
Abstract
Methanogenesis is the primary biogenic source of methane in the atmosphere and a key contributor to climate change. The long-standing dogma that methanogenesis originated within the Euryarchaeota was recently challenged by the discovery of putative methane-metabolizing genes in members of the Bathyarchaeota, suggesting that methanogenesis may be more phylogenetically widespread than currently appreciated. Here, we present the discovery of divergent methyl-coenzyme M reductase genes in population genomes recovered from anoxic environments with high methane flux that belong to a new archaeal phylum, the Verstraetearchaeota. These archaea encode the genes required for methylotrophic methanogenesis, and may conserve energy using a mechanism similar to that proposed for the obligate H2-dependent methylotrophic Methanomassiliicoccales and recently described Candidatus ‘Methanofastidiosa’. Our findings indicate that we are only beginning to understand methanogen diversity and support an ancient origin for methane metabolism in the Archaea, which is changing our understanding of the global carbon cycle.
Similar content being viewed by others


Methanogenesis performed by anaerobic archaea represents the largest biogenic source of methane on Earth1–3. This process is a key component of the global carbon cycle and plays an important role in climate change, owing to the high warming potential of methane4. Methanogenic archaea are widespread in nature and have been detected in a diverse range of environments including wetlands, rice agriculture, landfills, hydrothermal vents, ruminants and termites1,2. Traditionally, all methanogens belonged to the archaeal phylum Euryarchaeota and until recently were classified into seven orders1,3,5 (Methanobacteriales, Methanococcales, Methanomicrobiales, Methanosarcinales, Methanocellales, Methanopyrales and Methanomassiliicoccales). Substrates for methane production include H2/CO2 (hydrogenotrophic), acetate (acetoclastic) and methylated compounds (methylotrophic)5. Hydrogenotrophic methanogenesis is the most widespread1, being found in all methanogenic orders with the exception of Methanomassiliicoccales, and it has been suggested that this is the ancestral form of methane production6. Although the prevailing source of atmospheric methane is through direct acetate cleavage7, only members of the Methanosarcinales are capable of acetoclastic methanogenesis5,7. Methylotrophic methanogens are found in the orders Methanosarcinales, Methanobacteriales and Methanomassiliicoccales, and can be classified in two groups based on the presence or absence of cytochromes. Methylotrophs without cytochromes are obligately H2-dependent, while those that possess cytochromes (that is, members of the Methanosarcinales), can also oxidize methyl groups to CO2 via a membrane-bound electron transport chain5.
Our knowledge of methanogenic substrate utilization and energy conservation is still incomplete but is rapidly expanding through the development of new molecular techniques such as genome-centric metagenomics. This includes the discovery of hydrogenotrophic methanogenesis genes encoded in Methanosaeta8, a genus originally thought to be strictly acetoclastic, the hypothesized mode of energy metabolism in Methanonomassiliiccocales5,9, and the recently described sixth class of euryarchaeotal methanogens, Candidatus ‘Methanofastidiosa’, which is restricted to methanogenesis through methylated thiol reduction10. The hypothesis that methane metabolism originated early in the evolution of the Euryarchaeota11 has recently been challenged following the discovery of putative methane metabolism in the archaeal phylum Bathyarchaeota (formerly Miscellaneous Crenarchaeota Group)12,13. Two near-complete genomes belonging to this phylum contain distant homologues of the genes necessary for hydrogenotrophic and methylotrophic methanogenesis, including the methyl-coenzyme M reductase complex (MCR)12. These findings raise the possibility that further divergent methanogenic lineages await discovery.
Discovery of a divergent mcrA cluster
To enrich for novel methanogenic diversity, triplicate anaerobic digesters were inoculated with a mixture of samples sourced from natural (rumen and lake sediment) and engineered (anaerobic digesters and lagoon) environments, and supplied with alpha cellulose. The reactors showed stable methane production and were sampled over time. Six metagenomes comprising a total of 111 Gb were generated and co-assembled14 (Supplementary Table 1), and differential coverage binning recovered 68 substantially to near-complete population genomes, including four containing mcrA genes. The homologues in three of these genomes are closely related to recognized methanogenic lineages in the Euryarchaeota (94–99% amino acid (aa) identity), while the fourth genome (V1) encodes an mcrA that is divergent from extant sequences (∼68% aa identity). This divergent mcrA was used to screen the Sequence Read Archive (SRA)15, and closely related mcrA genes (85–100% aa identity, Supplementary Table 2) were identified in three metagenomic data sets obtained from high methane flux habitats: an anaerobic digester treating palm oil mill effluent (Malaysia)16, an iso-alkane degrading methanogenic enrichment culture from tailings ponds (Mildred Lake Settling Basin, Canada)17, and formation waters of coalbed methane wells (CD-8 and PK-28) located within the Surat Basin (Queensland, Australia)12 (Supplementary Table 1). Subsequent assembly and binning of these data sets led to the recovery of four additional population genomes (V2–V5, Table 1) containing mcrA sequences similar to the one found in V1. All five divergent mcrA-containing genomes are near-complete (90.3–99.1%) with low contamination (<1%), with the exception of V5, which is moderately complete (60.2%) with no detectable contamination. The estimated size of these genomes is relatively small (1.2–1.7 Mb), with a tight GC content distribution (55.4–58.1%) and number of predicted genes ranging between 1,248 and 1,758 (Table 1).
Phylogenetic analyses of the mcrA and 16S rRNA genes from these population genomes confirmed the novelty of this group (Fig. 1). The mcrA genes form a distinct cluster divergent from Euryarchaeota, the recently described Bathyarchaeota, and an environmental clade (recovered from Coal Oil Point seep field, COP)12 (Fig. 1a). The 16S rRNA gene tree shows that these sequences cluster with environmental clones belonging to the loosely defined Terrestrial Miscellaneous Crenarchaeota Group (TMCG), which currently has no genomic representation (Fig. 1b).

a, McrA protein tree showing monophyletic clustering of the divergent McrA sequences from V1–V4 (red) outside known euryarchaeotal and bathyarchaeotal methanogenic lineages. b, 16S rRNA gene tree showing the placement of V1–V5 (bolded) with environmental sequences classified as the Terrestrial Miscellaneous Crenarchaeota Group (TMCG), using the Bathyarchaeota 16S rRNA sequences as the outgroup. Bootstrap values were calculated via non-parametric bootstrapping with 100 replicates, and are represented by circles.
A new archaeal phylum, Verstraetearchaeota
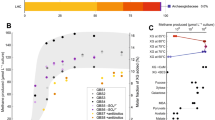
A genome tree comprising the five divergent mcrA-containing population genomes and 346 publicly available archaeal genomes was inferred using a concatenated set of 122 archaeal-specific marker genes. This tree confirms the placement of the population genomes in an archaeal phylum that is sister to the Crenarchaeota and Korarchaeota (Fig. 2a and Supplementary Fig. 1), for which we propose the name Candidatus ‘Verstraetearchaeota’. Comparative genomic analysis revealed that these genomes have an average amino acid identity (AAI) of 41 ± 1.5% to other Archaea, supporting their grouping as a separate phylum18. Genome tree phylogeny, AAI and 16S rRNA sequence identity suggest that these genomes represent two distinct genera within the Verstraetearchaeota (Fig. 2a and Supplementary Table 2), with V1–V3 belonging to one genus and V4–V5 belonging to the second. V1 and V2, and V4 and V5 are highly similar to one another (99 and 97% AAI, respectively), and probably represent different strains of the same species. We propose the genus names Candidatus ‘Methanomethylicus’ for V1–V3 and Candidatus ‘Methanosuratus’ for V4–V5.

a, Genome tree illustrating the placement of V1–V5 relative to 346 archaeal genomes, using the DPANN superphylum as the outgroup. The tree was inferred using the maximum-likelihood method with a concatenated set of 122 archaeal-specific marker genes, and bootstrap values were calculated using non-parametric bootstrapping with 100 replicates (represented by circles). Methanogenic Euryarchaeota are expanded and colour-coded based on order-level classification. b, Presence/absence of genes involved in methane metabolism and energy conservation for euryarchaeotal methanogens, the Verstraetearchaeota (V1–V5) and Bathyarchaeota (BA1–BA2). Annotation was primarily based on classification of genes into KO groups, and a functional complex consisting of multiple subunits was considered present if ≥80% of the genes comprising the complex were detected. Colour coding on the right-hand side of the figure reflects the genome tree classification (MMI: Methanomicrobiales; MS: Methanosarcinales; MCe: Methanocellales; MMa: Methanomassiliicoccales; MF: Candidatus ‘Methanofastidiosales’; MB: Methanobacteriales; MCo: Methanococcales; MP: Methanopyrales; V: Verstraetearchaeota; B: Bathyarchaeota).
Methane metabolism
A KEGG orthology (KO) based comparison of the four near-complete Verstraetearchaeota genomes (V1–V4; Table 1) was conducted against 156 selected archaeal genomes (Supplementary Fig. 2). At this global metabolic scale, Verstraetearchaeota resemble the Methanomassiliicoccales, C. ‘Methanofastidiosa’ and Bathyarchaeota, and are distinct from traditional euryarchaeotal methanogens (Supplementary Fig. 2). The V1–V4 genomes share 458 gene orthologues, with an additional 195 shared among V1–V3. The V4 genome contains 147 genes for which no homologues could be identified in the other genomes, consistent with the phylogenetic placement of this genome in a separate genus (Fig. 2a and Supplementary Fig. 3).
Metabolic reconstruction of the near-complete Verstraetearchaeota genomes revealed the presence of key genes associated with methylotrophic methanogenesis (Figs 2b, 3a, and Supplementary Table 3). These include a complete methyl-coenzyme M reductase complex (MCR; mcrABG and ancillary genes mcrCD) with conserved binding sites for coenzyme M, coenzyme B and F430 cofactors (Supplementary Fig. 4), and genes required for methane production from methanol (mtaA) and methanethiol (mtsA). In addition, a variable complement of genes for methylamine utilization (mtbA, mtmBC, mtbBC, mttBC and ramA) was identified across the genomes (Figs 2b, 3a and Supplementary Table 3). However, due to high sequence similarity, it is difficult to infer the substrate specificity of mtaA and mtbA, and it has been suggested that mtaA is capable of functionally replacing mtbA in methylamine utilization5. Subunit H of the tetrahydromethanopterin S-methyltransferase (mtrH) is also present in the Verstraetearchaeota and co-located with a gene encoding a methylamine-specific corrinoid protein, similar to the mtrH gene found in the Bathyarchaeota12,19, suggesting it plays a role in methylamine:coenzyme M methyl transfer activity. The remaining genes for hydrogenotrophic and acetoclastic methanogenesis are missing from the genomes, consistent with obligate H2-dependent methylotrophic methanogenesis. The contigs containing methanogenesis-related genes were independently found in all of the Verstraetearchaeota genomes and have sequence composition characteristics typical of their respective genomes (Supplementary Fig. 5).

a, Pathways for methylotrophic methanogenesis, hydrogen cycling and suggested energy conservation mechanisms. The first mechanism is shown in blue and entails heterodisulfide reduction by HdrABC–MvhABDG coupled to ferredoxin reoxidation by an Fpo-like (dark blue) or Ehb (light blue) complex. The second mechanism involves HdrD coupled directly to the Fpo-like complex (light green), and the third possibility links HdrD to an FAD hydrogenase (dark green). b, Other metabolic pathways including potential sugar and amino acid utilization, an incomplete TCA cycle and a nucleotide salvage pathway. Black arrows indicate genes that were found in all near-complete Verstraetearchaeota genomes (V1–V4), and grey arrows represent genes that are present in only a subset of the genomes (coloured circles indicate in which subset of genomes they are found). Dashed light grey arrows show parts of a pathway that are missing in all genomes.
The Verstraetearchaeota appear to have three possible mechanisms for the reduction of heterodisulfide (CoM-SS-CoB), formed during the final step of methanogenesis (Fig. 3a). The first mechanism couples the exergonic H2-dependent reduction of heterodisulfide to ferredoxin reduction, and involves a heterodisulfide reductase (HdrABC) and a putative cytoplasmic F420-non-reducing hydrogenase (MvhADG and MvhB)3 (Fig. 3a). Divergent homologues of the genes encoding this electron-bifurcating complex are found co-located in the Verstraetearchaeota (Supplementary Table 3). In addition, homologues of the membrane-bound NADH-ubiquinone oxidoreductase (Nuo), which show high sequence similarity to the F420-methanophenazine oxidoreductase (Fpo) found in Methanosarcinales20, are present and form an Fpo-like complex capable of re-oxidizing reduced ferredoxin, with concomitant translocation of protons or sodium ions across the cytoplasmic membrane (Fig. 3a). Similar to Methanomassiliicoccales5,9 and Methanosaeta thermophila21, the subunits required for binding and oxidation of NADH (NuoEFG) or F420 (FpoFO) are missing from the Verstraetearchaeota, supporting the use of ferredoxin as an electron donor. Reduced ferredoxin can also be re-oxidized by the energy-conserving hydrogenase B (Ehb) found in all genomes, as suggested for Methanosphaera stadtmanae1 and the recently described C. ‘Methanofastidiosum methylthiophilus’10. The proton gradient generated by the Fpo-like or Ehb complex can drive adenosine triphosphate (ATP) synthesis via an archaeal-type ATP synthase, thereby coupling methanogenesis to energy conservation and enabling internal hydrogen cycling (Fig. 3a). The second mechanism for heterodisulfide reduction uses HdrD, which is present in three copies in all Verstraetearchaeota genomes. Although HdrD is conventionally linked to HdrE (ref. 2), the latter is missing from the genomes, an observation that has also been made for the Methanomassiliicoccales9, C. ‘Methanofastidiosum methylthiophilus’10 and Bathyarchaeota12. Instead, HdrD may interact directly with the Fpo-like complex and together function as an energy-converting ferredoxin:heterodisulfide oxidoreductase5. Alternatively, the last mechanism could use hdrD, which is co-located with a gene encoding a cytoplasmic flavin adenine dinucleotide-containing dehydrogenase (glcD), similar in sequence to d-lactate dehydrogenase. This mechanism may allow the reduction of heterodisulfide to be coupled to lactate utilization (Fig. 3a), as suggested for the Bathyarchaeota12 and Archaeoglobus fulgidis22.
Fermentative metabolism
In addition to methane metabolism, the Verstraetearchaeota appear to be capable of utilizing sugars as a carbon source and generating acetyl-CoA via the Embden–Meyerhof–Parnas (EMP) pathway and pyruvate-ferredoxin oxidoreductase (Por) (Fig. 3b and Supplementary Table 4). In contrast to V1–V3, V4 contains multiple polysaccharide and sugar transporters, yet lacks phosphoglycerate mutase (Apg) and enolase (Eno), suggesting that V4 may use an alternative or divergent form of the pathway for the conversion of 3-phosphoglycerate to phosphoenolpyruvate. All genomes also encode an intermediate type II/III ribulose 1,5-biphosphate carboxylase/oxygenase (RuBisCO) (Supplementary Fig. 6), which may function in a nucleotide salvage pathway generating 3-phosphoglycerate from adenosine monophosphate (AMP) that can enter the EMP pathway to produce acetyl-CoA (refs 23–25) (Fig. 3b). The presence of multiple genes encoding peptide and amino-acid transporters suggests that the Verstraetearchaeota are capable of importing these compounds, which can then be degraded to keto-acids by various endopeptidases (PepB, PepD, PepP and PepT), glutamate dehydrogenase (Gdh) and aminotransferases (AspB, ArgD, GabT, IlvE, GlmS and HisC)26,27. The keto-acids could be converted to acetyl-CoA by 2-oxoacid:ferredoxin oxidoreductases (Kor, Ior, Por and Vor) and aldehyde-ferredoxin oxidoreductases (Aor)12,26 (Fig. 3b and Supplementary Table 4). Acetyl-CoA, generated through the various metabolic processes, can be converted to acetate via an archaeal-type adenosine diphosphate (ADP)-forming acetate synthetase (Acd)28, which would allow the Verstraetearchaeota to produce energy via substrate-level phosphorylation. Although this is unusual for microorganisms involved in methane metabolism, the ability to carry out complex fermentation has also been detected in the Bathyarchaeota12 and could suggest substrate diversification for these newly discovered methanogens.
Environmental distribution
To assess the environmental distribution of the Verstraetearchaeota, we searched for related 16S rRNA and mcrA genes in publicly available databases. Full-length 16S rRNA gene sequences in Greengenes most closely related to V1–V3 were detected in two habitats, a mesophilic methanogenic granular sludge reactor29 and deep sediment of the freshwater Lake Pavin (France)30. Those most closely related to V4–V5 were found in the Niibori petroleum reservoir31 and Zavarzin thermal spring32 (Fig. 1b). Most other clones related to Crenarchaeota and found in hot spring sediments are basal to the Verstraetearchaeota. Partial-length gene sequences identified in the SRA suggest that the environmental distribution of the Verstraetearchaeota extends to wetlands, sediments (lakes and river estuaries), soils and hydrocarbon-rich environments (fracture water, petroleum seep, mud volcanoes, subduction zone and deep-sea vents) (Supplementary Table 5). Common characteristics of the habitats in which Verstraetearchaeota are found include anoxic conditions, high methane fluxes and a likelihood for increased concentrations of methylated compounds.
Evolution of methanogenesis
To examine the evolutionary history of methane metabolism in the Archaea, protein trees were constructed for all subunits of the MCR complex and ancillary proteins (Fig. 4 and Supplementary Figs 7, 8). In all cases, the Verstraetearchaeota form a robust monophyletic group, as do the seven traditional and one recently described euryarchaeotal methanogenic orders, the Bathyarchaeota and the COP environmental clade12, which is consistent with the genome tree and rules out recent lateral gene transfer (LGT) of the MCR complex between orders or phyla. Furthermore, higher-level associations corresponding to the traditional ‘Class I’ and ‘Class II’ groupings of euryarchaeotal methanogens are present and support vertical inheritance13, with the exception of an ancient lateral transfer of mrt from Methanococcales to some Methanobacteriales. The Verstraetearchaeota are not robustly associated with any other methanogenic lineage in the gene trees, which precludes our ability to distinguish between vertical inheritance and ancient LGT from a euryarchaeotal donor. However, given that this lineage has all the core genes required for methanogenesis and associated energy conservation, it is unlikely that methanogenesis in the Verstraetearchaeota could be a result of LGT. We therefore predict that methanogenesis pre-dates the origin of the Euryarchaeota13 and that this trait will be found in as yet undiscovered lineages across the Archaea, supporting methane metabolism as one of the most ancient forms of microbial metabolism.

The trees were inferred using the McrABG found in the Verstraetearchaeota, euryarchaeotal methanogens, Bathyarchaeota and an environmental data set from Coal Oil Point. Bootstrap values were determined using non-parametric bootstrapping with 100 replicates and are represented by circles. Nodes with <75% bootstrap support were collapsed. Lineages are collapsed (depicted as wedges), and colour-coded labelling is according to the genome tree phylogeny in Fig. 2 (‘Class I’ methanogens: Methanobacteriales (MB), Methanococcales (MCo) and Methanopyrales; ‘Class II’ methanogens: Methanomicrobiales (MMi), Methanosarcinales (MS) and Methanocellales (MCe); Methanomassiliicoccales (MMa); Candidatus ‘Methanofastidiosales’ (MF); Verstraetearchaeota (V); Bathyarchaeota (B); Coal Oil Point (COP)).
C. ‘Methanomethylicus mesodigestum’ (gen. nov., sp. nov.)
Methanomethylicus mesodigestum (L. n. methanum, methane; L. adj. methylicus, methyl, referring to the methylotrophic methane-producing metabolism; L. pref. meso, middle, highlighting the preference for mesophilic habitats; L. v. digestus, form of digest, undergo digestion, pertaining to the anaerobic digester environment). This organism is inferred to be capable of methylotrophic methanogenesis, is non-motile, and not cultivated, represented by near-complete population genomes V1 (type strain) and V2 obtained from anaerobic digesters (Table 1).
C. ‘Methanomethylicus oleusabulum’ (gen. nov., sp. nov.)
Methanomethylicus oleusabulum (L. pref. methanum, methane; L. adj. methylicus, methyl, referring to the methylotrophic methane-producing metabolism; L. pref. oleum, oil; L. n. sabulum, sand, representing the oil sands environment). This organism is inferred to be capable of methylotrophic methanogenesis, is non-motile, and not cultivated, represented by a near-complete population genome V3 obtained from an iso-alkane degrading enrichment culture from tailings ponds (Table 1).
C. ‘Methanosuratus petracarbonis’ (gen. nov., sp. nov.)
Methanosuratus petracarbonis (L. pref. methanum, methane, referring to the methylotrophic methane-producing metabolism; suratus, referring to the Surat Basin where the genomes were found; L. n. petra, rock, representing the formation water habitat; L. n. carbo carbonis, coal, carbon, pertaining to the coalbed methane well environment). This organism is inferred to be capable of methylotrophic methanogenesis, is non-motile, and not cultivated, represented by population genomes V4 (near-complete) and V5 (moderately complete) obtained from formation water of coalbed methane wells (Table 1).
Description of Methanomethyliaceae (fam. nov.)
The description is the same as for the genus Methanomethylicus. Suff, -aceae, ending to denote a family. Type genus: Candidatus ‘Methanomethylicus’, gen. nov.
Description of Methanomethyliales (ord. nov.)
The description is the same as for the genus Methanomethylicus. Suff. -ales, ending to denote an order. Type family: Methanomethyliaceae, fam. nov.
Description of Methanomethylia (class nov.)
The description is the same as for the genus Methanomethylicus. Suff. -ia, ending to denote a class. Type order: Methanomethyliales, ord. nov.
Description of Verstraetearchaeota (phyl. nov.)
We propose the name Verstraetearchaeota for this phylum; Verstraete, recognizing the contributions of Professor Willy Verstraete (Centre for Microbial Ecology and Technology, Ghent University, Belgium) to the development and application of engineered microbial ecosystems such as anaerobic digesters; suff. -archaeota, ending to denote an archaeal phylum.
Methods
Anaerobic digester sample collection, DNA extraction and library preparation
Triplicate cellulose-degrading anaerobic digesters (biological replicates n = 3) were inoculated with a mixture sourced from natural (rumen and lake sediment) and engineered (anaerobic lagoon, granules and several digesters) environments33. The digesters were operated for 362 days with continual monitoring of reactor performance parameters and microbial community dynamics. Samples for DNA extraction and metagenomic sequencing were taken at day 96 and day 362 (total of six samples), centrifuged at 14,000g for 2 min to collect the biomass, and snap-frozen in liquid nitrogen before storing at −80 °C until further processing. DNA was extracted from these samples using the MP-Bio Fast DNA Spin Kit for Soil (MP Biomedicals), following the manufacturer's instructions, and stored at −20 °C until sequencing. Libraries for metagenomic sequencing were prepared as described previously and sequenced using one-third of an Illumina HiSeq2000 flowcell lane each14.
Metagenome assembly and population genome binning
The six metagenomes (2 × 150 bp paired-end reads, 111 Gb) generated from the cellulose-degrading anaerobic digesters were quality filtered, trimmed and combined into a large co-assembly using CLC Genomics Workbench v6 (CLC Bio)14. Population genomes were recovered from the co-assembly based on differential coverage profiles, kmer signatures and GC content using GroopM v2.0 (ref. 34), and manually refined using the ‘refine’ function in GroopM, resulting in 101 population genomes14 (PRJNA284316). One of the genomes (referred to as V1) contained an mcrA sequence with low similarity to euryarchaeotal mcrA sequences (∼68% aa identity).
Raw reads from a metagenome constructed from an anaerobic digester treating palm oil mill effluent16 (ERR276848, 18.1 Gb) were processed using SeqPrep to remove adaptor sequences and merge overlapping reads (https://github.com/jstjohn/SeqPrep.git), and Nesoni for quality trimming with a quality score threshold of 20 (https://github.com/Victorian-Bioinformatics-Consortium/nesoni.git). Processed reads were assembled using the de novo assembly algorithm in CLC Genomics Workbench v7, with a word size of 25 and bubble size of 50. Contigs larger than 2 kb were binned into population genomes using VizBin (ref. 35). This resulted in a population genome (referred to as V2) that contained an mcrA sequence identical to the one found in V1 (100% aa identity).
Metagenomes produced from iso-alkane-degrading enrichment cultures (SRR1313242–43, 22.4 Gb, technical replicates n = 2) and toluene-degrading microbial communities (SRR942927, SRR943311–13 and SRR943315, 85.6 Gb, technical replicates n = 5) from tailings ponds17 (Mildred Lake Settling Basin, Canada) were processed using CLC Genomics Workbench v8 for quality trimming and adapter removal. The metagenomes were combined into one large co-assembly using the de novo assembly algorithm in CLC with default parameters and minimum contig size of 500 bp. Binning of the contigs into population genomes was performed using MetaBAT (ref. 36), which resulted in 96 genomes, including one (referred to as V3) with an mcrA sequence highly similar to the V1 mcrA (95% aa identity).
Metagenomes generated from formation waters of coal bed methane wells (CD-8 (19 Gb), PK-28 (7.9 Gb)) were processed with SeqPrep and Nesoni for adapter removal, merging and quality trimming. The processed reads for each metagenome were assembled individually using the de novo assembly algorithm in CLC Genomics Workbench v6.5, with a word size of 25 and bubble size of 50. Contigs larger than 500 bp were binned into population genomes using DBB v1.0.0, which groups contigs based on coverage and tetranucleotide frequencies (https://github.com/dparks1134/DBB). Each of the data sets produced a population genome that contained an mcrA sequence similar to the V1 mcrA, that is, V4 for CD-8 (88% aa identity) and V5 for PK-28 (86% aa identity).
Population genome quality assessment
The five population genomes were manually refined by including unbinned contigs from the metagenomes based on Blastp (ref. 37) v2.2.30+ homology to the other genomes, and removing contaminating contigs based on coverage, GC, tetranucleotide frequency and taxonomic profiles of the genes using RefineM (https://github.com/dparks1134/refinem). Scaffolding between contigs was performed using the ‘roundup’ function in FinishM (https://github.com/wwood/finishm) and GapCloser (ref. 38). Completeness, contamination and strain heterogeneity estimates were calculated based on the presence of lineage-specific single copy marker genes using CheckM (ref. 39). The number of tRNA genes within each genome was calculated using tRNA-scan with default search parameters40.
Screening of SRA using Verstraetearchaeota mcrA sequences
A collection of environmental public metagenomes were downloaded from the SRA database, based on metadata from SRAdb (ref. 41), and processed using a custom bio-gem bio-sra (https://github.com/wwood/bioruby-sra/). SRA format files were converted to FASTQ using sra-tools (http://ncbi.github.io/sra-tools/) and HMMER (ref. 42) was used with an mcrA model to identify reads that probably represent mcrA genes. Each read was translated into six reading frames using OrfM (https://github.com/wwood/OrfM.git) and only reads with an e-value <1 e−5 were retained. Homology comparison between these reads and a data set of 144 mcrA genes (including Verstraetearchaeota, Bathyarchaeota and Euryarchaeota mcrA) was performed using Blastp v2.2.30+. Alignments with a bitscore >50 and a top hit to one of the V1–V4 mcrA sequences were tabulated to explore the environmental distribution of the Verstraetearchaeota. For each SRA metagenome, the number of hits with ≥85% similarity to a V1–V4 mcrA sequence and the average % amino acid identity of these hits were calculated. Total counts were reported for all metagenomes belonging to one SRA BioProject.
Screening of SRA using Verstraetearchaeota 16S rRNA sequences
The collection of metagenomes downloaded from the SRA database was mapped to the GreenGenes (ref. 43) v2013_08 99% dereplicated database of 16S rRNA sequences using BWA-MEM (ref. 44) with default parameters, and converted to a sorted indexed BAM file using samtools45. The SRA bam file was filtered twice using the ‘filter’ function in BamM (https://github.com/Ecogenomics/BamM.git) to only retain hits with ≥90% and ≥97% identity to a 16S rRNA sequence. Reads mapping to the GreenGenes IDs that most closely resemble the V1–V4 16S rRNA sequences were identified in the bam file. The total number of hits was calculated for all SRA BioProject metagenomes.
Genome tree phylogeny
An archaeal genome tree was constructed using the Genome Tree DataBase (GTDB v1.1.0, https://github.com/Ecogenomics/GTDBNCBI.git) with a concatenated set of 122 achaeal-specific conserved marker genes inferred from genomes available in NCBI (accessed July 2015) using GenomeTreeTK (https://github.com/dparks1134/GenomeTreeTK) (Supplementary Table 6). Marker genes were identified across 346 good-quality archaeal genomes and the Verstraetearchaeota genomes (V1–V5) using hidden Markov models (HMMs), and a phylogenetic genome tree was constructed from the concatenated alignments using FastTree (ref. 46) v2.1.8 under the WAG and GAMMA models. The tree was decorated with taxonomic information using tax2tree (ref. 47), and bootstrap support values were calculated via non-parametric bootstrapping of the multiple alignment data set (100 replicates) with GenomeTreeTK v0.0.14. The genome tree was visualized in ARB (ref. 48), rooted using the DPANN superphylum as an outgroup, and modified for publication in Adobe Illustrator.
16S rRNA gene phylogeny
The 16S rRNA genes identified in V1–V5 and Bathyarchaeota genomes12,27 were aligned with full-length 16S rRNA sequences in the GreenGenes database43 classified as Crenarchaeota, Bathyarchaeota (former Miscellaneous Crenarchaeota Group), Terrestrial Hot Spring Crenarchaeota Group (THSCG) and Aigarchaeota. The multiple sequence alignment from SSU-ALIGN (ref. 49) v0.1 was used to generate a phylogenetic tree using FastTree v2.1.8, and bootstrap support values were determined using 100 non-parametric bootstrap replicates. The 16S rRNA gene tree was visualized in ARB, rooted using Bathyarchaeota as an outgroup, and modified for publication in Adobe Illustrator.
Functional gene tree phylogeny
Amino acid sequences for functional genes of interest (mcr and rbc) were extracted from the Verstraetearchaeota (V1–V5), Bathyarchaeota (BA1 and BA2, if present), Methanomassiliicoccales and Methanofastidiosa genomes. These sequences were used to search for homologues across a database derived from genomes in the Integrated Microbial Genomes (IMG) database50 (accessed February 2015) using the ‘Blastp’ function in Mingle (https://github.com/Ecogenomics/mingle.git). The IMG database was dereplicated to 50 taxa per genus in a manner that aims to retain taxonomic diversity. A phylogenetic tree for the aligned sequences was constructed using FastTree v2.1.8, and bootstrap support values were determined with non-parametric bootstrapping (100 replicates). The gene trees were rooted at midpoint, decorated with taxt2tree and visualized in ARB. Trees were modified for publication in Adobe Illustrator, and nodes with <75% bootstrap support were collapsed in the McrABG trees.
Genome annotation and metabolic reconstruction
Contigs were annotated using PROKKA (ref. 51) v1.11.db20151014, and genes (putatively) involved in core metabolic pathways were verified using NCBI's Blast webserver52, homology search with Blastp v2.2.30+ across the IMG database v4.1, Pfam domains53, and gene trees. The consensus annotation was used for metabolic reconstruction. Additionally, a selection of 247 publically available genomes in the NCBI database belonging to Euryarchaeota, Crenarchaeota, Thaumarchaeota, Bathyarchaeota, Korarchaeota and Verstraetearchaeota were annotated by searching against Uniref100 (accessed October 2015) using the ‘Blastp’ function in Diamond v0.7.12 (https://github.com/bbuchfink/diamond.git). The top hit for each gene with an e-value <1 e−3 was mapped to the KO database54,55 using the Uniprot ID mapping files. Hits to each KO were summed to produce a count table for all genomes. This KO table was used in a principle component analysis (PCA) for overview comparison of metabolic profiles. It also formed the basis for the heatmap showing presence of absence of genes involved in methane metabolism. The PCAs and heatmap were generated with the Vegan56 and RColorBrewer57 packages in RStudio v3.2.0.
Comparison of MCR complex sequence active site conservation and conformation
The tertiary structures of McrA, McrB and McrG subunits from the Candidatus ‘Methanomethylicus mesodigestum'AD1 genome was predicted using I-TASSER v4.3 using the software default parameters58. The confidence scores (C-scores) of the top models were 0.69, 0.55 and 0.74, respectively, for the McrA, McrB, and McrG subunits. Protein–ligand binding sites were verified with the I-TASSER associated COACH package59, which showed coenzyme M, coenzyme B and cofactor F430 binding sites60 to be identical between V1 and the Methanopyrus kandleri MCR crystal structure (PDB ID: 1E6V). Superimposing the crystal structure of the AD1 MCR onto the Methanopyrus kandleri MCR crystal structure resulted in global structural alignment scores (TM-scores) for McrA, McrB and McrG of 0.98.1, 98.2 and 98.3, respectively. The root-mean-square deviations (RMSDs) of the TM-aligned residues for McrA, McrB and McrG subunits were 0.63, 0.51 and 0.86 Å, respectively.
Data availability
The sequences have been deposited at the NCBI Sequence Read Archive (Whole Genome Submission) under BioProject ID PRJNA321438 with accession nos. MAGS00000000 (V1), MAGS00000000 (V2), MAGS00000000 (V3), MAGS00000000 (V4) and MAGW00000000 (V5).
References
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W. & Hedderich, R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591 (2008).
Hedderich, R. & Whitman, W. B. in The Prokaryotes Vol. 4 (eds Rosenberg, E. et al.) 662 (Springer, 2013).
Thauer, R. K. et al. Hydrogenases from methanogenic Archaea, nickel, a novel cofactor and H2 storage. Ann. Rev. Biochem. 79, 507–536 (2010).
IPCC Climate Change 2014: Synthesis Report (eds Core Writing Team, Pachauri, R.K. & Meyer, L.A.) (Cambridge Univ. Press, 2014).
Lang, K. et al. New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of ‘Candidatus Methanoplasma termitum’. Appl. Environ. Microbiol. 81, 1338–1352 (2015).
Bapteste, E., Brochier, E. & Boucher, Y. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea 1, 353–363 (2005).
Fournier, G. P. & Gogarten, J. P. Evolution of acetoclastic methanogenesis in Methanosarcina via horizontal gene transfer from cellulolytic Clostridia. J. Bacteriol. 190, 1124–1127 (2008).
Rotaru, A.-E. et al. A new model for electron flow during anaerobic digestion: direct interspecies electron transfer to Methanosaeta for the reduction of carbon dioxide to methane. Energy Environ. Sci. 7, 408–415 (2014).
Borrel, G. et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genomics 15, 1–23 (2014).
Nobu, M. K., Narihiro, T., Kuroda, K., Mei, R. & Liu, W.-T. Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J. 10, 2478–2487 (2016).
Gribaldo, S. & Brochier-Armanet, C. The origin and evolution of Archaea: a state of the art. Phil. Trans. R. Soc. Lond. B 361, 1007–1022 (2006).
Evans, P. N. et al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350, 434–438 (2015).
Borrel, G., Adam, P. S. & Gribaldo, S. Methanogenesis and the Wood–Ljungdahl pathway: an ancient, versatile and fragile association. Genome Biol. Evol. 8, 1706–1711 (2016).
Vanwonterghem, I., Jensen, P. D., Rabaey, K. & Tyson, G. W. Genome-centric resolution of microbial diversity, metabolism and interactions in anaerobic digestion. Environ. Microbiol. 18, 3144–3158 (2016).
Leinonen, R., Sugawara, H. & Shumway, M. The sequence read archive. Nucleic Acids Res. 39, 19–21 (2011).
Bala, J. D., Lalung, J. & Ismail, N. Palm oil mill effluent (POME) treatment ‘Microbial communities in an anaerobic digester’: a review. Int. J. Sci. Res. Publ. 4, 1–24 (2014).
Siddique, T. et al. Long-term incubation reveals methanogenic biodegradation of C5 and C6 iso-alkanes in oil sands tailings. Environ. Sci. Technol. 49, 14732–14739 (2015).
Konstantinidis, K. T. & Tiedje, J. M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264 (2005).
Lloyd, K. G. et al. Predominant archaea in marine sediments degrade detrital proteins. Nature 496, 215–218 (2013).
Baumer, S. et al. The F420H2 dehydrogenase from Methanosarcina mazei is a redox-driven proton pump closely related to NADH dehydrogenases. J. Biol. Chem. 275, 17968–17973 (2000).
Welte, C. & Deppenmeier, U. Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim. Biophys. Acta 1837, 1130–1147 (2014).
Hocking, W. P., Stokke, R., Roalkvam, I. & Steen, I. H. Identification of key components in the energy metabolism of the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidis by transcriptome analyses. Front. Microbiol. 5, 95 (2014).
Tabita, F. R. et al. Function, structure and evolution of the RuBisCO-like proteins and their RuBisCO homologs. Microbiol. Mol. Biol. Rev. 71, 576–599 (2007).
Sato, T., Atomi, H. & Imanaka, T. Archaeal type III RuBisCOs function in a pathway for AMP metabolism. Science 315, 1003–1006 (2007).
Castelle, C. J. et al. Genomic expansion of domain Archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 25, 690–701 (2015).
Seitz, K. W., Lazar, C. S., Hinrichs, K.-U., Teske, A. P. & Baker, B. J. Genomic reconstruction of a novel, deeply branched sediment archaeal phylum with pathways for acetogenesis and sulfur reduction. ISME J. 10, 1696–1705 (2016).
Lazar, C. S. et al. Genomic evidence for distinct carbon substrate preferences and ecological niches of Bathyarchaeota in estuarine sediments. Environ. Microbiol. 18, 1200–1211 (2015).
Musfeldt, M. & Schonheit, P. Novel type of ADP-forming acetyl coenzyme A synthetase in hyperthermophilic Archaea: heterologous expression and characterization of isoenzymes from the sulfate reducer Archaeoglobus fulgidus and the methanogen Methanococcus jannaschii. J. Bacteriol. 184, 636–644 (2002).
Yashiro, Y. et al. Methanoregula formicica sp. nov., a methane-producing archaeon isolated from methanogenic sludge. Int. J. System. Evol. Microbiol. 61, 53–59 (2011).
Borrel, G. et al. Stratification of Archaea in the deep sediments of a freshwater meromictic lake: vertical shift from methanogenic to uncultured archaeal lineages. PLoS ONE 7, e43346 (2012).
Kobayashi, H. et al. Phylogenetic diversity of microbial communities associated with the crude-oil large-insoluble-particle and formation-water components of the reservoir fluid from a non-flooded high-temperature petroleum reservoir. J. Biosci. Bioeng. 113, 204–210 (2012).
Rozanov, A. S. et al. Molecular analysis of the benthos microbial community in Zavarzin thermal spring (Uzon Caldera, Kamchatka, Russia). BMC Genomics 15, S12 (2014).
Vanwonterghem, I. et al. Deterministic processes guide long-term synchronised population dynamics in replicate anaerobic digesters. ISME J. 8, 2015–2028 (2014).
Imelfort, M. et al. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ 2, e603 (2014).
Laczny, C. C. et al. VizBin—an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome 3, 1 (2015).
Kang, D. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165 (2015).
Altschul, S. F., Gisch, W., Miller, W., Meyers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 1–6 (2012).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Fichant, G. A. & Burks, C. Identifying potential tRNA genes in genomic DNA sequences. J. Mol. Biol. 220, 659–671 (1991).
Zhu, Y., Stephens, R. M., Meltzer, P. S. & Davis, S. R. SRAdb: query and use public next-generation sequencing data from within R. BMC Bioinformatics 14, 1–4 (2013).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Soo, R. M. et al. An expanded genomic representation of the phylum Cyanobacteria. Genome Biol. Evol. 6, 1031–1045 (2014).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618 (2011).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004).
Nawrocki, E. P. Structural RNA Homology Search and Alignment Using Covariance Models PhD thesis, Washington Univ. Saint Louis (2009).
Markowitz, V. M. et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40, 115–122 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Marchler-Bauer, A. et al. CDD: NCBI's conserved domain database. Nucleic Acids Res. 43, 222–226 (2014).
Finn, R. D. et al. The Pfam protein families database. Nucleic Acids Res. 42, 222–230 (2014).
Kanehisa, M. & Goto, S. KEGG Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, 199–205 (2014).
Vegan: community ecology package (Oksanen, J. et al., 2013); http://CRAN.R-project.org/package=vegan
RColorBrewer: ColorBrewer Palettes (Neuwirth, E., 2011); http://CRAN.R-project.org/package=RColorBrewer
Roy, A. K. & Zhang, Y. I-TASSER A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010).
Yang, J., Roy, A. K. & Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 29, 2588–2595 (2013).
Ermler, U., Grabarse, W., Shima, S., Goubeaud, M. & Thauer, R. K. Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science 278, 1457–1462 (1997).
Acknowledgements
This study was supported by the US Department of Energy's Office of Biological Environmental Research (award no. DE-SC0010574), and the Commonwealth Scientific and Industrial Research Organisation (CSIRO) Flagship Cluster ‘Biotechnological solutions to Australia's transport, energy and greenhouse gas challenges’. I.V. acknowledges support from the University of Queensland International Scholarship, and P.D.J. acknowledges support from the Australian Meat Processor Corporation (2013/4008 Technology Fellowship). P.H. and I.V. acknowledge support from the Australian Research Council Laureate Program entitled ‘Reconstructing the universal tree and network of life’. G.W.T. acknowledges support by the University of Queensland Vice-Chancellor's Research Focused Fellowship. The authors thank the sequencing facilities at the Australian Centre for Ecogenomics and Institute for Molecular Biosciences (University of Queensland, Australia) for library preparation and sequencing.
Author information
Authors and Affiliations
Contributions
I.V. designed and performed the anaerobic digestion study, analysed the data and wrote the paper. P.E. assisted with the metabolic analysis and performed protein modelling. D.P. assisted with the binning of the formation water metagenomes and the mcrA screening of the SRA database. P.D.J. was involved in the design of the anaerobic digestion study and helped run the reactors. B.J.W. performed the binning of the palm oil mill effluent metagenome and assisted with the 16S rRNA gene screening of the SRA database. P.H. helped interpret the phylogenetic analyses and wrote the paper. G.W.T. oversaw the project, was involved in the design of the anaerobic digestion study, helped interpret the data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information
Supplementary Figures 1-8, Supplementary Tables 1-6 (PDF 1229 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Vanwonterghem, I., Evans, P., Parks, D. et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat Microbiol 1, 16170 (2016). https://doi.org/10.1038/nmicrobiol.2016.170
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/nmicrobiol.2016.170
This article is cited by
-
Unraveling the phylogenomic diversity of Methanomassiliicoccales and implications for mitigating ruminant methane emissions
Genome Biology (2024)
-
The majority of microorganisms in gas hydrate-bearing subseafloor sediments ferment macromolecules
Microbiome (2023)
-
Variations in the archaeal community and associated methanogenesis in peat profiles of three typical peatland types in China
Environmental Microbiome (2023)
-
Mcr-dependent methanogenesis in Archaeoglobaceae enriched from a terrestrial hot spring
The ISME Journal (2023)
-
Diversity and function of methyl-coenzyme M reductase-encoding archaea in Yellowstone hot springs revealed by metagenomics and mesocosm experiments
ISME Communications (2023)
