
Abstract
Chemical reactions are manifestations of the dynamics of molecular valence electrons and their couplings to atomic motions. Emerging methods in attosecond science can probe purely electronic dynamics in atomic and molecular systems1,2,3,4,5,6. By contrast, time-resolved structural-dynamics methods such as electron7,8,9,10 or X-ray diffraction11 and X-ray absorption12 yield complementary information about the atomic motions. Time-resolved methods that are directly sensitive to both valence-electron dynamics and atomic motions include photoelectron spectroscopy13,14,15 and high-harmonic generation16,17: in both cases, this sensitivity derives from the ionization-matrix element18,19. Here we demonstrate a time-resolved molecular-frame photoelectron-angular-distribution (TRMFPAD) method for imaging the purely valence-electron dynamics during a chemical reaction. Specifically, the TRMFPADs measured during the non-adiabatic photodissociation of carbon disulphide demonstrate how the purely electronic rearrangements of the valence electrons can be projected from inherently coupled electronic–vibrational dynamics. Combined with ongoing efforts in molecular frame alignment20 and orientation21,22, TRMFPADs offer the promise of directly imaging valence-electron dynamics during molecular processes without involving the use of strong, highly perturbing laser fields23.
Similar content being viewed by others
Main
Figure 1 provides a conceptual overview of our method. Carbon disulphide, CS2, is a molecule that exhibits all the features generic to polyatomic dynamics: vibrational mode coupling, conical intersections, spin conversion and photodissociation. As such, its non-adiabatic photodissociation reaction CS2(X)+h ν(201.2 nm)→CS2*(C)→CS(X)+S(1D,3P), shown in Fig. 1, provides an excellent test. In this work we combine experimental measurements with theory to demonstrate how the TRMFPAD images the evolution of the valence-electronic structure of an excited-state wavepacket during the complex, coupled electron–nuclear processes inherent to chemical reactions. TRMFPADs probe both nuclear and electronic degrees of freedom through the photoionization matrix elements,  . These matrix elements describe how the initial wavepacket, and its subsequent evolution in time (Ψi(t)), is projected onto the ionization continuum, consisting of the cation (Ψ+) and photoelectron (Ψe) states, through dipole coupling (
. These matrix elements describe how the initial wavepacket, and its subsequent evolution in time (Ψi(t)), is projected onto the ionization continuum, consisting of the cation (Ψ+) and photoelectron (Ψe) states, through dipole coupling ( ) with the laser field (E; refs 18, 24). In the case of polyatomic molecules, Ψi(t) and Ψ+ are composed of coupled electronic and vibrational components (see Supplementary Information). The significance of the TRMFPAD can be understood by considering the intimate relationship between Ψe and the electronic part of Ψi(t). For example, under the well-known Born (plane-wave) approximation, Ψe is simply the spatial Fourier transform of the ionized orbital. Although this approximation is too crude, it highlights the direct link between the evolving valence-electronic components of Ψi(t) (Fig. 1c) and the outgoing photoelectron Ψe (Fig. 1a). The d(t) play a role in any ionization-based experiment: both time/kinetic-energy-resolved photoelectron spectroscopy (TRPES) and time-, energy- and angle-resolved measurements (TRMFPADs) are defined by d(t) (ref. 18); in high-harmonic generation both strong-field ionization and electron recombination are described by similar matrix elements19.
) with the laser field (E; refs 18, 24). In the case of polyatomic molecules, Ψi(t) and Ψ+ are composed of coupled electronic and vibrational components (see Supplementary Information). The significance of the TRMFPAD can be understood by considering the intimate relationship between Ψe and the electronic part of Ψi(t). For example, under the well-known Born (plane-wave) approximation, Ψe is simply the spatial Fourier transform of the ionized orbital. Although this approximation is too crude, it highlights the direct link between the evolving valence-electronic components of Ψi(t) (Fig. 1c) and the outgoing photoelectron Ψe (Fig. 1a). The d(t) play a role in any ionization-based experiment: both time/kinetic-energy-resolved photoelectron spectroscopy (TRPES) and time-, energy- and angle-resolved measurements (TRMFPADs) are defined by d(t) (ref. 18); in high-harmonic generation both strong-field ionization and electron recombination are described by similar matrix elements19.

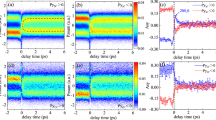
a,b, TRMFPADs (a) resulting from the wavepacket that evolves on the excited-state potential-energy surface (b). Dissociation to form free CS+S products occurs along the asymmetric stretch coordinate, through conical intersections to lower-lying singlet and triplet states. c, Left, Valence electronic wavefunctions in the initial linear geometry, of dominant Σu+ character. Right, The appearance of Πg character on wavepacket evolution. The time evolution of the MFPAD (a) directly reflects the evolution in valence-electronic character (c).
The CS2(C) electronic state is a manifold of quasi-bound vibrational levels, dissociative along the asymmetric stretch coordinate. Experimentally, a wavepacket on the C state was prepared by a pump laser pulse, then probed by a time-delayed ionization laser pulse25. We employed full three-dimensional momentum vector photoelectron imaging, in combination with molecular alignment, to simultaneously obtain energy-resolved and angle-resolved data (see Methods). In Fig. 2 we show our experimental results probing the dissociation of CS2(C). The energy-resolved TRPES data are shown for three photoelectron kinetic-energy regions, corresponding to projection of the wavepacket onto different cation vibrational states (further details in Supplementary Information). A quasi-periodic oscillation is observed in c, the highest-kinetic-energy window, corresponding to projection onto the vibrationless cation ground state. The oscillation is due to the coupling between stretch and bend vibrations in the excited-state wavepacket. However, the observation of the wavepacket oscillation becomes obscured in TRPES channels corresponding to projection onto higher vibrational levels in the ion, a and b. Importantly, the TRMFPADs still show—in all three energy windows—coherent wavepacket motion due to the evolving valence-electron character, as illustrated in Fig. 1.

a–c, TRPES from three different energy windows and their associated TRMFPADs at t=100, 500 and 900 fs (±50 fs) pump–probe time delays. The solid line through the TRPES data is the result of a fit including both biexponential and oscillatory terms; the dashed line below the data points shows only the oscillatory component of the fit (multiplied by 5 for emphasis). Statistical errors  in the TRPES data are of the order of the diameter of the plot marker circles, or less. The TRMFPADs are obtained by fitting an expansion in spherical harmonics to the angle-resolved photoelectron data; raw data are shown as a function of θ overlaid on the three-dimensional plots with statistical error bars (
in the TRPES data are of the order of the diameter of the plot marker circles, or less. The TRMFPADs are obtained by fitting an expansion in spherical harmonics to the angle-resolved photoelectron data; raw data are shown as a function of θ overlaid on the three-dimensional plots with statistical error bars ( ). The energy window in c corresponds to projection onto the vibrationless cation and shows a pronounced oscillation in both the TRPES and TRMFPADs. By contrast, in the energy windows in a and b, corresponding to projection onto vibrationally excited cation states, the TRPES—mainly reflecting vibrational dynamics—shows decreasing sensitivity to the wavepacket oscillation. In all three cases, however, the coherent oscillation remains observed in the TRMFPADs, demonstrating the sensitivity of this observable to the purely valence-electronic dynamics.
). The energy window in c corresponds to projection onto the vibrationless cation and shows a pronounced oscillation in both the TRPES and TRMFPADs. By contrast, in the energy windows in a and b, corresponding to projection onto vibrationally excited cation states, the TRPES—mainly reflecting vibrational dynamics—shows decreasing sensitivity to the wavepacket oscillation. In all three cases, however, the coherent oscillation remains observed in the TRMFPADs, demonstrating the sensitivity of this observable to the purely valence-electronic dynamics.
In the TRPES data, differences in the observed spectrum as a function of energy are due to projection onto different cation vibrational states. Because vibrational transitions have a nuclear coordinate dependence, different final vibrational states will ‘observe’ the wavepacket differently as the molecule stretches, bends and dissociates to products; hence the oscillation patterns observed in the TRPES are highly dependent on the vibrational part of Ψ+ (ref. 26). Furthermore, at higher internal energies, the cation vibrational states are unresolved and the energy-resolved signal, being an incoherent sum over these, has its oscillations averaged out, as seen in Fig. 2a and b.
These data reveal both the difference between the TRPES and TRMFPAD measurements and the underlying valence-electron dynamics. The wavepacket has an oscillation period of ∼1 ps, corresponding to the ∼33 cm−1 level spacing between the two quasi-bound, dissociative vibrational states populated by the pump laser pulse. As the wavepacket evolves on the potential-energy surface, the molecule explores different nuclear geometries resulting in changing valence-electronic character, as shown in Fig. 1. The valence-electronic wavefunction of the system therefore evolves on the timescale of nuclear motion, and the observed changes in the TRMFPAD reveal this electronic evolution. The independence of the period and phase of the TRMFPAD oscillation from the final vibrational state demonstrates that the MFPAD observables disentangle the vibrational and electronic motions of this reacting molecule.
In the following we develop a general model for TRMFPADs, which emphasizes the relationship between the TRMFPADs and the excited-state non-adiabatic wavepacket. The free electron, Ψe, is a scattering wavefunction expressed as an expansion, with amplitudes and phases, in spherical scattering waves24,27. Although TRPES measurements are in principle sensitive to both the electronic and vibrational components of Ψi(t) through their dependence on d(t), the angle-averaging implicit in TRPES obscures the details of Ψe. Angle-resolved measurements in the molecular frame—TRMFPADs—provide the most detailed information on Ψe and, hence, the evolving valence-electronic structure of Ψi(t). Theoretical studies previously suggested the possibility, in non-adiabatic wavepacket dynamics, that angle-resolved measurements will be useful28,29. So far, however, experimental studies have been restricted to corroborating excited-state dynamics inferred from TRPES studies and ab initio dynamics calculations14,15.
In general, we can write the TRMFPAD as an expansion in spherical harmonic functions Y L M(θ,ϕ), with expansion coefficients βL M(t), often termed ‘anisotropy parameters’. This can further be written in terms of the time-independent ionization channels using the excited-state wavepacket expansion coefficients Cn(t) and ‘eigenstate’ expansion coefficients βL M(n,n′) (see Supplementary Information):

In this form it is clear how the TRMFPAD, described by the βL M(t), responds to the evolving wavepacket defined by the Cn(t). In particular we note that the βL M(t) can be normalized to be independent of the integrated photoelectron yield, so the angular forms of the TRMFPADs are independent of both the population decay and vibrational Franck–Condon overlap factors, which primarily determine the form of the energy-resolved TRPES (as in Fig. 2, left).
In general, the exact angular structure of the TRMFPAD depends on both the magnitudes and phases of the d(t) (see Supplementary Information), ab initio methods for which are in development but remain very challenging for polyatomics29. Therefore, for simplicity, we apply symmetry arguments to determine the allowed photoionization channels and work in the linear geometry (corresponding to  symmetry). This model provides a route to understanding the key aspects of the TRMFPAD. The initially prepared wavepacket has Σu+ electronic character, which projects, on ionization, to the cation X(Πg) state and photoelectron wavefunctions of πg symmetry. The wavepacket at later times will develop mixed Σu++Πg electronic character, enabling further projection onto photoelectron wavefunctions of σu+ and δu symmetries. To gain insight into the time evolution of the MFPAD, we set all symmetry-allowed radial matrix elements to be non-zero, and normalize such that the u continuum carries 20% of the magnitude of the g continuum (matrix elements are listed in Supplementary Table S1). The radial matrix elements are used to calculate the βL M(n,n′); we then use equation (1) to calculate the TRMFPADs. The Cn(t) were set as appropriate for the prepared wavepacket, composed of two quasi-bound vibrational levels separated by ∼33 cm−1, as shown in Fig. 3a. The coherent superposition of these levels corresponds to the wavepacket motion shown in Fig. 1, and is projected onto the photoelectron continuum by the probe laser pulse at Δt.
symmetry). This model provides a route to understanding the key aspects of the TRMFPAD. The initially prepared wavepacket has Σu+ electronic character, which projects, on ionization, to the cation X(Πg) state and photoelectron wavefunctions of πg symmetry. The wavepacket at later times will develop mixed Σu++Πg electronic character, enabling further projection onto photoelectron wavefunctions of σu+ and δu symmetries. To gain insight into the time evolution of the MFPAD, we set all symmetry-allowed radial matrix elements to be non-zero, and normalize such that the u continuum carries 20% of the magnitude of the g continuum (matrix elements are listed in Supplementary Table S1). The radial matrix elements are used to calculate the βL M(n,n′); we then use equation (1) to calculate the TRMFPADs. The Cn(t) were set as appropriate for the prepared wavepacket, composed of two quasi-bound vibrational levels separated by ∼33 cm−1, as shown in Fig. 3a. The coherent superposition of these levels corresponds to the wavepacket motion shown in Fig. 1, and is projected onto the photoelectron continuum by the probe laser pulse at Δt.

a, The wavepacket, composed of two quasi-bound, dissociating vibrational states, Ψi(t)=C1(t)ψ1+C2(t)ψ2, is prepared by a pump laser pulse (not illustrated). The probe laser pulse projects the wavepacket onto the ionization continuum at Δt and detection of the photoelectron yields TRMFPADs. b, Calculated TRMFPADs based on the symmetry-allowed photoelectron wavefunctions for CS2(C) photodissociation, using equation (1). The coordinate system for photoionization is also illustrated. The calculation includes a convolution with the experimental molecular-frame axis distribution; full details are given in Supplementary Information. The TRMFPADs show a coherent oscillation, imaging the valence-electronic character of the excited state as the prepared wavepacket evolves. The initial Σu+ electronic character is seen in the TRMFPADs at 100 fs. At 500 fs, the wavepacket has developed Πg electronic character, leading to the peaked distribution shown. By 900 fs, coherent evolution has led to a re-appearance of Σu+ electronic character.
In Fig. 3b we show the calculated TRMFPADs for an aligned sample. The TRMFPADs show large changes in intensity along the z axis, and smaller changes in the perpendicular direction. In good agreement with the experimental data shown in Fig. 2, the TRMFPADs show a coherent oscillation that directly reflects the evolving valence-electronic structure. For example, at t=500 fs, the MFPAD is peaked along the z axis, reflecting the appearance of Πg electronic character in the excited-state wavepacket, as illustrated in Fig. 1c. The symmetry-based calculations show how the evolution in valence-electronic character is projected onto the TRMFPADs independently of the coupled vibrational dynamics.
Our combined experimental and theoretical results demonstrate how TRMFPADs can provide deep insight into valence-electron dynamics. We anticipate that developments in the ab initio calculation of both excited-state wavepacket evolution and photoionization dynamics, combined with burgeoning methods for molecular alignment and orientation, will reveal unprecedented details of valence-electronic dynamics during complex molecular processes such as chemical reaction.
Methods
Energy-, time- and three-dimensional angle-resolved photoelectron data were recorded from a transiently aligned sample of CS2. The sample (0.5% in He), introduced through a 1 kHz pulsed nozzle (Even-Lavie), was non-adiabatically aligned using an intense 805 nm pulse (100 fs, 4×1012 Wcm−2). Femtosecond pump–probe measurements were carried out within the picosecond window of the rotational half-revival, during which the molecules were fixed in space14. The initial wavepacket in the C(1Σu+) electronic state was prepared by a pump pulse of 201.2 nm radiation, corresponding energetically to the barrier to linearity in the C state and chosen to lie between two of the quasi-bound vibrational states; both states were encompassed by the ∼200 cm−1 bandwidth of the pulse. Neighbouring pairs of quasi-bound states are of the form (m1ν1+m2ν2) and ([m1−1]ν1+[m2+1]ν2), where ν1 and ν2 are the (symmetric) stretch and bend vibrational modes and m1 and m2 are (unassigned) quanta in each mode. The two levels populated by the probe pulse are separated by ∼33 cm−1. A time-delayed femtosecond probe pulse of 268.3 nm radiation was used to probe the sample through ionization. The cross-correlation width of the pump and probe laser pulses was 125 fs. All laser pulses were linearly polarized with parallel polarization vectors, such that the alignment and pump–probe measurements were all made in the same reference frame (that is, same z axis). Full three-dimensional momentum vector imaging of the photoelectrons was employed to obtain the TRPES and TRMFPADs. Images were built up on an event-by-event basis using an imaging photoelectron spectrometer with a triple-stack microchannel plate detector backed by a crossed delay-line anode; three-dimensional momentum distributions were reconstructed from the (x, y, time-of-flight) data obtained experimentally. Further details of this technique are given in refs 14, 15.
References
Sansone, G. et al. Electron localization following attosecond molecular photoionization. Nature 465, 763–766 (2010).
Haessler, S. et al. Attosecond imaging of molecular electronic wavepackets. Nature Phys. 6, 200–206 (2010).
Smirnova, O. et al. High harmonic interferometry of multi-electron dynamics in molecules. Nature 460, 972–977 (2009).
Uiberacker, M. et al. Attosecond real-time observation of electron tunnelling in atoms. Nature 446, 627–632 (2007).
Drescher, M. et al. Time-resolved atomic inner-shell spectroscopy. Nature 419, 803–807 (2002).
Goulielmakis, E. et al. Real-time observation of valence electron motion. Nature 466, 739–743 (2010).
Zewail, A. H. Four-dimensional electron microscopy. Science 328, 187–193 (2010).
Sciaini, G. et al. Electronic acceleration of atomic motions and disordering in bismuth. Nature 458, 56–59 (2009).
Barwick, B., Flannigan, D. J. & Zewail, A. H. Photon-induced near-field electron microscopy. Nature 462, 902–906 (2009).
Reckenthaeler, P. et al. Time-resolved electron diffraction from selectively aligned molecules. Phys. Rev. Lett. 102, 213001 (2009).
Barty, A. et al. Ultrafast single-shot diffraction imaging of nanoscale dynamics. Nature Photon. 2, 415–419 (2008).
Bressler, C. et al. Femtosecond XANES study of the light-induced spin crossover dynamics in an iron(II) complex. Science 323, 489–492 (2009).
Blanchet, V., Zgierski, M. Z., Seideman, T. & Stolow, A. Discerning vibronic molecular dynamics using time-resolved photoelectron spectroscopy. Nature 401, 52–54 (1999).
Bisgaard, C. Z. et al. Time-resolved molecular frame dynamics of fixed-in-space CS2 molecules. Science 323, 1464–1468 (2009).
Gessner, O. et al. Femtosecond multidimensional imaging of a molecular dissociation. Science 311, 219–222 (2006).
Itatani, J. et al. Tomographic imaging of molecular orbitals. Nature 432, 867–871 (2004).
Wörner, H. J., Bertrand, J. B., Corkum, P. B. & Villeneuve, D. M. High-harmonic homodyne detection of ultrafast chemical dynamics. Nature 466, 604–607 (2010).
Stolow, A. & Underwood, J. G. Time-resolved photoelectron spectroscopy of nonadiabatic dynamics in polyatomic molecules. Adv. Chem. Phys. 139, 497–583 (2008).
Le, A-T., Lucchese, R. R., Tonzani, S., Morishita, T. & Lin, C. D. Quantitative rescattering theory for high-order harmonic generation from molecules. Phys. Rev. A 80, 013401 (2009).
Lee, K. F., Villeneuve, D. M., Corkum, P. B., Stolow, A. & Underwood, J. G. Field-free three-dimensional alignment of polyatomic molecules. Phys. Rev. Lett. 97, 173001 (2006).
Ghafur, O. et al. Impulsive orientation and alignment of quantum-state-selected NO molecules. Nature Phys. 5, 289–293 (2009).
Holmegaard, L. et al. Photoelectron angular distributions from strong-field ionization of oriented molecules. Nature Phys. 6, 428–432 (2010).
Lezius, M., Blanchet, V., Ivanov, M. Y. & Stolow, A. Polyatomic molecules in strong laser fields: Nonadiabatic multielectron dynamics. J. Chem. Phys. 117, 1575–1588 (2002).
Dill, D. Fixed-molecule photoelectron angular distributions. J. Chem. Phys. 65, 1130–1133 (1976).
Townsend, D. et al. 1b2(1Σu+) excited state decay dynamics in CS2 . J. Chem. Phys. 125, 234302 (2006).
Resch, K., Blanchet, V., Stolow, A. & Seideman, T. Toward polyatomic wave packet decomposition: Final state effects. J. Phys. Chem. A 105, 2756–2763 (2001).
Cooper, J. & Zare, R. N. Angular distribution of photoelectrons. J. Chem. Phys. 48, 942–943 (1968).
Suzuki, Y., Stener, M. & Seideman, T. Multidimensional calculation of time-resolved photoelectron angular distributions: The internal conversion dynamics of pyrazine. J. Chem. Phys. 118, 4432–4443 (2003).
Arasaki, Y., Takatsuka, K., Wang, K. & McKoy, V. Time-resolved photoelectron spectroscopy of wavepackets through a conical intersection in NO2 . J. Chem. Phys. 132, 124307 (2010).
Acknowledgements
We thank Guorong Wu (NRC Canada) for assistance with the alignment experiments, and Vincent McKoy (Caltech) for helpful discussion.
Author information
Authors and Affiliations
Contributions
C.Z.B. and A.S. planned the experiments. C.Z.B. and O.J.C. recorded experimental data. P.H. developed theoretical models and calculated TRMFPADs. O.J.C. calculated ab initio potential-energy surfaces. C.Z.B. and P.H. analysed data. All authors discussed the data and modelling, and contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Information (PDF 1342 kb)
Rights and permissions
About this article
Cite this article
Hockett, P., Bisgaard, C., Clarkin, O. et al. Time-resolved imaging of purely valence-electron dynamics during a chemical reaction. Nature Phys 7, 612–615 (2011). https://doi.org/10.1038/nphys1980
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys1980
This article is cited by
-
Probing electron localization during molecular dissociation by femtosecond strong-field ion momentum spectroscopy
Communications Physics (2023)
-
X-ray induced Coulomb explosion imaging of transient excited-state structural rearrangements in CS2
Communications Physics (2023)
-
Real-space subfemtosecond imaging of quantum electronic coherences in molecules
Nature Photonics (2022)
-
Geometric and electronic structure probed along the isomerisation coordinate of a photoactive yellow protein chromophore
Nature Communications (2020)
-
Spatiotemporal imaging of valence electron motion
Nature Communications (2019)
