
Abstract
Manipulation of the molecular-axis distribution is an important ingredient in experiments aimed at understanding and controlling molecular processes1,2,3,4,5,6. Samples of aligned or oriented molecules can be obtained following the interaction with an intense laser field7,8,9, enabling experiments in the molecular rather than the laboratory frame10,11,12. However, the degree of impulsive molecular orientation and alignment that can be achieved using a single laser field is limited13 and crucially depends on the initial states, which are thermally populated. Here we report the successful demonstration of a new technique for laser-field-free orientation and alignment of molecules that combines an electrostatic field, non-resonant femtosecond laser excitation14 and the preparation of state-selected molecules using a hexapole2. As a unique quantum-mechanical wavepacket is formed, a large degree of orientation and alignment is observed both during and after the femtosecond laser pulse, which is even further increased (to 〈cosθ〉=−0.74 and 〈cos2θ〉=0.82, respectively) by tailoring the shape of the femtosecond laser pulse. This work should enable new applications such as the study of reaction dynamics or collision experiments in the molecular frame, and orbital tomography11 of heteronuclear molecules.
Similar content being viewed by others

Main
The outcome of molecular collision experiments is strongly affected by the angular anisotropies in the initial molecular axis distribution. In bimolecular and molecule–surface collisions, collision cross-sections sensitively depend on the relative arrangement of the collision partners1,2. Likewise, photon–molecule collisions such as X-ray diffraction and photodissocation experiments aimed at the elucidation of molecular structure or photochemical activity depend on and can benefit from angular confinement of the sample3,4. The two most important moments of the molecular axis distribution are the ‘alignment’ (〈cos2θ〉) and ‘orientation’ (〈cosθ〉), where θ is the angle between the molecular axis and a reference axis.
First attempts to orient and align molecules relied on electrostatic fields. A hexapole electric field can be used to state-select polar molecules and orient them through their first-order Stark effect2,5 using a moderate field strength. The orientation is limited by the selected state. ‘Brute-force orientation’ uses a strong homogeneous electrostatic field and relies on the second- and higher-order Stark effect6. It requires molecules with a large dipole moment and extremely high electric-field strengths.
As a part of extensive efforts aimed at achieving laser-controlled chemistry15,16,17, laser-controlled alignment8 has attracted considerable attention. Suitably chosen laser fields can exert torques on molecules, exploiting the interaction of the laser field with the molecular polarizability. Both adiabatic alignment, where molecules are exposed to a slowly varying laser field18, and non-adiabatic (impulsive) alignment, where molecules align after receiving a kick by a short laser pulse7, have been successfully implemented. The advantage of the latter is that it leads to a periodic alignment under field-free conditions, when the rotational wavepacket excited by the short laser pulse rephases. This is desirable for applications where the presence of a strong laser field interferes. The impulsive alignment that can be achieved using a single laser pulse is intrinsically limited13 but can be improved by applying a laser-pulse sequence13,19 or an appropriately tailored laser-pulse profile20,21.
The availability of aligned molecules has sparked the emergence of new techniques such as tomographic imaging of electronic orbitals11, and the observation of multicentre interference in high-harmonic generation as a tool for structural determination12. Further improvements in the alignment of molecules are likely to enable new applications, such as diffractive imaging at emerging X-ray free-electron lasers such as LCLS at Stanford, XFEL at Hamburg and SCSS at Spring-822,23.
The progress on laser-controlled orientation has thus far been modest. Single-colour laser fields with a waveform that is symmetric with respect to the laser polarization axis cannot create ‘head-versus-tail’ order. Laser-driven orientation by two-colour phase-locked laser excitation24, by terahertz half-cycle pulses25 or by combining electrostatic and pulsed non-resonant laser fields14,26 has been proposed. Only the last method has been experimentally demonstrated on OCS9,27 and on HXeI (ref. 28) and iodobenzene29 molecules. The degree of orientation that can be achieved very sensitively depends on the rotational temperature of the molecular sample. In this letter, we present experiments where laser field-free orientation (of the nitric oxide (NO) molecule) is achieved by combining short-pulse laser control, hexapole quantum state selection and d.c. orientation in an electrostatic field through the first-order Stark effect. Strong revivals of both orientation and alignment are produced, which are furthermore significantly enhanced by tailoring the laser pulse profile by the application of a sigmoidal phase mask.
The experimental set-up is shown in Fig. 1 and is described in detail in the Methods section. Experiments on impulsive alignment were performed with the polarization of the 800 nm pump pulse in the plane of the 2D detector, and the polarization of the 400 nm probe pulse parallel to the detector axis30. This arrangement minimizes the role of both geometrical (that is, the dependence of ionization processes on the angle between the molecular axis and the laser polarization) and dynamical alignment by the probe-laser pulse. Figure 2a shows a N2+ image recorded without the pump pulse, where the observed 2D distribution is isotropic. When the pump pulse precedes the probe pulse (Fig. 2b–d), rapid modifications of the 2D projection are observed as a function of the pump–probe time delay, including a planar delocalization of the molecule perpendicular to the pump-laser polarization (τ=19.6 ps, Fig. 2c) and an alignment along the laser polarization (τ=20.1 ps, Fig. 2d). The alignment 〈cos2θ〉 is closely related to the value of 〈cos2θ2D〉, which can be directly retrieved from the images, where θ2D is the angle of the measured velocity with respect to the vertical (laser polarization) axis. The dependence of 〈cos2θ2D〉 on the pump–probe time delay is plotted in Fig. 2e. Strong alignment revivals spaced by approximately 5 ps are observed, indicating that a rotational wavepacket has been produced that freely evolves after the laser excitation has ended. The Raman process that leads to the formation of this wavepacket involves transitions ΔJ=±1 or 2, where J is the angular momentum. As ΔJ=±2 transitions are dominant at high intensity31, as confirmed by the Fourier spectrum in Fig. 2f, the observed rephasing period is Trev≈1/(4B c)=5 ps, where B is the rotational period of NO. Note that the oscillation for τ<0 originates from a small pre-pulse, occurring 21.5 ps before the main pulse.

State-selected NO molecules are prepared using a hexapole focusing field. An 800 nm pulse is used to impulsively align and orient the molecules. A 400 nm pulse is used to probe the induced rotational dynamics by Coulomb explosion. The fragments produced by the 400 nm pulse are detected by a velocity map imaging spectrometer (VMIS) and recorded with a CCD (charge-coupled device) camera. Both pulses are linearly polarized and their polarization axes are controlled by means of half-wave plates. A computer-controlled pulse shaper can be used to modify the 800 nm pulse profile, enabling the molecular orientation to be enhanced.

a–d, Bottom panels: N2+ images recorded for different pump–probe time delays τ using a 1 mJ (400 nm) probe-laser pulse polarized along the detector axis, which fragments the molecules after they have interacted with a 300 μJ, 90 fs (800 nm) pump laser polarized parallel to the plane of the detector. Top panels: Corresponding three-dimensional angular distributions computed numerically by solving the time-dependent Schrödinger equation (TDSE). e, Time-dependent evolution of the 〈cos2θ2D〉 (black points and line) obtained from the images (see text). The theoretical evolution (red line) of the impulsive alignment 〈cos2θ〉 calculated by solving the TDSE for a 90 fs pulse with an intensity of 4×1013 W cm−2 and taking into account a pre-pulse of 5×1012 W cm−2 at a time delay of τ=−21.5 ps is also shown. f, Fourier spectrum of the averaged value 〈cos2θ2D〉 shown in e, revealing the beat frequencies involved in the time-dependent alignment.
To determine the experimental value of 〈cos2θ〉, the time dependence of 〈cos2θ2D〉 was compared with that of 〈cos2θ〉 calculated by solving the TDSE (see Supplementary Information). The time dependence of 〈cos2θ〉 depends on the kick strength (laser intensity and pulse duration; see Supplementary Equation (7)) and on the temperature of the sample. As we start from a single rotational state (that is, T=0 K), we can infer the actual degree of molecular alignment by using the measured pulse duration and adjusting only the strength of the laser field to match the measured structure. The best agreement was found for an intensity of 4×1013 W cm−2 (see Fig. 2e) and enables us to conclude that 〈cos2θ〉 reaches a maximum of 0.76. As confirmed in further experiments and calculations where the pulse energy was increased, this is the maximum impulsive alignment that can be achieved by a single laser pulse. This is the first observation of the theoretical prediction of the saturation of alignment13. Unlike experiments performed at room temperature or in a rotationally cold sample, when working with a sample of state-selected molecules saturation of alignment happens at an intensity where ionization of the molecules remains small.
Figure 2b, which was recorded away from a revival time, shows hints of a complicated structure in the angular distribution. This suggests that, using a detection scheme that enables full retrieval of the three-dimensional angular distribution (such as the use of femtosecond extreme-ultraviolet pulses that dissociatively ionize the aligned molecules30), it may be possible to fully reconstruct the amplitudes and phases of the rotational wavepacket produced.
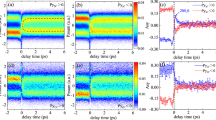
The experiments shown in Fig. 2 reveal molecular alignment but no orientation. To investigate impulsive orientation, experiments were carried out with the pump-laser polarization parallel to both the detector axis and the electrostatic field, and the polarization of the probe pulse at an angle of 30∘ with respect to this axis. This enables us to distinguish between fragments with a recoil velocity away from or towards the 2D detector30. The measured 2D velocity distribution consists of two spots, with a relative intensity defined by the molecular orientation. Note that alignment can also be reconstructed in this geometry, although it is more influenced by the probe-laser pulse. In Fig. 3a–f, N2+ fragment images are shown as a function of the pump–probe time delay. In the upper part fragments are detected where the N atom initially pointed downwards, whereas fragments where the N atom pointed upwards are detected below. The dominant N2+ detection clearly oscillates between the upper and the lower part of the detector. This periodic inversion of the asymmetry indicates that the pump pulse produces a rotational wavepacket with an orientation that periodically alternates. Similar to the previous discussion on impulsive alignment, we can extract 〈cosθ2D〉 (see Fig. 3g), and a corresponding 〈cosθ〉, which is obtained from a TDSE simulation for a pump-pulse intensity of 4×1013 W cm−2. As only quantum beats between adjacent levels ΔJ=±1 contribute to the impulsive orientation, the full revival period is Trev≈1/(B c)=20 ps. The degree of orientation reaches a maximum value of |〈cosθ2D〉|=0.4, which according to the TDSE simulation corresponds to |〈cosθ〉|=0.6. As for the alignment, the quantitative relation between 〈cosθ2D〉 and 〈cosθ〉 is a complex function of the initial orientation in the d.c. field, the dynamic and geometric alignment and orientation created by the probe laser, and the angle between the pump and probe lasers.

a–f, N2+ images recorded for a pump polarization parallel to the detector axis and a probe polarization at an angle of 30∘ with respect to the pump polarization, using the same pulse energies and durations as in Fig. 2. g,h, Time-dependent evolution of 〈cosθ2D〉 and 〈cos2θ2D〉 (black line and points) obtained from the images (see text). The theoretical evolution of the impulsive alignment 〈cos2θ〉 and orientation 〈cosθ〉 obtained by solving the TDSE for a 90 fs pump of intensity 4×1013 W cm−2 by taking into account a pre-pulse of 5.012 W cm−2 at −21.5 ps is shown as well (red line).
Fourier-transform-limited (FTL) pulses are not optimal for accomplishing impulsive alignment and orientation. On the basis of the parity mixing in the initial state we can show (see Supplementary Information) that, in the case of perfect alignment, 〈cosθ〉=0.88 would be possible at the d.c. field strength used in the experiment. However, TDSE simulations indicate that impulsive orientation by an FTL pulse saturates at 〈cosθ〉=−0.6. Increasing the laser intensity does not improve the orientation and furthermore leads to a high degree of ionization. It has been theoretically demonstrated that field-free alignment of diatomic molecules can be increased when a sigmoidal phase mask is applied20. A sigmoidal function introduces a phase jump described by three parameters, namely an amplitude A, a laser frequency ν0 where the jump occurs and a parameter s describing the slope of the phase function around ν0:

The corresponding temporal profile consists of a long leading edge that pre-aligns the molecules before a main kick pulse. As the main pulse interacts with a sample of aligned molecules, the excitation leads to a better alignment than achieved with only the FTL pulse20. To explore the utility of sigmoidal phase patterns for achieving molecular orientation and alignment, a feedback-loop-controlled experiment was performed using a simplex optimization algorithm. The three parameters characterizing the sigmoidal phase pattern were varied to minimize 1−|〈cosθ2D〉| at τ=20.1 ps, where the FTL pulse created a minimum in 1−|〈cosθ2D〉|. Figure 4a,b shows N2+ images recorded with the FTL pulse and with the best sigmoidal phase function found by the simplex method, respectively. The orientation obtained with the shaped laser pulse clearly exceeds that obtained with the FTL pulse. This is further illustrated in Fig. 4c, which shows the result of integrating the images along the vertical (x axis), and in Fig. 4d, where a comparison is shown between the orientation obtained without the pump pulse, with an FTL pulse and with the optimally shaped laser pulse. A comparison of the experiment with TDSE results using the experimental parameters of the sigmoidal phase function (A=6π, ν0=0.375 THz and s=−0.0012 THz) enables us to estimate 〈cosθ〉=−0.74, meaning that seven molecules are facing one side for every molecule facing the other side. According to a calculation performed under the same conditions using an evolutionary algorithm with an unrestricted parameterization of the phase, this value can further be increased to 〈cosθ〉=−0.84. As shown in Fig. 4g, the optimized laser field consists of two long and weak pulses before the main pulse that contain approximately half of the energy of the main pulse. The two pulses pre-orient the molecule before the main kick, similar to what was shown in ref. 20 for alignment. Consequently, an improved rephasing of the rotational wave packet at the full revival period is achieved. The orientation revival is accompanied by a strong revival of the alignment (〈cos2θ〉=0.82).

a,b, N2+ images recorded at a time delay of τ=20.1 ps after the interaction of NO molecules with a 300 μJ, 90 fs FTL pulse (a) and a shaped laser pulse where a sigmoidal phase mask has been applied (b). c, Intensity integration of the images (A, in black, and B, in red) along the x axis. d, Evolution of 1−|〈cosθ2D〉| without a pump pulse, with the FTL pulse and with the sigmoidal phase function that leads to the optimization of the orientation, for five consecutive experimental images. The sigmoidal phase function clearly improves the orientation of the molecules and increases |〈cosθ2D〉| to 0.5. e,f, Time-dependent evolution (black line and points) and theoretical evolution (red lines) of 〈cosθ2D〉 and 〈cos2θ2D〉 obtained from the images recorded using the sigmoidal phase mask. g, Comparison of an experimental cross-correlation of the optimized pump pulse with an FTL pulse (black points) and the calculated pulse profile when applying the sigmoidal phase mask (red line).
To achieve 〈cosθ〉=−0.74 a d.c. electric field of 13 kV cm−1 and a shaped laser pulse reaching a peak intensity of ∼2×1013 W cm−2 were used. Calculations suggest that on increasing the d.c. field to 50 kV cm−1 and allowing for peak intensities (within the shaped laser pulse) up to 3×1013 W cm−2 (which is still below the intensity of the FTL pulses used in this study), and using an unrestricted parameterization of the phase, the value of 〈cosθ〉 can be increased to 0.964. At this point, 99.2% of the molecules are pointing in the same direction, within a narrow cone that has a full-width at half-maximum of 23∘.
As hexapole state selection has been demonstrated on a variety of polar symmetric and near-asymmetric top molecules, there exists a large number of molecules that are amenable to this technique. Hence, samples of impulsively oriented molecules can be prepared where the degree of orientation is high enough to enable application in further experiments that may lead to new insights in the understanding of molecules and their fundamental interactions. In addition, these results may progress the field of ultracold molecules, where orientation of polar molecules in an optical trap could assist in the formation of a superlattice or a Bose–Einstein or Fermi–Dirac condensate.
Methods
The first step of the experiment consists of preparing the molecular sample in one selected state. A mixture of NO (10%) in argon at a stagnation pressure of 1.8 bar is expanded into a vacuum chamber with a pulsed nozzle valve operated at a repetition rate of 12.5 Hz. After passing through a skimmer placed 5 cm from the nozzle, molecules enter into a 1 m hexapole state selector composed of six hexagonally placed rods raised to a 9 kV voltage that alternates in sign. The radial electric field along the length of the hexapole induces a Stark shift of the molecular rotational levels. Molecules that experience a negative Stark shift (high-field seeking) are pushed toward the rods (defocused) and molecules that experience a positive Stark shift (low-field seeking) are focused by the electric field at a position that depends on their internal state (for a review, see ref. 2). In our experiment, the hexapole focuses NO molecules that reside in the upper low-field-seeking upper-lambda ɛ=−1 component (see Supplementary Information) of the J=1/2 state at the centre of the VMIS. NO molecules in the lower ɛ=+1 component of J=1/2 are deflected away. Owing to the supersonic cooling of the pulsed nozzle expansion the higher excited rotational levels of NO are only weakly populated and pass essentially undeflected along the axis of the hexapole owing to their much smaller Stark effect. At the centre of the VMIS, the state-selected molecules experience a d.c. electric field of 13 kV cm−1 between the repeller and the extractor that induces a constant orientation of 〈cosθ〉=−0.30 for the low-field-seeking state selected here, where θ is defined as the angle between the electrostatic field and the molecular NO axis.
In the second step of the experiment, impulsive molecular orientation/alignment is achieved by excitation with a short laser pulse and then probed through Coulomb explosion of the molecules by a second pulse. Both pulses are obtained from a linearly polarized Ti:sapphire femtosecond amplifier (30 mJ, 70 fs at a repetition rate of 25 Hz). The probe beam is frequency doubled in a 3 mm potassium dihydrogen phosphate crystal. After doubling, the pulse energy is approximately equal to 1 mJ. Before passing through a delay line, which enables us to vary the pump–probe delay, the pump beam is sent through a 4-f dispersion line where a 640 pixel spatial light modulator (SLM from JenOptiK) is placed at the Fourier plane. The SLM device enables us to modify the spectral phase of the pump electric field, which alters the temporal profile of the pulse. The two beams are recombined collinearly using a dichroic mirror and focused into the VMIS chamber by an f=20 cm achromatic lens. The laser beams intersect the molecular sample at the centre of the VMIS, where charged fragments originating from Coulomb explosion of the molecules by the 400 nm pulse are projected onto a position-sensitive detector consisting of a dual microchannel plate followed by a phosphor screen. A 20 ns, 1.6 kV gating pulse is applied on the microchannel plate to selectively detect fragments of a certain mass/charge ratio arriving on the detector. The 2D velocity distribution on the phosphor screen is recorded by a CCD camera and then transferred to an acquisition computer. To remove the effect of background contamination, a subtraction routine was used to subtract fragments measured in the absence of the pulsed NO beam during every second laser shot.
Change history
25 March 2009
NOTE: In the version of this Letter originally published online, the author list of ref. 29 was incorrect. It has now been corrected in all versions of this Letter.
References
Kuipers, E. W., Tenner, M. G., Kleyn, A. W. & Stolte, S. Observation of steric effects in gas–surface scattering. Nature 334, 420–422 (1988).
Parker, D. H. & Bernstein, R. B. Oriented molecule beams via the electrostatic hexapole: Preparation, characterization, and reactive scattering. Ann. Rev. Phys. Chem. 40, 561–595 (1989).
Rischel, C. et al. Femtosecond time-resolved X-ray diffraction from laser-heated organic films. Nature 390, 490–492 (1997).
Rakitzis, T. P., Van den Brom, A. J. & Janssen, M. H. M. Directional dynamics in the photodissociation of oriented molecules. Science 303, 1852–1854 (2004).
Brooks, P. R. Reactions of oriented molecules. Science 193, 11–16 (1976).
Loesch, H. J. & Remscheid, A. Brute force in molecular reaction dynamics: A novel technique for measuring steric effects. J. Chem. Phys. 93, 4779–4790 (1990).
Rosca-Pruna, F. & Vrakking, M. J. J. Experimental observation of revival structures in picosecond laser-induced alignment of I2 . Phys. Rev. Lett. 87, 153902 (2001).
Stapelfeldt, H. & Seideman, T. Colloquium: Aligning molecules with strong laser pulses. Rev. Mod. Phys. 75, 543–557 (2003).
Goban, A., Minemoto, S. & Sakai, H. Laser field-free molecular orientation. Phys. Rev. Lett. 101, 013001 (2008).
Litvinyuk, I. V. et al. Alignment-dependent strong field ionization of molecules. Phys. Rev. Lett. 90, 233003 (2003).
Itatani, J. et al. Tomographic imaging of molecular orbitals. Nature 432, 867–871 (2004).
Kanai, T., Minemoto, S. & Sakai, H. Quantum interference during high-order harmonic generation from aligned molecules. Nature 435, 470–474 (2005).
Leibscher, M., Averbukh, I. Sh. & Rabitz, H. Molecular alignment by trains of short laser pulses. Phys. Rev. Lett. 90, 213001 (2003).
Cai, L., Marango, J. & Friedrich, B. Time-dependent alignment and orientation of molecules in combined electrostatic and pulsed nonresonant laser fields. Phys. Rev. Lett. 86, 775–778 (2001).
Zare, R. N. Laser control of chemical reactions. Science 279, 1875–1879 (1998).
Judson, R. S. & Rabitz, H. Teaching lasers to control molecules. Phys. Rev. Lett. 68, 1500–1503 (1992).
Assion, et al. Control of chemical reactions by feedback-optimized phase-shaped femtosecond laser pulses. Science 282, 919–922 (1998).
Sakai, H. et al. Controlling the alignment of neutral molecules by a strong laser field. J. Chem. Phys. 110, 10235–10238 (1999).
Bisgaard, C. Z., Poulsen, M. D., Peronne, E., Viftrup, S. S. & Stapelfeldt, H. Observation of enhanced field-free molecular alignment by two laser pulses. Phys. Rev. Lett. 92, 173004 (2004).
Rouzée, A., Hertz, E., Lavorel, B. & Faucher, O. Towards the adaptive optimization of field-free molecular alignment. J. Phys. B 41, 074002 (2008).
Suzuki, T., Sugawara, Y., Minemoto, S. & Sakai, H. Optimal control of nonadiabatic alignment of rotationally cold N2 molecules with the feedback of degree of alignment. Phys. Rev. Lett. 100, 033603 (2008).
Neutze, R., Wouts, R., Van der Spoel, D., Weckert, E. & Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 406, 752–757 (2000).
Chapman, H. N. et al. Femtosecond diffractive imaging with a soft-X-ray free-electron laser. Nature Phys. 2, 839–843 (2006).
Vrakking, M. J. J. & Stolte, S. Coherent control of molecular orientation. Chem. Phys. Lett. 271, 209–215 (1997).
Machholm, M. & Henriksen, N. E. Field-free orientation of molecules. Phys. Rev. Lett. 87, 193001 (2001).
Friedrich, B. & Herschbach, D. Enhanced orientation of polar molecules by combined electrostatic and nonresonant induced dipole forces. J. Chem. Phys. 111, 6157–6160 (1999).
Sakai, H., Minemoto, S., Nanjo, H., Tanji, H. & Suzuki, T. Controlling the orientation of polar molecules with combined electrostatic and pulsed, nonresonant laser fields. Phys. Rev. Lett. 90, 083001 (2003).
Baumfalk, R., Nahler, N. H. & Buck, U. Photodissociation of oriented HXeI molecules in the gas phase. J. Chem. Phys. 114, 4755–4758 (2001).
Holmegaard, L. et al. Laser-induced alignment and orientation of quantum-state-selected large molecules. Phys. Rev. Lett. 102, 023001 (2009).
Gijsbertsen, et al. Direct determination of the sign of the NO dipole moment. Phys. Rev. Lett. 99, 213003 (2007).
Meijer, A. S. et al. Controlling rotational state distributions using two-pulse stimulated Raman excitation. Phys. Rev. A 76, 023411 (2007).
Acknowledgements
We acknowledge P. Johnsson for discussions and technical assistance provided during this project. This work is part of the research program of the ‘Stichting voor Fundamenteel Onderzoek der Materie (FOM)’, which is financially supported by the ‘Nederlandse organisatie voor Wetenschappelijk Onderzoek (NWO)’. Financial support by the Marie Curie Research Training Networks ‘XTRA’ is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
Supplementary Informations (PDF 1202 kb)
Rights and permissions
About this article
Cite this article
Ghafur, O., Rouzée, A., Gijsbertsen, A. et al. Impulsive orientation and alignment of quantum-state-selected NO molecules. Nature Phys 5, 289–293 (2009). https://doi.org/10.1038/nphys1225
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys1225
This article is cited by
-
Single-shot measurement of few-cycle optical waveforms on a chip
Nature Photonics (2022)
-
Field-free molecular orientation by delay- and polarization-optimized two fs pulses
Scientific Reports (2020)
-
Molecular movie of ultrafast coherent rotational dynamics of OCS
Nature Communications (2019)
-
All-optical field-free three-dimensional orientation of asymmetric-top molecules
Nature Communications (2018)
-
Isotope-selective ionization utilizing field-free alignment of isotopologues using a switched nanosecond laser pulse
Applied Physics B (2018)
