (1993). Topography of serotonin neurons in the dorsal raphe nucleus that send axon collaterals to the rat prefrontal cortex and nucleus accumbens. Brain Res 624: 188–198.
van Reekum CM, Urry HL, Johnstone T, Thurow ME, Frye CJ, Jackson CA et al (2007). Individual differences in amygdala and ventromedial prefrontal cortex activity are associated with evaluation speed and psychological well-being. J Cogn Neurosci 19: 237–248.
Varga V, Szekely AD, Csillag A, Sharp T, Hajos M (2001). Evidence for a role of GABA interneurones in the cortical modulation of midbrain 5-hydroxytryptamine neurones. Neuroscience 106: 783–792.
Vertes RP (2004). Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse 51: 32–58.
Volkow ND, Wang GJ, Ma Y, Fowler JS, Wong C, Ding YS et al (2005). Activation of orbital and medial prefrontal cortex by methylphenidate in cocaine-addicted subjects but not in controls: relevance to addiction. J Neurosci 25: 3932–3939.
Weiss JM, Goodman PA, Losita BA, Corrigan S, Charry JM, Bailey WH (1981). Behavioral depression produced by an uncontrollable stressor: relationship to norepinephrine, dopamine, and serotonin levels in various regions of rat brain. Brain Res Rev 3: 36–97.
Will MJ, Der-Avakian A, Bland ST, Grahn RE, Hammack SE, Sparks PD et al (2004). Electrolytic lesions and pharmacological inhibition of the dorsal raphe nucleus prevent stressor potentiation of morphine conditioned place preference in rats. Psychopharmacology (Berl) 171: 191–198.
Will MJ, Watkins LR, Maier SF (1998). Uncontrollable stress potentiates morphine's rewarding properties. Pharmacol Biochem Behav 60: 655–664.
Yoon KW, Covey DF, Rothman SM (1993). Multiple mechanisms of picrotoxin block of GABA-induced currents in rat hippocampal neurons. J Physiol 464: 423–439.
You ZB, Tzschentke TM, Brodin E, Wise RA (1998). Electrical stimulation of the prefrontal cortex increases cholecystokinin, glutamate, and dopamine release in the nucleus accumbens: an in vivo microdialysis study in freely moving rats. J Neurosci 18: 6492–6500.
Acknowledgements
This research was supported by US National Institute on Drug Abuse grants DA013159 (SFM) and DA023329 (RRR).
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
We declare that, except for income received from our primary employers and the above-mentioned funding, no further financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Rozeske, R., Der-Avakian, A., Bland, S. et al. The Medial Prefrontal Cortex Regulates the Differential Expression of Morphine-Conditioned Place Preference Following a Single Exposure to Controllable or Uncontrollable Stress. Neuropsychopharmacol 34, 834–843 (2009). https://doi.org/10.1038/npp.2008.34
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2008.34
Keywords
This article is cited by
-
Distinct Effects of Social Stress on Working Memory in Obsessive-Compulsive Disorder
Neuroscience Bulletin (2021)
-
Stress in adolescence and drugs of abuse in rodent models: Role of dopamine, CRF, and HPA axis
Psychopharmacology (2014)
-
The Activation of NMDA Receptor–ERK Pathway in the Central Amygdala is Required for the Expression of Morphine-Conditioned Place Preference in the Rat
Neurotoxicity Research (2011)
