
Abstract
Low-dimensional electron systems, as realized in layered materials, often tend to spontaneously break the symmetry of the underlying nuclear lattice by forming so-called density waves1; a state of matter that at present attracts enormous attention2,3,4,5,6. Here we reveal a remarkable and surprising feature of charge density waves, namely their intimate relation to orbital order. For the prototypical material 1T-TaS2 we not only show that the charge density wave within the two-dimensional TaS2 layers involves previously unidentified orbital textures of great complexity. We also demonstrate that two metastable stackings of the orbitally ordered layers allow manipulation of salient features of the electronic structure. Indeed, these orbital effects provide a route to switch 1T-TaS2 nanostructures from metallic to semiconducting with technologically pertinent gaps of the order of 200 meV. This new type of orbitronics is especially relevant for the ongoing development of novel, miniaturized and ultrafast devices based on layered transition metal dichalcogenides7,8.
Similar content being viewed by others

Main
Among the various transition metal dichalcogenides (TMDs), 1T-TaS2 stands out because of its particularly rich electronic phase diagram as a function of pressure and temperature9. This phase diagram not only features incommensurate, nearly commensurate and commensurate charge density waves (CDWs), but also pressure-induced superconductivity below 5 K. In addition to this, it was proposed early on that the low-temperature commensurate CDW (C-CDW), which is illustrated in Fig. 1a, c, also features many-body Mott physics10. Experimental evidence for the presence of Mott physics in 1T-TaS2 has indeed been obtained recently by time-resolved spectroscopies, which observed the ultrafast collapse of a charge excitation gap, which has been interpreted as a fingerprint of significant electron–electron interactions11,12,13.
 supercell structure of 1T-TaS2.
supercell structure of 1T-TaS2.
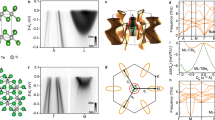
a,c, View along the c-axis and parallel to the ab-plane, respectively (a, b and c are lattice vectors of the undistorted  crystal structure). The Ta-displacements indicated by red arrows in a are mainly parallel to the ab-planes, resulting in clusters containing 13 Ta-sites. Red dashed lines in c highlight the breathing of the S-sites perpendicular to the ab-plane. b, Schematic of the Brillouin zone. d, LDA supercell band structure along the high-symmetry direction in the Brillouin zone shown in b. Grey lines indicate the unreconstructed band structure. e, Unfolded band structure. The thickness of bands measures the spectral weight at this position. Arrows labelled 1 and 2 indicate the gaps at Γ and between M and K, which are commonly named the Mott gap and CDW gap, respectively.
crystal structure). The Ta-displacements indicated by red arrows in a are mainly parallel to the ab-planes, resulting in clusters containing 13 Ta-sites. Red dashed lines in c highlight the breathing of the S-sites perpendicular to the ab-plane. b, Schematic of the Brillouin zone. d, LDA supercell band structure along the high-symmetry direction in the Brillouin zone shown in b. Grey lines indicate the unreconstructed band structure. e, Unfolded band structure. The thickness of bands measures the spectral weight at this position. Arrows labelled 1 and 2 indicate the gaps at Γ and between M and K, which are commonly named the Mott gap and CDW gap, respectively.
Even though the above scenario for the C-CDW is widely accepted, important experimental facts remain to be understood: the very strong suppression of the C-CDW with external pressure is puzzling. Already above 0.6 GPa, the C-CDW is no longer stable, although nesting conditions, band widths and lattice structure remain essentially unchanged. It is also not clear how ordered defects within the C-CDW, which emerge in the nearly commensurate phase (NC-CDW) on heating14,15 and do not cause significant changes in the bandwidths, can render the electron–electron on-site interaction U completely ineffective16. In the following we will show that all these issues are explained consistently in terms of orbital textures that are intertwined with the CDW. Furthermore, we demonstrate that this new twist to the physics of CDWs also provides a new and powerful device concept for future applications based on TMDs.
An important feature of the C-CDW is its partially disordered stacking along the c-axis: characterizing the relative alignment of the C-CDW in adjacent ab-planes in terms of stacking vectors TS, which connect the central Ta-sites in successive layers (Fig. 2e, f), the c-axis stacking is given by an alternation of TS = 1c with a random choice from the three symmetry equivalent vectors TS = 2a + c, 2b + c or −2(a + b) + c (refs 17, 18). The effect of the different TS on the electronic structure has so far remained unexplored. Our highly efficient DFT approach (see Methods), however, enables us to study these effects for the first time within the local density approximation (LDA).

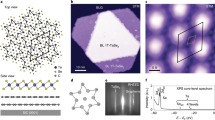
a, ARPES data of 1T-TaS2 measured at 1 K. b, XRD intensity of 1T-TaS2 measured along the l-direction for the C-CDW and NC-CDW phase. The in-plane q-vector corresponds to hq = 16/13, kq = 1/13 for the C-CDW phase at 100 K and hq = 1.248, kq = 0.068 for the NC-CDW phase at 230 K. The single reflection observed for the NC-CDW implies a well-ordered stacking along c, fully consistent with previous results15. On entering the C-CDW phase, the XRD pattern changes markedly and two broad peaks appear owing to the presence of an alternating c-axis stacking (see text). A numerical simulation of the XRD (solid line) further reveals a finite correlation length of the alternating c-axis stacking of approximately 10 × c. c,d, Calculated band structure for TS = 2a + c and TS = 1c, respectively. To facilitate the comparison with ARPES data shown in a the bandweights have been convolved with a Lorentzian shaped resolution function. e,f, Visualization of the stacking with TS = 2a + c and TS = 1c, respectively. Red arrows indicate the stacking vectors that connect the central Ta-sites (shown in red) in successive layers.
For our ab initio calculations, we chose two superstructures with TS = 1c and TS = 2a + c, respectively, which represent the two metastable types of stacking found experimentally. The calculated total energy for both superstructures differs by only a few meV, implying that these two stacking types are indeed metastable. Figure 1d shows the band structures for the C-CDW with TS = 1c and the undistorted lattice, where the band dispersions are shown along high-symmetry directions of the Brillouin zone of the unmodulated structure (Fig. 1b). In good agreement with previous DFT results19, the band structure in the plane through Γ, M and K is fully gapped and only along Γ–A does a Fermi level crossing occur. This yields a one-dimensional metal, in line with recent reports20. A major disadvantage of such supercell calculations is, however, that the additional backfolded bands only indicate for which momenta (k) and energies (ω) electronic states exist. There is no information about the spectral weight of these states. Exactly this information is provided by the so-called spectral function A(k, ω) (ref. 21), which is a physically relevant quantity that can be accessed experimentally by, for example, angle-resolved photoemission spectroscopy (ARPES).
An approximation for A(k, ω) within DFT can be obtained using the so-called unfolding procedure22. The result of this unfolding procedure for the band structure shown in Fig. 1d is presented in Fig. 1e, where the thickness of the individual bands reflects the spectral weight |A(k, ω)|2. It is clearly visible that only a small fraction of the reconstructed band structure carries substantial spectral weight. Unlike the bands in Fig. 1d, the unfolded band structure allows one to clearly identify dispersing bands and, correspondingly, the size and location of energy gaps. Specifically, our calculation yields two gaps around the Fermi level EF, indicated by arrows in Fig. 1e. The gap at Γ is usually interpreted as a Mott gap due to the electron–electron interaction U (refs 13, 23), whereas the gap between M and K fits well to the Fermi surface nesting vector and is therefore commonly assigned to a CDW gap due to the electron–phonon coupling. It is very important to note here that our calculations do not include any on-site interactions U. The fact that the gap at Γ is obtained without including U is therefore surprising and already implies that this gap cannot be caused by Mott physics alone.
In Fig. 2 we compare the unfolded LDA band structures for TS = 1c and TS = 2a + c, which yields the first major result of this study: the marked dependence of the electronic structure on the CDW stacking. As can be observed in Fig. 2c, d, essential features of the low-energy electronic structure depend critically on the stacking. Most notably the bands around the Γ point are strongly affected. Whereas for TS = 2a + c the in-plane dispersions show EF-crossings along Γ–K and Γ–M, both crossings are completely absent for TS = 1c. Therefore, a transition from an in-plane metal to an in-plane semiconductor occurs as a function of TS. Considering that the ab-planes of 1T-TaS2 are usually thought to realize strongly two-dimensional metallic systems, these are most surprising results.
The comparison of the two DFT models to the ARPES data in Fig. 2a shows that the calculation for TS = 2a + c describes the experiment fairly well for binding energies below −0.3 eV. However, it is also obvious that both DFT models fail to describe the electronic states close to EF and, in particular, the pseudo-gap observed experimentally24. Including an on-site U at Ta does not cure this discrepancy, implying that these effects are beyond LDA + U. Indeed, the strong TS-dependence of the electronic structure suggests that these deviations are related to the stacking disorder in the real material18, which cannot be taken into account by our DFT models. Although this issue certainly deserves scrutiny in future work, here we focus on determining the origin of the marked effects of TS.
To this end we calculated the charge density distribution of the uppermost occupied states in real space. As can be seen in Fig. 3, for both stackings a complex orbital texture within the ab-plane emerges. This is the second major result of this work: the discovery of an orbital texture that is intertwined with a CDW. Note that not only the occupancy of a certain type of orbital changes spatially, also the symmetry of the orbitals clearly changes from site to site, resulting in a complex orbital ordering pattern.

a,b, The band structure near EF with the highest occupied band highlighted in red. The energy window used to calculate the energy-resolved electron density is indicated by the green area. c,d, A complex orbital texture emerges within the ab-plane for TS = 1c (c) and TS = 2a + c (d). e,f, View of the ac-plane corresponding to the areas in c and d. e, For TS = 1c significant hopping is allowed only along the c-direction. f, A substantial ab-component of the hopping appears for TS = 2a + c.
The orbital structure for TS = 1c permits significant charge hopping only along c, as illustrated in Fig. 3e. In other words, the charges flow along orbital stripes along c, corresponding to the quasi-one-dimensional character of the uppermost band in Figs 3a and 2d. This drastically changes in the case of TS = 2a + c. Now there is a significant ab-component of the hopping, as illustrated in Fig. 3f, so that the uppermost band attains a larger in-plane dispersion and crosses EF along Γ–M and Γ–K (Figs 3b and 2c).
These results have important consequences: first, the comparison of the ARPES data and the DFT results shows that the Γ gap is obtained on a quantitative level without an on-site repulsion U on Ta. Although this result does not exclude the presence of electron–electron interactions, it strongly argues against U being the main cause for the Γ gap. Instead, the band structure calculations reveal that this gap is for a large part due to the interlayer hybridization. The latter is in turn mostly caused by the 3z2 − r2-type orbitals pointing along c—that is, the Γ gap is directly coupled to the orbital texture. This result naturally explains the ultrafast response of this gap observed in time-resolved ARPES experiments, as the disruption and reordering of the electronic orbitals can evolve on much faster timescales than the lattice.
Second, the presence of orbital textures explains the strong pressure dependence of the CDW order in 1T-TaS2, because through the orbital texture pressure has a large effect on the stability of CDW phases with different stackings in a way that goes well beyond traditional nesting scenarios. We also stress that the crucial role of the interlayer interactions is verified experimentally by the data in Fig. 2b, which shows that the stacking changes completely across the C-CDW/NC-CDW transition. This change in stacking together with our DFT results also rationalizes the collapse of the gap at Γ on entering the NC-CDW with warming.
Third, and most importantly, the relative orientation of the orbitals in adjacent ab-planes has a spectacular effect on the band dispersions. This can be readily understood in terms of the overlap integrals, which depend critically on the relative orientation of the orbitals in adjacent layers along c (Fig. 2e, f). The above immediately yields a new device concept, which employs metastable orbital orders for controlling the electronic structure of nanostructures: as illustrated in Fig. 4 for a bilayer of 1T-TaS2, switching between the metastable orbital configurations causes a complete semiconductor–metal transition. In other words, by changing the orbital order in the direction perpendicular to the layers, one can control the conductivity parallel to the layers. Even though this effect has not yet been observed directly, a very recent experiment provides first evidence that this might be achieved reversibly and on ultrafast timescales using optical laser pulses25.

a,b, Calculated band structure for a bilayer of 1T-TaS2 with TS = 1c (a) and TS = 2a + c (b). A semiconductor-to-metal transition takes place on changing the stacking of the two layers. c, Orbital order corresponding to the semiconducting (top) and metallic state (bottom).
Orbital textures hence enable one to manipulate the band dispersion and gap structure of 1T-TaS2 in a very effective way. Therefore, they provide a new route to tailor and switch the electronic properties of TMDs, possibly on the femtosecond timescale. This concept of orbitronics may hence enable the creation of novel, small and ultrafast electronics, greatly increasing the potential for TMDs in future device applications.
Methods
DFT.
The DFT calculations were done using the FPLO14 package26, which has been developed at the IFW Dresden and supports unfolding of the band structure as outlined in ref. 22. Owing to its small basis size it allows extremely efficient calculations of large supercells, which makes the presented calculations numerically affordable. The supercell structure for TS = 1c was derived from refs 15, 27. To simulate different layer stackings we transformed the hexagonal supercell into a triclinic supercell without changing the atomic displacements. It is important to note that a structural relaxation, which starts from the undistorted lattice, rapidly converges against the experimentally observed superstructure. Furthermore, the present DFT calculations reproduce the so-called Mott and CDW gap on a quantitative level. This verifies our approach and shows that the present DFT models capture the important interactions present in the real material.
XRD.
XRD data were obtained on beamline BW5 at the Deutsches Elektronensynchrotron (DESY). We mounted a high-quality single crystal of 1T-TaS2 in a displex helium cryostat sitting in an Euler cradle. The presented XRD data was obtained by measuring the diffracted intensity of the superstructure reflection while scanning along the crystallographic l-direction for two different temperatures.
ARPES.
ARPES measurements were conducted at the 13-ARPES endstation on beamline UE112PG2 at the Berlin Synchrotron (BESSY). We used p-polarized light with a 96 eV photon energy so that the final state crystal momentum at normal emission corresponds to the Γ point28. The sample temperature was kept at 1 K.
References
Grüner, G. Density Waves in Solids Vol. 89 (Addison-Wesley, 1994).
Chang, J. et al. Direct observation of competition between superconductivity and charge density wave order in Y Ba2Cu3O6.67 . Nature Phys. 8, 871–876 (2012).
Ghiringhelli, G. et al. Long-range incommensurate charge fluctuations in (Y, Nd)Ba2Cu3O6+x . Science 337, 821–825 (2012).
Da Silva Neto, E. H. et al. Ubiquitous interplay between charge ordering and high-temperature superconductivity in cuprates. Science 343, 393–396 (2014).
De la Cruz, C. et al. Magnetic order close to superconductivity in the iron-based layered LaO1−xFxFeAs systems. Nature 453, 899–902 (2008).
Mühlbauer, S. et al. Skyrmion lattice in a chiral magnet. Science 323, 915–919 (2009).
Wang, Q. H., Kalantar-Zadeh, K., Kis, A., Coleman, J. N. & Strano, M. S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nature Nanotech. 7, 699–712 (2012).
Chhowalla, M. et al. The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nature Chem. 5, 263–275 (2013).
Sipos, B. et al. From Mott state to superconductivity in 1T-TaS2 . Nature Mater. 7, 960–965 (2008).
Fazekas, P. & Tosatti, E. Electrical, structural and magnetic properties of pure and doped 1T-TaS2 . Phil. Mag. B 39, 229–244 (1979).
Hellmann, S. et al. Ultrafast melting of a charge-density wave in the Mott insulator 1T-TaS2 . Phys. Rev. Lett. 105, 187401 (2010).
Petersen, J. C. et al. Clocking the melting transition of charge and lattice order in 1T-TaS2 with ultrafast extreme-ultraviolet angle-resolved photoemission spectroscopy. Phys. Rev. Lett. 107, 177402 (2011).
Perfetti, L. et al. Time evolution of the electronic structure of 1T-TaS2 through the insulator–metal transition. Phys. Rev. Lett. 97, 067402 (2006).
Wu, X. L. & Lieber, C. M. Hexagonal domain-like charge density wave phase of 1T-TaS2 determined by scanning tunneling microscopy. Science 243, 1703–1705 (1989).
Spijkerman, A., de Boer, J. L., Meetsma, A., Wiegers, G. A. & van Smaalen, S. X-ray crystal-structure refinement of the nearly commensurate phase of 1T-TaS2 in (3 + 2)-dimensional superspace. Phys. Rev. B 56, 13757–13767 (1997).
Ang, R. et al. Real-space coexistence of the melted Mott state and superconductivity in Fe-substituted 1T-TaS2 . Phys. Rev. Lett. 109, 176403 (2012).
Tanda, S., Sambongi, T., Tani, T. & Tanaka, S. X-ray study of charge density wave structure in 1T-TaS2 . J. Phys. Soc. Jpn 53, 476–479 (1984).
Nakanishi, K. & Shiba, H. Theory of three-dimensional orderings of charge-density waves in 1T-TaX2 (X: S, Se). J. Phys. Soc. Jpn 53, 1103–1113 (1984).
Bovet, M. et al. Interplane coupling in the quasi-two-dimensional 1T-TaS2 . Phys. Rev. B 67, 125105 (2003).
Darancet, P., Millis, A. J. & Marianetti, C. A. Three-dimensional metallic and two-dimensional insulating behavior in octahedral tantalum dichalcogenides. Phys. Rev. B 90, 045134 (2014).
Damascelli, A., Hussain, Z. & Shen, Z-X. Angle-resolved photoemission studies of the cuprate superconductors. Rev. Mod. Phys. 75, 473–541 (2003).
Ku, W., Berlijn, T. & Lee, C-C. Unfolding first-principles band structures. Phys. Rev. Lett. 104, 216401 (2010).
Rossnagel, K. On the origin of charge-density waves in select layered transition-metal dichalcogenides. J. Phys. Condens. Matter 23, 213001 (2011).
Pillo, T. et al. Remnant Fermi surface in the presence of an underlying instability in layered 1T-TaS2 . Phys. Rev. Lett. 83, 3494–3497 (1999).
Stojchevska, L. et al. Ultrafast switching to a stable hidden quantum state in an electronic crystal. Science 344, 177–180 (2014).
Koepernik, K. & Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 59, 1743–1757 (1999).
Brouwer, R. Incommensurability in Crystal Structures PhD thesis, Rijksuniversiteit Groningen (1978)
Rossnagel, K., Rotenberg, E., Koh, H., Smith, N. V. & Kipp, L. Continuous tuning of electronic correlations by alkali adsorption on layered 1T-TaS2 . Phys. Rev. Lett. 95, 126403 (2005).
Acknowledgements
This work was financially supported by the German Research Foundation under grant DFG-GRK1621. J.T. and J.G. gratefully acknowledge financial support by the German Research Foundation through the Emmy Noether program (grant GE 1647/2-1). Y.I.J. and P.A. were supported by US Department of Energy grant DE-FG02-06ER46285. We thank K. Rossnagel for fruitful discussions.
Author information
Authors and Affiliations
Contributions
J.G. and P.A. conceived the research project on 1T-TaS2. T.R., J.T., Y.I.J., M.v.Z. and J.G. conducted the synchrotron experiments. H.B. grew the single crystals. T.R., J.T. and K.K. performed the DFT calculations. T.R., J.T. and J.G. analysed the results and developed the concept of orbitronics. T.R., J.T., K.K., M.v.Z., P.A., B.B. and J.G. prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Ritschel, T., Trinckauf, J., Koepernik, K. et al. Orbital textures and charge density waves in transition metal dichalcogenides. Nature Phys 11, 328–331 (2015). https://doi.org/10.1038/nphys3267
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys3267
This article is cited by
-
Dualistic insulator states in 1T-TaS2 crystals
Nature Communications (2024)
-
Endotaxial stabilization of 2D charge density waves with long-range order
Nature Communications (2024)
-
Orbital-selective charge-density wave in TaTe4
npj Quantum Materials (2023)
-
Order-disorder phase transition driven by interlayer sliding in lead iodides
Nature Communications (2023)
-
Dirac nodal arc in 1T-VSe2
Communications Materials (2023)
