
Abstract
A rapid method for temporally and spatially controlled CRISPR-mediated gene knockout in vertebrates will be an important tool to screen for genes involved in complex biological phenomena like regeneration. Here we show that in vivo injection of CAS9 protein–guide RNA (gRNA) complexes into the spinal cord lumen of the axolotl and subsequent electroporation leads to comprehensive knockout of Sox2 gene expression in SOX2+ neural stem cells with corresponding functional phenotypes from the gene knockout. This is particularly surprising considering the known prevalence of RNase activity in cerebral spinal fluid, which apparently the CAS9 protein protects against. The penetrance/efficiency of gene knockout in the protein-based system is far higher than corresponding electroporation of plasmid-based CRISPR systems. We further show that simultaneous delivery of CAS9–gRNA complexes directed against Sox2 and GFP yields efficient knockout of both genes in GFP-reporter animals. Finally, we show that this method can also be applied to other tissues such as skin and limb mesenchyme. This efficient delivery method opens up the possibility for rapid in vivo genetic screens during axolotl regeneration and can in principle be applied to other vertebrate tissue systems.
Similar content being viewed by others

Introduction
The advent of CRISPR (clustered regularly interspaced short palindromic repeats) technology has transformed the prospects of studying gene function via gene deletion in numerous cell-based and biological systems.1–3 The development of diverse delivery and expression systems have been applied to generate knockout cell lines or to develop comprehensive screening systems in cultured cells, while direct injection of Cas9 mRNA or protein with associated gRNAs have been used to elicit efficient gene knockout during embryogenesis with germline transmission.4–6
Recently, a tissue-specific Cas9 expression system for somatic tissue gene knockout was described in zebrafish.7 Using transgenesis to induce tissue-specific knockouts is a powerful method to study tissue-specific gene function, but the time scales of generating transgenic animals could present some limitations for rapidly screening larger gene sets. Furthermore, depending on the temporal and spatial specificity of the promoter used, expression via a given promoter may initiate the gene knockout earlier than desired or in unwanted tissues. Therefore, a rapid, acute, means to spatially and temporally direct gene knockout would be valuable for studying genetically modified cells in somatic tissues and for screening larger gene sets for functional analysis in somatic tissues.
Here, we find that injection of CAS9 protein–gRNA complexes targeting Sox2 or GFP into the cerebral spinal fluid of the axolotl (Ambystoma mexicanum) spinal cord followed by electroporation yields comprehensive knockout of Sox2 or GFP in the SOX2+ neural stem cells (NSCs) of the axolotl spinal cord with the gene knockout being inherited after tail amputation in the proliferative spinal cord NSCs. Comparison to electroporation of plasmid-based delivery systems showed that the protein-based systems yielded a much higher efficiency in producing cells with the knockout phenotype. The regeneration phenotype of these Sox2-gRNA-electroporated spinal cords shows remarkable similarity to those animals in which Sox2 had been comprehensively deleted via injection of the mixture of Cas9 mRNA and gRNAs in eggs, showing high effectiveness of the electroporation method. Co-electroporation of CAS9&GFP–gRNA and CAS9&Sox2–gRNA complexes showed no decrease in the efficiency and distribution of individual gene knockout in the targeted cells, opening an alternative and easy way for multiplex gene knockouts to study gene function. Electroporation could also be used to elicit gene knockout in other tissues such as satellite cells, bone, dermal mesenchymal and epidermis showing the general utility of this method.
Results
Electroporation of CAS9 protein–gRNA complexes yields more efficient knockout of a GFP-reporter gene compared with Cas9/gRNA-expression vectors
Delivery of expression vectors via electroporation has gained broad utility for studying vertebrate organogenesis at temporal and spatial controlled manner, such as during axolotl regeneration, zebrafish fin regeneration, mouse brain development and chicken development.8–11 Therefore, we tested electroporation as a method to efficiently express CRISPR components in somatic tissues to obtain tissue-specific knockout phenotypes. The axolotl spinal cord represents a tissue context in which a defined set of cells lining the lumen, and definable by SOX2+ expression, have been shown to take up plasmids at high efficiency and therefore represented a good system to test whether electroporation of plasmids encoding Cas9 and gRNA, or even electroporation of CAS9 protein–gRNA complexes directly would yield high-efficiency knockout in somatic cells.
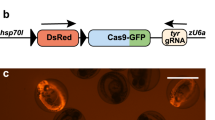
We first assessed the efficiency of GFP gene knockout after electroporation of a recently described vector-based system for Cas9/gRNAs expression,12 versus the electroporation of CAS9 protein–gRNA complexes. We injected the pCAGGS-Cas9 and CMV-dsRed-RGR-GFP-gRNA12 plasmids (for details see materials and methods) against GFP into the spinal cord lumen of the ubiquitous GFP-expressing transgenic axolotl13 followed by a rapid series of square electrical pulses, with the electrodes placed in two orientations (Figure 1a). The delivery of CRISPR plasmids (Figure 1b) yielded a knockdown of GFP expression in ~60% of dsRed-expressing (Figure 1c,d), a tracer gene in the gRNA-expression cassette (Figure 1b, lower panel), SOX2+ spinal cord NSCs in the CAGGS-GFP transgenic animals. The cells lacking GFP expression corresponded closely to those cells expressing the dsRed. (Figure 1c). However, within the entire spinal cord in the electroporated region only ~20% of SOX2+ NSCs lose GFP expression (Figure 1e) due to the presence of many cells that showed no expression of the plasmids. This observation indicated that the electroporation efficiency of the relatively large vector (pCAGGS-Cas9≈10 kb) into the nucleus of the SOX2+ NSCs might be the main determinant limiting the percentage of cells that could be genetically modified.

Knockout of GFP in the axolotl spinal cord NSCs through CRISPR plasmid electroporation. (a) A scheme of the axolotl spinal cord electroporation procedure. (b) Structure of CRISPR plasmids, pCAGGS-Cas9 (top) and CMV-dsRed-RGR-gRNA (bottom) used for electroporation. HDV, hepatitis delta virus ribozyme; HH, hammerhead ribozyme; NLS, nuclear localisation signal; pA, polyadenylation signal. (c) Fluorescence images of dsRed (red), GFP (green), immunofluorescence for SOX2 (white) combined with DAPI (blue) on 10-μm cryosections showing the loss of GFP expression in a subpopulation of dsRed-labelled spinal cord cells (arrows) at 20 days post electroporation of pCAGGS-Cas9 and CMV-dsRed-RGR-GFP-gRNA plasmids (pCAGGS-Cas9+RGR-GFP-gRNA, upper panel) compared with control (only RGR-GFP-gRNA plasmid, lower panel). Dotted lines define the spinal cord area. Scale bar, 100 μm. (d and e) Quantification of the percentage of (d) GFP+ cells over total electroporated dsRed+ SOX2+ spinal cord cells and (e) GFP+ cells over total spinal cord SOX2+ NSCs. Loss of GFP expression is observed in ~60% of NSCs that electroporated with the pCAGGS-Cas9 and CMV-dsRed-RGR-GFP-gRNA plasmids (Cas9+RGR-GFP-gRNA) compared with control (Ctr, only CMV-dsRed-RGR-GFP-gRNA plasmid), but in only ~20% cells over total spinal cord SOX2+ NSCs. Data are collected from three CRISPR plasmids-electroporated animals and three controls (four to five cross-cryosections per animals).
Given the limitations of plasmid delivery, we next attempted to deliver CAS9 protein–gRNA complexes via electroporation. These entities are much smaller than the corresponding plasmids and only need to reach the cytoplasm to be effective, so we hypothesised that a higher percentage of SOX2+ cells would receive the complexes and become genetically modified. A question was whether the high activity of RNases found in the cerebral spinal fluid, which normally prevents the effective electroporation of unprotected mRNAs, would confound this strategy or whether the CAS9 protein would protect the gRNAs from rapid destruction. Remarkably, electroporation of CAS9&GFP–gRNA complexes yielded a near complete loss of GFP expression (~90%) in SOX2+ spinal cord NSCs of CAGGS-GFP transgenic axolotls at 15-day post electroporation (Supplementary Figure S1A,B).
We further checked whether the knockout of the neutral GFP transgene in NSCs, via CAS9–gRNA electroporation, would affect axolotl spinal cord regeneration. In our CAGGS-GFP transgenic animals, the regenerating spinal cord can be visualised in whole-mount DIC-like stereomicroscopy via its contrast properties and also in the GFP fluorescence channel for its high-GFP expression of the densely packed spinal cord tissue (Figure 2a, right panel, red arrows). In line with the highly efficient GFP knockout in the developing spinal cord NSCs, we found that the GFP-signal in the regenerating spinal cord was no longer visible in the CAS9&GFP–gRNA-treated animals (Figure 2a, left, red arrows; Supplementary Figure S1C). Measuring of the length of 6-day-regenerated spinal cord and tails showed that CAS9&GFP–gRNA complexes do not delay regeneration process (Figure 2b). Cross-sections along the 6-day regenerating tail demonstrated that the ‘mature’ part (resembling the uninjured portion of the spinal cord anterior to the amputation plan) and newly regenerated spinal cord of CAS9&GFP–gRNA complexes-electroporated animals displayed a normal structure and harboured a comparable number of cells to control transfected spinal cord (Figure 2c,d). However, fluorescence imaging in the GFP channel showed that in the CAS9&GFP–gRNA-transduced spinal cord SOX2+ cells specifically lacked GFP expression (Figure 2c). Quantification along the mature and regenerating portions of the tail showed a consistent percentage of SOX2+ cells lacking GFP expression. In a series of sections along the mature and regenerating spinal cord, the percentage of SOX2+ cells that retained GFP expression (no knockout) constantly ranged from 5.5 to 12.4% (Figure 2e), indicating the normal contribution of GFP− NSCs to regenerated spinal cord. PCR cloning and sequencing of the GFP genomic locus of CAS9&GFP–gRNA-treated spinal cord confirmed that majority of the cloned PCR products include modifications at the target site (Supplementary Figure S1D). Our above results showed that electroporation of CAS9 protein–gRNA complexes is a very effective and non-toxic approach to knockout the GFP transgene in axolotl spinal cord NSCs.

Knockout of GFP in the axolotl spinal cord NSCs through CAS9–gRNA complex electroporation. (a) DIC (upper panel) and GFP fluorescence (middle panel) images of 6-day regenerates from CAGGS-GFP transgenic axolotls injected with CAS9&GFP–gRNA (left panel) or control (CAS9&Tyr-gRNA, right panel) into the spinal cord lumen and then electroporation. The regenerating spinal cord area, depicted by rectangles, is shown at a higher magnification in the lower panel. Note the clear loss of GFP expression in the regenerating spinal cord tube treated with CAS9&GFP–gRNA as compared with the control. In the CAGGS-GFP transgenic axolotls, GFP is expressed at a much higher level in the mature muscle than in other tissues or in the newly formed immature myofibers. Dotted lines indicate the amputation planes; arrows indicate the spinal cord; dpA, day post amputation. Scale bar, 1 mm. (b) Quantification of the length of 6-day-regenerated spinal cord (solid shapes) and tails (empty shapes) electroporated with CAS9&GFP–gRNA (triangles, n=20) or control CAS9&Tyr-gRNA (Ctr, diamonds, n=12). Error bars, s.d. (c) GFP fluorescence (green) and immunofluorescence for SOX2 (red) combined with DAPI (blue) on 10-μm cross-cryosections show the massive loss of GFP expression in NSCs along the spinal cord from mature ('M') to regenerated ('R') regions of 6-day tail regenerates treated with CAS9&GFP–gRNA compared with the control. The spinal cord areas (squares) are showed at higher magnification as single channel or merged images. Scale bar, 200 μm. (d and e) Quantification of (d) the number of nuclei per cross-section in the tubular spinal cord and (e) the percentage of GFP+ SOX2+ cells over total SOX2+ spinal cord cells of 6-day regenerates at different positions along their length from axolotls treated with CAS9&GFP–gRNA (triangles, n=5) or CAS9&Tyr-gRNA control (diamonds, n=5). Each scale on the x-axis represents ~800 μm of length. M, mature uninjured tail region anterior to the amputation plane; R, regenerate posterior to the amputation plane. Error bars, s.d.; ***P<0.001.
Efficient and broad knockout of the Sox2 gene in spinal cord after electroporation
The results from our GFP-reporter animal, which contains multiple GFP copies in the genome. suggested that the electroporation approach may be useful to generate homozygous knockouts of endogenous genes in the spinal cord. We recently characterised the penetrant knockout of the axolotl Sox2 gene after injection of embryos with Cas9 mRNA/gRNAs. Animals harbouring widespread loss of SOX2 expression survived to larval stages, and amputation of tails showed impaired amplification of SOX2+ cells during spinal cord regeneration.14 We next asked if electroporation of CAS9 protein–gRNA complexes made with the same Sox2–gRNAs would yield knockout of SOX2 expression and a similar phenotype.
Indeed, after electroporation of CAS9&Sox2–gRNA complexes, we found loss of SOX2 expression along the spinal cord, with occasional cells retaining expression (Figure 3a; Supplementary Figure S2A). Quantification of SOX2 immunofluorescence in electroporated animals showed that most of the animals displayed knockout for SOX2 expression in >75% of luminal, TUJ1-negative spinal cord cells (Figure 3b). Amputation of the tail of axolotls treated with CAS9 protein–Sox2–gRNA complexes resulted in an impaired outgrowth of the ependymal tube into the tail blastema, with a depletion of spinal cord cells over the length of the regenerate, similar to the phenotype observed when knocking out Sox2 from embryonic stages (Figure 3c, arrows; Figure 3d–f).14 PCR cloning and sequencing of the Sox2 genomic locus of CAS9&Sox2–gRNA-transfected spinal cord showed that majority of the cloned PCR products include modifications at the target site (Supplementary Figure S2B). In contrast, CRISPR plasmid electroporation only led to very low penetrance of Sox2 knockout (Supplementary Figure S2C–E). These results showed that through electroporation we can achieve penetrant enough knockout of spinal cord NSCs to elicit functional phenotypes.

Knockout of Sox2 in the axolotl spinal cord NSCs through CAS9–gRNA complex electroporation. (a) Immunofluorescence for SOX2 (red) and TUJ1 (green) combined with DAPI (blue) on cross-sections show a massive loss of SOX2 immunoactivity in NSCs adjacent to the amputation plane when electroporated with CAS9&Sox2–gRNA (upper panel) compared with CAS9&Tyr-gRNA control (lower panel) at 15 days post electroporation (dpE). The spinal cord areas (squares) are shown at higher magnification as single channel or merged images. Note that SOX2 (labelling NSCs) and TUJ1 (labelling neurons) expression are mutually exclusive in the control spinal cord (lower panel). Arrows indicate the SOX2+ cells in the lateral line. Scale bar, 100 μm. (b) Quantification of the percentage of SOX2+ cells over total non-neuronal (TUJ1−) spinal cord cells adjacent to the amputation plane from axolotls electroporated with CAS9&Sox2–gRNA (circles, n=21) or CAS9&Tyr-gRNA control (diamonds, n=21). Error bars, s.d.; ***P<0.001. (c) Bright field (BF) images of 6-day regenerates. Left: CAS9&Sox2–gRNA; right: CAS9&Tyr-gRNA control. Lower panels show higher magnification images of regenerating spinal cord area (rectangles). In CAS9&Sox2–gRNA-treated axolotls (left lower panel), the spinal cord tube (arrows) that extends into the blastema is significantly shorter and not so clear compared with the control (right lower panel). Dashed lines indicate the amputation planes. Scale bar, 1 mm. (d) Quantification of the length of 6-day-regenerated spinal cord (solid shapes) and tails (empty shapes) electroporated with CAS9&Sox2–gRNA (circles, n=22) or CAS9&Tyr-gRNA control (Ctr, diamonds, n=12). The control data set is same as in Figure 1c. Note that although there is no significant difference of the overall length of the tail, the length of regenerated spinal cord reduced significantly when treated with CAS9&Sox2–gRNA compared with the control. Error bars, s.d.; ***P<0.001. (e) Immunofluorescence for SOX2 (red) and GFAP (green) combined with DAPI on cross-sections along the spinal cord from mature ('M') to regenerated ('R') region of 6-day tail regenerates electroporated with CAS9&Sox2–gRNA (upper panel) or CAS9&Tyr-gRNA control (lower panel). Right lane is the lower-magnification image showing an overview of the tail structure. Note that in the tail blastema, knockout of Sox2 causes reduced cell number in the spinal cord of the CAS9&Sox2–gRNA-treated animals compared with the control. Towards the end of the regenerate ('R2') in the CAS9&Sox2–gRNA-treated animals, GFAP+ spinal cord cells are intermingled within the mesenchyme cells, instead of organising into a tubular structure as in the control. Scale bars, 100 μm. (f) Quantification of the number of nuclei per cross-section in the tubular spinal cord of the 6-day regenerate at different positions along its length from axolotls treated with CAS9&Sox2–gRNA (triangles, n=5) or CAS9&Tyr-gRNA control (diamonds, n=5). Each scale on the x-axis represents ~800 μm. M, mature uninjured tail region anterior to the amputation plane; R, regenerate posterior to the amputation plane. Error bars, s.d.; **P<0.005, ***P<0.001.
Multiplex knockout of genes by electroporation
We next asked if the simultaneous delivery of CAS9&GFP–gRNA and CAS9&Sox2–gRNA complexes would allow both genes to be efficiently knocked out. After mixing of complexes, injection and electroporation, we observed an efficient knockout of both genes in the mature and regenerating spinal cord (Figure 4a). Quantification showed that GFP and Sox2 CAS9–gRNA complex-mediated double-gene knockout yielded a similar knockout efficiency for each individual gene compared with the electroporation of the individual complexes (Figure 4b,c). Moreover, the majority (over 90%) of the targeted cells lose the expression of both GFP and SOX2 (Figure 4d). These results demonstrated that the CAS9 protein–gRNA electroporation technique could achieve efficient multiplex gene knockout.

GFP and Sox2 double-gene knockout in the axolotl spinal cord NSCs through CAS9–gRNA complex electroporation. (a) GFP fluorescence (green) and immunofluorescence for SOX2 (red) combined with DAPI (blue) on cross-sections show a massive loss of GFP and SOX2 expression in NSCs in the mature ('M') and regenerated ('R') region of 10-day tail blastema treated with CAS9&GFP/Sox2–gRNAs (upper panel) compared with the CAS9&Tyr-gRNA control (lower panel). Dotted lines define the spinal cord area; arrows indicate dorsal root ganglia (DRG). Note that the comprehensive loss of GFP and SOX2 occurs only in the spinal cord area but not in the DRG or elsewhere in the tail of CAS9&GFP/Sox2–gRNAs-treated samples compared with the control. CA, cartilage; NT, notochord. Scale bar, 100 μm. (b and c) Quantification of the percentage of SOX2+ (b) and GFP+ (c) cells over total non-neuronal spinal cord cells at 15 days post electroporation of CAS9&GFP/Sox2–gRNA (circles in b, triangles in c, n=6) or CAS9&Tyr-gRNA control (diamonds, n=6). Neurons in the spinal cord are determined by their location and by the shape of the DAPI-stained nuclei as described in the Materials and methods. Error bars, s.d.; ***P<0.001. (d) Graph shows that the majority (94%) of the targeted NSCs lose both GFP and SOX2 expression (GFP− SOX2−) within in the cell population harbouring at least one gene deletion. Only very minority (6%) shows either GFP (GFP− SOX2+) or Sox2 (GFP+ SOX2−) single gene knockout.
Electroporation mediated gene knockout can be applied to other cell types
Finally, we asked whether the electroporation method can be applied to other axolotl cell types. We were encouraged to examine knockout in other cell types since we also observed downregulation of GFP in the tail epidermis after spinal cord electroporation with the CAS9&GFP–gRNA complexes (Supplementary Figure S3A). This knockout in the epidermis occurs because the penetration of the microinjection needle through the side of the animal is necessary to gain access to the spinal cord lumen, and sufficient CAS9–gRNA complexes were deposited on the skin surface to elicit knockout of GFP in the epidermis. We next chose the axolotl limb as a target, since it is composed of multiple tissue types that can be targeted by electroporation. We injected the mature limb with the CAS9&GFP–gRNA complexes followed by electroporation. Subsequent amputation resulted in a regenerated limb harbouring a significant number of cells lacking GFP expression (Figure 5a,b′). Based on the muscle satellite cell marker PAX7 expression, the morphology and the location of the cells, we could identify the absence of GFP expression in satellite cells, bone, dermal mesenchyme and skin cells (Figure 5b,b′) in limbs treated with CAS9&GFP–gRNA complexes, but not in the control. The lower rate of affected cells in the limb blastema compared with the tail spinal cord reflects the organisation of the limb where the mesenchymal progenitors are diffusely distributed over large area of muscle and skin. Therefore achieving a uniformly high concentration of complex in the limb prior to electroporation is more difficult compared with injection into the highly defined lumen of the spinal cord.

GFP knockout in other cell types in the axolotl through CAS9–gRNA complex electroporation. (a and a′) GFP fluorescence (green), PAX7 immunofluorescence (red), DAPI (blue) and DIC images of cross-sections from 21-day regenerating limb treated with CAS9&GFP–gRNAs (a) and CAS9&Tyr-gRNA (control, a′). Boxed areas (number 1–4 in a and a′) are shown at higher magnification in b and b′. Scale bar, 200 μm. (b and b′) Images show the loss of GFP expression in a subpopulation of satellite cells identified by PAX7 staining (first row, arrows), as well as the cells at bone region (second row, dotted line area), dermal mesenchyme (third row, arrows) and epidermis (fourth row, dotted line area), which were identified based on the cell morphology and location, in the limbs treated with CAS9&GFP–gRNAs (b) compared with the control (b′). Red lines indicate the location of basal lamina that separates the epidermis (Epi) from the Dermis (Der).
Discussion
In this study, we have successfully established an acute high-efficiency, tissue-specific gene knockout method via CAS9 protein electroporation in diversified tissues in axolotl, particularly in the spinal cord NSCs. We further demonstrated the application of this technique to knockout the endogenous Sox2 gene in the spinal cord, which caused the expected spinal cord regeneration defect in axolotls. Finally, we demonstrated the feasibility of multiple-gene knockout. In this method, the sequestering of the gRNA into the CAS9 complex apparently protects it from degradation within the spinal cord fluid that is enriched with RNase15 (J-FF, unpublished results). The stability due to the pre-assembly of CAS9 protein and gRNA even in the absence of genomic DNA,16–18 together with the composite negative charge of CAS9 protein–gRNA complexes19 are likely to be the key features allowing highly efficient gene modification via electroporation of the complexes into the spinal cord NSCs. Compared with a previously published technique using lipidic transfection reagents to deliver CAS9–gRNA complexes into murine otic tissue,19 which caused a 20% GFP transgene knockout in vivo, we obtain a highly efficient (over 90%) knockout of GFP transgene in spinal cord NSCs.
We observed a much higher knockout efficiency using the CAS9–gRNA complexes, compared with CRISPR plasmid electroporation. The delivery of the CRISPR plasmids to knockout target gene via electroporation had previously been reported in the mouse retina and post-mitotic neurons.20,21 In our case, the efficiency of CRISPR plasmid-mediated Sox2 knockout was not sufficient to obtain a spinal cord regeneration defect in axolotls due to the limited number of transfected cells. The disadvantages of plasmid electroporation, compared with CAS9–gRNA complexes electroporation have several dimensions. First, the delivery of the CRISPR plasmids to the target cells is not as efficient for the CAS9 protein–gRNA complexes. The pCAGGS-Cas9 plasmid (≈10 kb) used in this study is more than 10 times the molecular weight of CAS9 protein–gRNA complex, limiting its access to cells. In addition, we co-electroporated pCAGGS-Cas9 and CMV-dsRed-RGR-gRNA plasmids requiring uptake of both plasmids, which further decreased the knockout efficiency. As the DsRed marker gene was only on the gRNA-expression plasmid, we currently do not know which percentage of the DsRed-positive cells were co-expressing the Cas9 plasmid. This requirement for co-electroporation may explain the lower efficiency of knockout in which 60% of DsRed-expressing cells were GFP-negative as compared with over 90% of NSCs being GFP-negative in the CAS9–gRNA complexes electroporated samples. Further optimisation of the level and/or ratio of transfected Cas9 versus gRNA might be required to improve the performance of plasmid-mediated gene knockout efficiency.
When we knocked out the GFP transgene and the Sox2 gene simultaneously, we found the individual gene knockout efficiency was almost identical to conditions in which the CAS9–gRNA complexes were electroporated singly. Previously, multiple-gene knockout was achieved by injecting the mixture of Cas9 mRNA and several gRNAs into fertilised eggs to create multigene-modified mice and zebrafish,6,22 or via an expression of a vector incorporating multiple U6 promoters-gRNA cassettes in zebrafish.23 In the latter method, the chaining together of multiple genes on a single plasmid yields increased plasmid sizes, which would limit electroporation efficiency of somatic tissues. Here, the parallel electroporation of CAS9–gRNA complexes is a simple method to elicit multiple-gene knockout in a tissue-specific way. This makes the technique particularly powerful for the functional screening of multiple genes.
Here, we have demonstrated target gene knockout via CAS9–gRNA complex electroporation in axolotl. However, in principle, this direct delivery of the CAS9–gRNA complexes could be applied in many other species, since this technique breaks the species limitation by avoiding the use of a promoter to drive Cas9/gRNA-expression. This technique will be particularly useful for the species that still lack a proper RNA polymerase III system to drive gRNA expression.
Materials and methods
CRISPR plasmids construction
For gRNA expression, we used a ribozyme-based vector system,12 in which the gRNA is flanked by two individual ribozyme sequences (ribozyme–gRNA–ribozyme (RGR)). Ribozymes undergo self-catalyzed cleavage to precisely release the gRNA molecule. We generated plasmids CMV:dsRed-RGR-GFP-gRNA and CMV:dsRed-RGR-Sox2-gRNA by inserting the synthesised RGR-GFP-gRNA and RGR-Sox2-gRNA gene fragments (IDT, see Supplementary Materials and Methods) into the vector CMV: dsRed2-Triplex plasmid (Addgene #55200)12 opened with AgeI and XmaI. In the final plasmids, the RGR-gRNA cassette is located after the dsRed and the Triplex sequence (see Figure 1b). Triplex is used to stabilize the dsRed mRNA after Ribozyme self-cleavage. The gRNA expression can be indirectly monitored by the expression of dsRed fluorescence driven by the constitutive CMV promoter in the targeted cells. To create pCAGGS-Cas9 plasmid, Cas9 gene followed by a nuclear localisation signal (Cas9-NLS) was PCR-amplified using plasmid MLM3613 (Addgene, Cambridge, MA, USA, #42251)24 as a template, and the primer pair—Cas9-for and Cas9-rev (see Supplementary Information). The PCR product was subcloned into NheI- and BamHI-opened pCAGGS vector with Gibson assembly protocol. All resulting plasmids were verified by sequencing.
CAS9 protein
To prepare CAS9-NLS nuclease, the Cas9-NLS-coding sequence was PCR-amplified using MLM3613 as a template with the primer pair—Cas9-for2 and Cas9-rev2. The PCR product was then cloned between the NotI and AscI sites in pOCC97, a vector derived from pET-28a (Novagen, Madison, WI, USA), which directs the expression of an N-terminal hexahistidine (His6)-maltose-binding protein (MBP) fusion to the target gene via a cleavable linker recognised by the human rhinovirus 3C protease. T7 Express pRARE cells (New England Biolabs, Ipswich, MA, USA) harbouring pOCC97-Cas9-NLS were induced (200 μM IPTG) for 16 h at 18 °C. After re-suspending the pelleted cells in Lysis Buffer (50 mM TrisCl, 1 M NaCl, 1 mM DTT pH 8), cells were lysed by two passes at 1,500 bar through an Emulsiflex microfluidizer (Avestin, Ottawa, ON, Canada). The extract was clarified by centrifugation, then the supernatant precipitated by adding polyethylenimine (0.25% final). After clarification by centrifugation, the supernatant was then precipitated by addition of solid ammonium sulphate to 70% saturation. The pelleted precipitate was resuspended in Lysis Buffer and the His6-tagged fusion protein purified by affinity chromatography using HisTrap FF columns (GE Healthcare, Waukesha, WI, USA). After elution with buffer containing 250 mM imidazole, 3C protease was added (1:100 wt/wt) and the protein was dialyzed overnight against dialysis buffer (20 mM HEPES pH 7.5, 150 mM KCl, 5% glycerol, 1 mM DTT). Cas9-NLS was separated from the cleaved His6-MBP tag by loading the dialyzed preparation onto a cation exchange column (HiTrap SP HP; GE Healthcare) equilibrated in dialysis buffer. After washing with the equilibration buffer, the column was developed with a linear gradient from 150 to 1000 mM KCl over 15 column volumes. The peak of Cas9NLS enzyme eluting at ~50 mS was concentrated and exchanged into storage buffer (20 mM HEPES, 150 mM KCl pH 7.5) using 30 kDa cut-off Amicon Ultra centrifugal filter units (Millipore, Billerica, MA, USA) then adjusted to 5 mg/ml before snap freezing as aliquots.
gRNA
The GFP-gRNA (targeting site: 5′- GGCCACAAGTTCAGCGTGTC-3′), Sox2–gRNA (targeting site: 5′- GGAGGTCTGCTGCGGGGCGG-3′) and Tyr-gRNA (targeting site: 5′- GGACTTCACTATCCCCTACT-3′, as control) described previously14 were used for the knockout of relevant target genes. The gRNAs were synthesised using MAXIscript T7 kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions.
Axolotl electroporation
All animal experiments were carried out in accordance with German animal welfare legislation. We mixed 1 μg/μl CAS9-NLS protein and 1 μg/μl gRNA and incubated at room temperature for 5 min to allow the formation of CAS9–gRNA complex. Electroporation of CAS9–gRNA complexes or CRISPR plasmids (mixture of 1 μg/μl RGR-gRNA and 1 μg/μl pCAGGS-Cas9-NLS plasmids, but only the RGR-gRNA plasmid for the control) to the spinal cord or limb was carried out as previously described.9,25 To improve the electroporation efficiency on roof and floor plate cells, one extra dorsal–ventral-orientated electroporation was performed to the spinal cords as illustrated in Figure 1a. Axolotl tails were amputated/collected at 14–20 days post electroporation. The removed tails were used for analysing the target gene knockout efficiency. Amputated axolotls were allowed to regenerate for 6 or 10 days, and then the gene knockout phenotype in tail regenerates was analysed. For limb experiments, amputation was carried out 7 days post electroporation. Limb-amputated animals were allowed to regenerate for 21 days for analysis. About 2–2.5 cm (from snout to tail tip) CAGGS-GFP transgenic or white axolotls were used for electroporation.
Genotyping
Spinal cords of 6-day or 10-day tail regenerates electroporated with CAS9–gRNA complex or PBS control were dissected for genomic DNA isolation. Genomic DNA preparation, genotyping PCR and sequencing of targeted genomic locus were carried out as previously described.14
Immunohistochemistry
Immunostaining analyses were carried out on 10-μm cross-cryosections of axolotl tail or limb samples using standard protocols. The following primary antibodies against the following antigens were used in this study: SOX2 (rabbit polyclonal antibody),26 TUJ1 (R&D systems, Minneapolis, MN, USA, MAB1195), NEUN (Millipore, MAB377), GFAP (Millipore, MAB360) and PAX7 (mouse monoclonal antibody, DSHB, Iowa City, IA, USA). Fluorescence images were acquired with a Zeiss Axio Observer microscope (Carl Zeiss, Oberkochen, Germany), using ×20 objectives. Bright field, DIC and fluorescence tail images were acquired with an Olympus stereomicroscope (Olympus, Tokyo, Japan). All images of GFP fluorescence or SOX2 immunofluorescence from axolotls electroporated with CAS9–gRNA complex and corresponding controls were acquired with identical exposure time.
Quantification and statistical analysis
The length of regenerated spinal cord or tail was measured as previously described.14 In the CAS9&Sox2–gRNA-electroporated tails, GFAP+ cells were quantified as total cells in regenerated spinal cord when tubular spinal cord structures are observed. To identify the non-neuronal spinal cord cells, TUJ1, NEUN immunostaining, cell nuclei location and morphology were applied to exclude neurons. Determined by DAPI-stained nuclei, neurons are located in the peripheral region of the spinal cord without direct contact to the lumen, and the nuclei of neurons have a round shape compared with the oval-shaped spinal cord NSC nuclei (see Figure 3a). Plotting of data and statistical analyses were performed using program Prism (GraphPad, La Jolla, CA, USA). Student’s t-test was used for P values calculation.
References
Doudna, J. A. & Charpentier, E. Genome editing. The new frontier of genome engineering with crispr-cas9. Science 346, 1258096 (2014).
Hsu, P. D., Lander, E. S. & Zhang, F. Development and applications of crispr-cas9 for genome engineering. Cell 157, 1262–1278 (2014).
Sander, J. D. & Joung, J. K. Crispr-cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 32, 347–355 (2014).
Gagnon, J. A. et al. Efficient mutagenesis by cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide rnas. PLoS ONE 9, e98186 (2014).
Sung, Y. H. et al. Highly efficient gene knockout in mice and zebrafish with rna-guided endonucleases. Genome Res. 24, 125–131 (2014).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by crispr/cas-mediated genome engineering. Cell 153, 910–918 (2013).
Ablain, J., Durand, E. M., Yang, S., Zhou, Y. & Zon, L. I. A crispr/cas9 vector system for tissue-specific gene disruption in zebrafish. Dev. Cell 32, 756–764 (2015).
Thummel, R. et al. Inhibition of zebrafish fin regeneration using in vivo electroporation of morpholinos against fgfr1 and msxb. Dev. Dyn. 235, 336–346 (2006).
Albors, A. R. & Tanaka, E. M. High-efficiency electroporation of the spinal cord in larval axolotl. Methods Mol. Biol. 1290, 115–125 (2015).
Nakamura, H., Katahira, T., Sato, T., Watanabe, Y. & Funahashi, J. Gain- and loss-of-function in chick embryos by electroporation. Mech. Dev. 121, 1137–1143 (2004).
Fei, J. F., Haffner, C. & Huttner, W. B. 3' utr-dependent, mir-92-mediated restriction of tis21 expression maintains asymmetric neural stem cell division to ensure proper neocortex size. Cell Rep. 7, 398–411 (2014).
Nissim, L., Perli, S. D., Fridkin, A., Perez-Pinera, P. & Lu, T. K. Multiplexed and programmable regulation of gene networks with an integrated rna and crispr/cas toolkit in human cells. Mol. Cell 54, 698–710 (2014).
Sobkow, L., Epperlein, H. H., Herklotz, S., Straube, W. L. & Tanaka, E. M. A germline gfp transgenic axolotl and its use to track cell fate: Dual origin of the fin mesenchyme during development and the fate of blood cells during regeneration. Dev. Biol. 290, 386–397 (2006).
Fei, J. F. et al. Crispr-mediated genomic deletion of sox2 in the axolotl shows a requirement in spinal cord neural stem cell amplification during tail regeneration. Stem Cell Rep. 3, 444–459 (2014).
Blank, A. & Dekker, C. A. Ribonucleases of human serum, urine, cerebrospinal fluid, and leukocytes. Activity staining following electrophoresis in sodium dodecyl sulphate-polyacrylamide gels. Biochemistry 20, 2261–2267 (1981).
Jiang, F., Zhou, K., Ma, L., Gressel, S. & Doudna, J. A. A cas9-guide rna complex preorganized for target DNA recognition. Science 348, 1477–1481 (2015).
Anders, C., Niewoehner, O., Duerst, A. & Jinek, M. Structural basis of pam-dependent target DNA recognition by the cas9 endonuclease. Nature 513, 569–573 (2014).
Nishimasu, H. et al. Crystal structure of cas9 in complex with guide rna and target DNA. Cell 156, 935–949 (2014).
Zuris, J. A. et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 33, 73–80 (2015).
Straub, C., Granger, A. J., Saulnier, J. L. & Sabatini, B. L. Crispr/cas9-mediated gene knock-down in post-mitotic neurons. PLoS ONE 9, e105584 (2014).
Wang, S., Sengel, C., Emerson, M. M. & Cepko, C. L. A gene regulatory network controls the binary fate decision of rod and bipolar cells in the vertebrate retina. Dev. Cell 30, 513–527 (2014).
Jao, L. E., Wente, S. R. & Chen, W. Efficient multiplex biallelic zebrafish genome editing using a crispr nuclease system. Proc. Natl Acad. Sci. USA 110, 13904–13909 (2013).
Yin, L. et al. Multiplex conditional mutagenesis using transgenic expression of cas9 and sgrnas. Genetics 200, 431–441 (2015).
Hwang, W. Y. et al. Efficient genome editing in zebrafish using a crispr-cas system. Nat. Biotechnol. 31, 227–229 (2013).
Mercader, N., Tanaka, E. M. & Torres, M. Proximodistal identity during vertebrate limb regeneration is regulated by meis homeodomain proteins. Development 132, 4131–4142 (2005).
Tapia, N. et al. Reprogramming to pluripotency is an ancient trait of vertebrate oct4 and pou2 proteins. Nat. Commun. 3, 1279 (2012).
Acknowledgements
We are grateful to Beate Gruhl, Sabine Mögel and Anja Wagner for outstanding animal care, Dr Yuka Taniguchi for drawing the axolotl cartoon and Dr Aida Rodrigo Albors for reading the manuscript. The work was supported by an HFSP program grant, DFG-274/3-2, DFG-274/3-3, DFG-274/2-3/SFB655 from Cells into Tissues, a CRTD Seed grant on Zn-finger nucleases and central funds from the DFG Research Center, CRTD, ERC Advanced Investigator Grant.
Author information
Authors and Affiliations
Contributions
J.-F.F. designed, performed and analysed the experiments and wrote the manuscript. D.K., M.S., P.M., Y.Z. and D.D. performed the experiments. E.M.T. provided funding, designed and analysed the experiments, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the npj Regenerative Medicine website (http://www.nature.com/npjregenmed)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Fei, JF., Knapp, D., Schuez, M. et al. Tissue- and time-directed electroporation of CAS9 protein–gRNA complexes in vivo yields efficient multigene knockout for studying gene function in regeneration. npj Regen Med 1, 16002 (2016). https://doi.org/10.1038/npjregenmed.2016.2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/npjregenmed.2016.2
