
Abstract
Atypical enteropathogenic Escherichia coli (aEPEC) is an umbrella term given to E. coli that possess a type III secretion system encoded in the locus of enterocyte effacement (LEE), but lack the virulence factors (stx, bfpA) that characterize enterohaemorrhagic E. coli and typical EPEC, respectively. The burden of disease caused by aEPEC has recently increased in industrialized and developing nations, yet the population structure and virulence profile of this emerging pathogen are poorly understood. Here, we generated whole-genome sequences of 185 aEPEC isolates collected during the Global Enteric Multicenter Study from seven study sites in Asia and Africa, and compared them with publicly available E. coli genomes. Phylogenomic analysis revealed ten distinct widely distributed aEPEC clones. Analysis of genetic variation in the LEE pathogenicity island identified 30 distinct LEE subtypes divided into three major lineages. Each LEE lineage demonstrated a preferred chromosomal insertion site and different complements of non-LEE encoded effector genes, indicating distinct patterns of evolution of these lineages. This study provides the first detailed genomic framework for aEPEC in the context of the EPEC pathotype and will facilitate further studies into the epidemiology and pathogenicity of EPEC by enabling the detection and tracking of specific clones and LEE variants.
Similar content being viewed by others

Atypical enteropathogenic Escherichia coli (aEPEC) is a globally emerging pathogen associated with acute and persistent diarrhoea in children1,2. Currently, aEPEC is defined by the presence of the locus of enterocyte effacement (LEE) pathogenicity island and the absence of other specific virulence determinants, including Shiga toxin (stx gene, which together with the LEE characterize enterohaemorrhagic E. coli, EHEC) and the plasmid-encoded bundle-forming pilus operon (bfp, which together with the LEE characterizes typical EPEC, tEPEC)3,4. Attempts to identify novel or known genes that explain the pathogenicity of aEPEC have largely failed1,5. In these studies, however, aEPEC has been treated as a single homogeneous group, whereas recent genomic analyses of other pathotypes of E. coli, such as enterotoxigenic E. coli (ETEC), indicate that they comprise multiple distinct lineages that have emerged in parallel via the horizontal acquisition of specific virulence determinants in the accessory genome6,7.
The LEE, which is the only known virulence determinant of aEPEC, is a 35 kb chromosomal pathogenicity island composed of 41 core genes organized into five operons8. It encodes a type III secretion system (T3SS), the intimin protein (Eae) and its translocated receptor (Tir), as well as translocons, chaperones, regulators and secreted effector proteins that are linked to virulence8–10. The hallmark histopathological trait of an EPEC infection is the formation of attaching and effacing lesions in the gut of the host as a consequence of cytoskeletal changes that result from the interaction of intimin with Tir4. The T3SS is a complex machine evolved from the bacterial flagellum11. Its constituent proteins form a needle-like structure, known as the ‘injectisome’ and are highly conserved to maintain the complex interactions required for T3SS functionality10–12. The T3SS enables virulence effector proteins encoded by genes located on the LEE and elsewhere in the accessory genome to be translocated into eukaryotic cells13,14.
The LEE is hypothesized to be transferred horizontally between E. coli of different chromosomal backgrounds9,15, but little is known about genetic variation within the LEE. Research based on six LEE sequences, not including aEPEC, suggested that different LEE component proteins are under different evolutionary pressures, with strong conservation of T3SS components and limited positive diversifying selection within other genes16. Another study centred on the LEE sequences from two aEPEC isolates also suggested conservation of the T3SS machinery and greater sequence variation in the effector genes17. Other studies have attempted to define LEE subtypes based on genetic variation in a handful of genes including eae, tir and three translocon genes espABD, but no definitive correlations have been identified between LEE subtypes and either EPEC or EHEC18,19.
Effectors encoded on the LEE and secreted by the T3SS disrupt host cell functions through a variety of mechanisms, thereby causing disease in the host and potentially increasing the fitness of the bacteria13. Indeed, it has been proposed that the initial role of the LEE T3SS apparatus was to transport flagellar components, but with the recruitment of the other LEE genes it has evidently adapted to deliver effectors directly to eukaryotic cells11. Non-LEE encoded (Nle) effectors secreted by the T3SS have a range of known virulence functions, including inhibition of the NF-κB cell-signalling pathway and host cell apoptosis20,21. Considerable variation exists within the Nle-efffector repertoire of EPEC and EHEC, however, with some evidence that a higher number of effectors per genome is associated with increased pathogenicity14.
In this study we investigated the evolution of aEPEC and the LEE through phylogenomic analysis of aEPEC isolates obtained during the Global Enteric Multicenter Study (GEMS) conducted in African and South Asian children with moderate-to-severe diarrhoea and matched asymptomatic controls22,23. We also incorporated publicly available genome sequences for EPEC (both tEPEC and aEPEC), EHEC (both O157 EHEC and non-O157 EHEC) and other E. coli reference genomes to provide a species-wide context for our study. Our analyses demonstrated the parallel emergence of multiple globally distributed aEPEC clones, through the acquisition of distinct LEE subtypes that are associated with distinct chromosomal backgrounds and insertion sites. These data have important implications for our understanding of the emergence of pathogenicity in E. coli and thus will facilitate future studies of EPEC epidemiology and virulence.
Results
Population structure of atypical EPEC
To investigate the population structure of aEPEC, we sequenced 196 novel isolates identified from the GEMS study22 and compared these to 171 publicly available E. coli genomes of diverse pathotypes and an E. albertii isolate (Supplementary Table 1). We used a mapping-based approach to construct a core genome phylogeny to model vertical evolution (see Methods), which revealed ten phylogenetically distinct aEPEC clusters or clonal groups (CGs) containing >5 isolates each (Fig. 1). Alternative core genome phylogenies inferred using a reference-free approach, with and without filtering for recombination, yielded near-identical tree topologies and recovered the same aEPEC clonal groups (Supplementary Note and Supplementary Fig. 1). CGs were named after their dominant multi-locus sequence types (STs)24 (Supplementary Table 1). The aEPEC isolates we analysed were originally identified by multiplex polymerase chain reaction (PCR) detection of eae but not bfpA or stx23. Genome analysis revealed the presence of the bfp operon with a divergent (beta) form of bfpA and per regulator genes in 11 GEMS isolates, which were reclassified as tEPEC (Fig. 1). Furthermore, as the LEE could conceivably have been non-functional in some isolates, we screened all GEMS isolates for their ability to secrete EspB and EspD with secretion assays confirming functionality of the encoded T3SS (Supplementary Methods, Supplementary Fig. 2).

A total of 359 E. coli genomes (258 aEPEC, 101 others) and 8 Shigella genomes were used to construct the tree, which was midpoint rooted. The pathotype for isolates carrying the LEE pathogenicity island is indicated in the outermost ring according to the key shown. The ten aEPEC clonal groups (CGs) discussed in the text are highlighted and named in accordance with the dominant sequence type (ST) according to the Achtman MLST scheme24. Two reference LEE-containing clones (tEPEC and O157 EHEC) are also shown.
The wide distribution of aEPEC within the E. coli core genome phylogeny confirms that aEPEC lineages have arisen on multiple occasions by acquiring the LEE pathogenicity island through horizontal gene transfer. This is consistent with the emergence of other E. coli pathotypes, such as ETEC6. Of the 258 aEPEC genomes we analysed, 184 (71%) fell into one of ten common aEPEC CGs comprising >5 genomes each, with the remaining genomes distributed among rarer clusters (≤5 genomes each). The ten aEPEC CGs exhibited within-clone nucleotide diversity of <0.06% amongst core genes, compared with >1% diversity between CGs and with other E. coli lineages (Supplementary Note). Four of the aEPEC CGs also contained isolates with additional virulence factors bfp or stx (Fig. 1). Based on the distribution of these virulence factors within the intra-clone phylogenies (Supplementary Fig. 3 and Supplementary Note), the most parsimonious scenario is that CG121 and CG10 are aEPEC clones, each formed by a single LEE acquisition event, with a subsequent bfpA acquisition event. CG3 contains multiple subclusters with bfpA (Supplementary Fig. 3), which could be explained by either loss of the bfp plasmid from some isolates or by frequent transfer of the plasmid into a permissive clonal background. A similar pattern was evident for stx within CG29 (Supplementary Fig. 3).
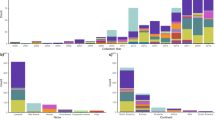
Rarefaction curves (Fig. 2a) indicate that additional sampling at the GEMS sites and elsewhere will probably reveal additional aEPEC clones, in addition to detecting further isolates belonging to the existing aEPEC clones and clusters. Most of the aEPEC clones we identified were present in all seven Asian and African GEMS sites (Fig. 2b) and were isolated in multiple years of the study (Fig. 2c), indicating that they are widely disseminated and able to persist in local human populations. Furthermore, eight aEPEC clones included aEPEC reference genomes isolated from Europe and/or America, suggesting these clones may be globally disseminated (for details see Supplementary Table 1). The greatest diversity of aEPEC was identified in the Asian GEMS sites, whereas the West African sites (The Gambia and Mali) showed the least diversity, with only five of the aEPEC clones detected for a period exceeding three months. This was probably due to the smaller sample size from this region (n = 46 isolates, compared with 77 from East Africa and 73 from Asia) (Supplementary Fig. 4).

a, Rarefaction curves illustrating the accumulation of aEPEC lineages (defined by RAMI and MLST) with increasing sample size, both overall (labelled aEPEC) and separately for the three major geographical regions where GEMS sites were located. b, Distribution of the ten major aEPEC CGs at each of the seven GEMS sites. c, Temporal spans (earliest to latest) showing when each of the ten major aEPEC CGs were isolated in the three broad regions of the GEMS study: West Africa (Mali and The Gambia), East Africa (Kenya and Mozambique) and South Asia (Bangladesh, India and Pakistan).
Evolution and population structure of the LEE
The LEE encodes the T3SS machinery and secreted proteins, which together form a complex system capable of manipulating host cells. Phylogenetic analysis based on eight genes (escCJNRSTUV, Supplementary Note) confirmed that all the LEE-encoded T3SS sequences extracted from our 170 novel isolates and 82 LEE-containing reference genomes belong to the E. coli T3SS (ETT1) cluster, which is a member of the Salmonella Pathogenicity Island 2 (SPI2) T3SS family11. Next we examined genetic variation across the full complement of 41 LEE genes (see Methods and Supplementary Note). Genes involved in the T3SS machinery showed greater sequence conservation (higher nucleotide similarity), and were under stronger purifying selection (lower dN/dS), than non-T3SS genes including eae, tir, the effector genes and the translocon genes, espA, espB and espD (Fig. 3).

Left: nucleotide similarity (box plots, error bars show value range: left axis) and dN/dS ratio (points: right axis) for the 41 LEE genes. Right: type III secretion system translocating effectors into a host cell and the intimin–Tir interaction that mediates the hallmark attaching and effacing lesion.
To investigate co-evolution of the LEE genes, we examined correlations between individual gene trees. This analysis indicated that variation in T3SS genes was tightly correlated with one another, while eae, tir and the genes encoding effectors and translocons varied more freely (Supplementary Fig. 5). Network analysis of the correlation data identified four sub-networks of co-evolving genes (Fig. 4). Sub-networks 1 and 2 were the largest and contained most of the genes that encode the T3SS machinery, regulators and the majority of chaperones. The genes in these two sub-networks were predominately located in the LEE1, LEE2 and LEE3 transcriptional operons (Fig. 4b). One effector gene, espG, was part of sub-network 1; the remaining effector genes, as well as eae, tir, two chaperone genes and six of the T3SS genes, formed two small sub-networks or were singletons (that is, they had evolutionary histories distinct from one another and from other genes). Adaptive selection within these genes was investigated in more detail (Supplementary Fig. 6 and Supplementary Note). The translocon genes (espA, espB and espD), the key genes involved in the formation of attaching–effacing lesions (namely eae and tir) and the effector genes (espF, espG and espZ) all had specific sites that were under strong positive (diversifying) selection and other sites that were under strong negative (purifying) selection.

a, Networks constructed from correlations between individual gene trees, plotted in Cytoscape 2.8.3 with a correlation cutoff of >0.90. Four tightly co-evolving gene networks were identified: sub-network 1 (20 genes), sub-network 2 (7 genes), sub-network 3 (2 genes) and sub-network 4 (3 genes). The remaining 9 genes are shown as singletons. Genes are coloured by functional group as shown in the key. b, Genetic organization of the LEE locus, showing the spatial distribution of genes in each sub-network. The five transcription operons of the LEE are shown at the top of the figure.
As the LEE gene-tree correlations were suggestive of recombination within the LEE, we used ClonalFrame25 to investigate vertical evolution and acquisition of the LEE in aEPEC. This revealed that although recombination has occurred at low rates across the entire LEE pathogenicity island, it most frequently affects eae, tir, the translocon and effector genes (Supplementary Fig. 7). Furthermore, our analyses revealed a deep-branching phylogenetic structure (Fig. 5), demarcating three distinct LEE lineages with an average nucleotide divergence of 1–4% within LEE lineages (similar to species-wide divergence between core chromosomal genes in E. coli or other species) and 4–7% between lineages (similar to the divergence typically encountered between homologous genes in related genera). LEE lineage 1 was composed entirely of novel aEPEC isolates, belonging to CG301 and CG378, while the previously characterized O157 EHEC and tEPEC isolates fell within the common LEE lineages 2 and 3 (Fig. 5). The three LEE lineages were further divided into 30 subtypes on the basis of their phylogeny (referred to hereafter as LEE-1, LEE-2, and so on). These LEE subtypes captured variation in individual LEE genes that is compatible with, but provides greater resolution than, previous subtyping analyses (Supplementary Figs 8 and 9, Supplementary Note).

a, Recombination-free phylogeny of the LEE was constructed via ClonalFrame analysis and used to identify 30 LEE subtypes. Branch lengths defining the three major lineages are truncated to allow resolution within lineages. True branch lengths are shown in the full tree (bottom left inset). Each LEE subtype is labelled with the number of isolates of that type identified and the frequency of three possible LEE insertion sites (coloured according to the key shown). LEE subtypes that contain tEPEC and EHEC isolates are highlighted. b, Frequencies of each Nle-effector gene in each LEE subtype shown as a heatmap, with dark red indicating the effector was detected in all isolates of that lineage and white indicating that the effector was not detected in any.
Association of LEE subtypes with distinct patterns of Nle-effector genes and LEE insertion sites
Screening for genes encoding known Nle-effector genes indicated that different LEE subtypes may be associated with different complements of effectors (Fig. 5 and Supplementary Fig. 10). Specifically, the distributions of most of the Nle-effector genes were significantly associated with the three LEE lineages (P < 0.05, Fisher's exact test with simulated P value based on 2,000 replicates; Supplementary Table 2) and with many of the LEE subtypes. Isolates within the well-characterized subtypes LEE-27 (carried by tEPEC E2348/69) and LEE-10 (O157 EHEC) harboured many of the known effector genes, such as nleB1 and nleE, which are thought to be co-transferred horizontally5,26. In contrast, subtypes belonging to the novel LEE lineage 1 (LEE-1 in CG378 and LEE-2 in CG301) carried few of the known Nle-effector genes. This probably reflects a discovery bias in Nle-effector screens to date, with the corollary that additional effectors may remain to be discovered among CG301 and CG378 strains.
The distribution of LEE subtypes among the different CGs and clusters is shown in Fig. 6. These data illustrate the numerous events in which distinct LEE subtypes were acquired by different E. coli isolates with distinct chromosomal backgrounds. The LEE can be inserted into one of three sites in the E. coli chromosome: tRNA-selC, tRNA-pheU and tRNA-pheV9. The most common site we found was tRNA-selC, accounting for half of all LEE insertions, in a range of chromosomal backgrounds (Figs 5 and 6, Supplementary Fig. 10). The other insertion sites were less frequent in terms of both overall number of isolates and the number of independent insertions. These three insertion sites were associated with the three LEE lineages (P = 0.0005, Fisher's exact test with a simulated P value based on 2,000 replicates) as follows: all LEE lineage 1 insertions occurred in tRNA-pheU, 20 of the 22 LEE subtypes in LEE lineage 3 were inserted in tRNA-selC, and LEE lineage 2 was inserted most frequently in either tRNA-pheU or tRNA-pheV (Fig. 5, Supplementary Table 3 and Supplementary Fig. 10). All isolates in the closely related groups O157 EHEC and CG335 (aEPEC) carried LEE-10 (LEE lineage 3) in tRNA-selC, consistent with a single shared acquisition event (Fig. 6), followed by the subsequent acquisition of stx to form the O157:H7 EHEC lineage. Most aEPEC clones were associated with a single LEE subtype and insertion site (Fig. 6 and Supplementary Fig. 10) except GC3, CG29, CG40 and CG517. The LEE variants clustered together within the intra-clone phylogenies (Supplementary Fig. 3), consistent with rare events resulting in replacement of the LEE locus. Notably CG3, CG40 and CG29 all had predominantly LEE-8 (LEE lineage 2) plus LEE subtypes from LEE lineage 3, suggesting that LEE-8 may be either unstable (displaced by other incoming LEE insertions) or promiscuous (frequently displacing existing LEE insertions).

The tree shows the E. coli core gene phylogeny (as in Fig. 1), collapsed into clusters and including the E. albertii outgroup. The distributions of the 30 LEE subtypes are shown as numbered boxes. Colours indicate lineage, as defined by the legend (LEE Linages); recombination-free LEE phylogeny is shown in the bottom left inset and in Fig. 5. The vertical lines indicate the proportion of the different LEE subtypes within the core lineages. Lineages that contain tEPEC and EHEC isolates are indicated. Numbers indicate the predominant subtypes; bars indicate relative frequencies of subtypes within each CG or cluster. The relative frequencies of LEE insertion sites within each cluster are also shown, according to the legend (insertion site). The number of isolates in each cluster is as indicated (n values, red bar graphs).
Discussion
For over a decade, aEPEC has been described as an emerging pathogen1,2. The term ‘emerging pathogen’ is commonly used to describe agents of infection whose incidence is increasing, either following transition to a new host population or in an existing population caused by changing epidemiological factors (which may or may not be identified). Our genomic analyses provide the first high-resolution elucidation of the population structure of the emerging pathogen aEPEC, revealing that aEPEC clones and additional phylogenetically distinct lineages have emerged on multiple occasions (Fig. 1 and Supplementary Table 1). Furthermore, our data show conclusively that these E. coli carry distinct variants of the LEE and non-LEE encoded effectors. This indicates that aEPEC have ‘emerged’ repeatedly in the evolutionary sense, in that they have evolved on many separate occasions via horizontal gene transfer. Our data indicate that previous studies where aEPEC was treated as a homogenous group5,19,22,27 are likely to have been confounded by the occurrence of multiple aEPEC lineages, which differ in their accessory gene content and associated pathogenic potential (Figs 5 and 6), obscuring the true impact of aEPEC. The identification of multiple distinct aEPEC CGs provides a strong rationale for more detailed subtyping of aEPEC in future studies and highlights the inadequacy of the current delineation of EPEC into two subgroups, tEPEC and aEPEC27. Importantly, our findings provide an opportunity to re-examine and refine epidemiological studies of diarrhoeal disease aetiology and the emergence of aEPEC as a diarrhoeal pathogen, by enabling the stratification of aEPEC into distinct clones to investigate whether observed increases in aEPEC infections are in fact due to the emergence of a particular clone or clones within defined human populations. These findings also provide a framework to identify and characterize putative virulence factors in the accessory genome of the clonal lineages. This analysis was beyond the scope of the current study.
Our data revealed diverse selective pressures acting on LEE genes. Those genes encoding immunogenic proteins that are exposed to and interact with the host have accumulated extensive genetic diversity both within and between the various LEE subtypes (Fig. 3 and 4, Supplementary Fig. 6). In contrast, the T3SS genes of the LEE have been far more limited in their evolution, consistent with smaller-scale studies of LEE variation16 and wider trends across the conserved families of T3SS11. This has important implications for subtyping schemes, as it indicates which genes have the greatest resolving power to distinguish LEE subtypes (Supplementary Fig. 8). The LEE gene variant data are available at https://github.com/katholt/srst2, which can be used with SRST2 or BLAST to assign LEE subtypes to short reads or assembled genome data, respectively. Our findings greatly expand the scale and resolution of previous schemes by encapsulating the evolution of the LEE as not a single genomic island that is stably maintained, but a dynamic region under complex and varied selection pressures to retain functionality of the T3SS while continuing to adapt and evolve in response to host defences.
Our finding that most aEPEC clones are associated with a single LEE subtype indicates that these clones typically descend from a common ancestor in which a single LEE acquisition event occurred (as opposed to being lineages that commonly receive and retain LEE insertions) and that the LEE is maintained during subsequent intercontinental clonal expansion and geographical dissemination (Figs 2 and 6). The maintenance of a single LEE subtype within each clone may be linked to the presence of a compatible complement of Nle-effector genes encoded elsewhere in the genome and secreted by the LEE-encoded T3SS, which is supported by our finding of an association between LEE subtypes and the repertoire of Nle-effector genes (Supplementary Table 2). The distribution of Nle-effector genes in our E. coli strains (Fig. 5 and Supplementary Fig. 10) also supports the contention that some of these genes are transferred together on genomic islands, such as PAI O122, which carries nleE and nleB1 and flanks certain LEE subtypes5,28,29. NleE and NleB1 have complementary roles in enabling the bacteria to persist in the host, as NleE (a cysteine methyltransferase) inhibits local inflammation21 and NleB1 is a novel glycosyltransferase that modifies host cell signalling proteins and inhibits apoptosis of infected cells20. These two effectors contribute significantly to the infection strategy common to attaching and effacing pathogens. Future lines of investigation will be to characterize the mobilization of Nle-effector genes, including co-transfer of these genes within the bacterial population, and to identify novel Nle-effectors within LEE lineage 1. Further, our analyses provide a framework for further work to identify and characterize novel adhesins and potentially toxins that may contribute to pathogenicity in different lineages of aEPEC.
In conclusion, our data elucidate the population structure of aEPEC and provide an in-depth analysis of its only known virulence determinant, the LEE pathogenicity island. Our findings highlight the existence of globally disseminated aEPEC clones that have acquired different LEE subtypes in their evolutionary histories, suggesting that the acquisition of functional LEEs has played a driving role in the expansion of these successful clones. Importantly, this study provides a possible explanation for the failure of earlier attempts to characterize atypical EPEC in terms of clinical disease symptoms or virulence genes and provides a genomic framework for future research that can take into account differences in chromosomal and LEE lineages, which will be critical for future studies into the emergence of EPEC.
Methods
Bacterial isolates and sequencing
A total of 196 putative atypical EPEC isolates from GEMS were analysed in this study22. The GEMS isolates were originally identified as aEPEC by PCR screening for the virulence markers eae, bfpA, hlyA and stx23. The isolates selected for sequencing were mostly from faecal samples in which aEPEC alone (or with Giardia lamblia) was the only pathogen detected, where a pure culture could be isolated and where the case and control status were matched by site. Isolates sequenced from the seven sites were 3 of 58 aEPEC from Bangladesh, 48 of 303 from India, 22 of 115 from Pakistan, 13 of 85 from The Gambia, 59 of 203 from Kenya, 33 of 83 from Mali, and 18 of 74 from Mozambique. A clinical aEPEC isolate from an infant with diarrhoea from the Royal Children's Hospital in Melbourne and an E. albertii isolate from the GEMS study were also included.
Genomic DNA was extracted with the Sigma GenElute Bacterial Genomic DNA Kit from purified bacterial cultures grown overnight at 37 °C according to the manufacturer's instructions. DNA quality was measured with a NanoDrop spectrophotometer (NanoDrop Technologies) and a DNA concentration of at least 50 ng μl–1 was used for each isolate. Illumina sequencing libraries were prepared, combined into pools of 96 uniquely tagged isolates30 and then sequenced on the Illumina Hiseq 2000 platform at the Wellcome Trust Sanger Institute to generate tagged paired-end reads of 100 bases in length.
An additional 170 publicly available commensal and pathogenic E. coli and Shigella reference genomes were included. Details of all genomes analysed are provided in Supplementary Table 1.
Construction of a core genome SNP alignment
Single nucleotide polymorphisms (SNPs) were identified by comparison to the E. coli reference genome O103:H2 12009 (a LEE-positive non-O157 EHEC isolate from Japan)31 (Supplementary Note), using the in-house mapping-based pipeline RedDog (https://github.com/katholt/RedDog).
RedDog uses Bowtie232 to map each read set to the reference and SamTools33 to call SNPs (Phred score ≥30, read depth ≥5x and <2*average depth). Consensus alleles at all SNP sites identified in the isolate collection were then extracted from each read set using SamTools33 (Phred score ≥20 and unambiguous; otherwise allele call set to unknown ‘–’). Core genes were defined as those annotated in the O103:H2 12009 genome and present at ≥90% coverage of gene length (by read mapping) with 99% conservation in all E. coli genomes in the test collection (a total of 1,810 core genes). SNP sites within these core genes were concatenated to make a core genome SNP alignment for phylogenetic analysis, comprising 198,660 SNPs.
Core genome phylogenetic analysis and recombination detection
Maximum likelihood (ML) trees were inferred using RAxML run five times with the generalized time-reversible (GTR) model and a gamma distribution to model site-specific rate variation34. One hundred bootstrap pseudo-replicate analyses were performed to assess support for the ML phylogeny. For each analysis, the final tree shown is that with the highest likelihood across all five runs, with ML estimates of branch length and confidence in major bipartitions calculated using the bootstrap values across all runs. Recombination filtering was performed using ClonalFrameML35, using the best RAxML tree as the starting tree. Phylogenetic lineages were defined using RAMI36 to identify clusters based on patristic distance. A cutoff distance of 0.00032 was selected as it differentiated the O157 EHEC (CG11) lineage from the aEPEC CG335 lineage, in agreement with published data. The lineage accumulation curves for RAMI clusters, using only data from the GEMS aEPEC isolates, were calculated separately for the three geographic regions Asia, West Africa and East Africa, using vegan in R (http://cran.r-project.org/web/packages/vegan/index.html).
Illumina reads were assembled using the de novo short read assembler Velvet and Velvet Optimiser37, annotated using Prokka38 using the proteins annotated in O103:H2 12009 as a primary reference and used to construct an alternative reference-free core gene alignment (Supplementary Note).
Multi-locus sequence typing (MLST)
MLST sequence types (ST) of the Achtman scheme24 (http://mlst.warwick.ac.uk/mlst/) were determined from the short read data using SRST239 for the GEMS isolates and BLAST for reference genomes.
Nucleotide diversity and selection analysis
The pairwise diversity for each gene was calculated using MEGA640. The resulting pairwise distance matrix was inverted to give the pairwise similarity in R. The dN/dS ratio within each alignment was calculated with the SeqinR package41. Positive finite ratio values were included in the ratio calculation.
Gene network analysis
An alignment for each of the extracted 41 individual LEE genes was constructed using Muscle42. A ML tree was created for each gene alignment using RAxML with a GTR model with Gamma Substitution and Invariant sites with 100 bootstraps34. The genetic distance with each gene tree was calculated in R using the ade4 package43. Pairwise correlations between resulting distance matrices were calculated using the pairwise Mantel Test. Co-evolution networks of the LEE genes were constructed from pairwise correlations in Cytospace 2.844. MCL clustering was performed with the inflation parameter set at 2.2. The cutoff edge weight value was set at a correlation of >0.90 (approximately one standard deviation above the mean value for all pairwise correlations).
Vertical evolution of the LEE
The LEE gene alignments were concatenated and analysed using ClonalFrame25. ClonalFrame was run three times with 200,000 burn-in and 400,000 posterior iterations each, sampling at every 1,000th iteration. Chain convergence was assessed using Gelmen–Rubin convergence statistics (implemented in the ClonalFrame GUI) and the run with the best convergence statistics was selected for the final analysis. The posterior trees were exported and a strict consensus tree was constructed from these using Dendroscope45. The posterior probability of recombination events determined by ClonalFrame analysis was extracted and the mean calculated for probability events.
Detecting the site of insertion of the LEE into the chromosome
BLAST analysis, using the housekeeping genes surrounding the three known tRNA insertion sites of LEE (selC, pheU and pheV) as query sequences, was undertaken to determine the LEE insertion site in each genome assembly.
Detection of genes encoding putative Nle-effector genes in the accessory genome
A sequence database of genes encoding known Nle-effector genes from both EHEC and tEPEC was created based on published works (listed in Supplementary Table 4). GEMS isolate read sets were screened for these effectors using SRST239 with default parameter settings, which identifies only close homologues with ≥90% identity and ≥90% coverage of the reference sequences. Reference genomes were screened against the same database with BLAST with ≥90% identity and ≥90% coverage. The resulting matrix of effector gene presence/absence was clustered in R using hierarchical clustering.
Accession numbers
Illumina reads and annotated assemblies for the novel GEMS isolates are available in the European Nucleotide Archive (ENA) under project no. ERP001141. Individual sample accessions are provided in Supplementary Table 1, which also includes accessions for all other genomes used in the analysis.
References
Ochoa, T. J. & Contreras, C. A. Enteropathogenic Escherichia coli infection in children. Curr. Opin. Infect. Dis. 24, 478–483 (2011).
Hernandes, R. T., Elias, W. P., Vieira, M. A. M. & Gomes, T. A. T. An overview of atypical enteropathogenic Escherichia coli. FEMS Microbiol. Lett. 297, 137–149 (2009).
Trabulsi, L. R., Keller, R. & Gomes, T. A. T. Typical and atypical enteropathogenic Escherichia coli. Emerg. Infect. Dis. 8, 1–6 (2002).
Kaper, J. B., Nataro, J. P. & Mobley, H. L. T. Pathogenic Escherichia coli. Nature Rev. Microbiol. 2, 123–140 (2004).
Bugarel, M., Martin, A., Fach, P. & Beutin, L. Virulence gene profiling of enterohemorrhagic (EHEC) and enteropathogenic (EPEC) Escherichia coli strains: a basis for molecular risk assessment of typical and atypical EPEC strains. BMC Microbiol. 11, 1–10 (2011).
Von Mentzer, A. et al. Identification of enterotoxigenic Escherichia coli (ETEC) clades with long-term global distribution. Nature Genet. 46, 1321–1326 (2014).
Croxen, M. A. et al. Recent advances in understanding enteric pathogenic Escherichia coli. Clin Microbiol. Rev. 26, 822–880 (2013).
Elliot, S. J. et al. The complete sequence of the locus of enterocyte effacement (LEE) from the enteropathogenic Escherichia coli E2348/69. Mol. Microbiol. 28, 1–4 (1998).
Müller, D. et al. Comparative analysis of the locus of enterocyte effacement and its flanking regions. Infect. Immun. 77, 3501–3513 (2009).
Hueck, C. J. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62, 379–433 (1998).
Abby, S. S. & Rocha, E. P. C. The non-flagellar type III secretion system evolved from the bacterial flagellum and diversified into host-cell adapted systems. PLoS Genet. 8, e1002983 (2012).
Gauthier, A., Thomas, N. A. & Finlay, B. B. Bacterial injection machines. J. Biol. Chem. 278, 25273–25276 (2003).
Raymond, B. et al. Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. Trends Microbiol. 21, 430–441 (2013).
Dean, P. & Kenny, B. The effector repertoire of enteropathogenic E. coli: ganging up on the host cell. Curr. Opin. Microbiol. 12, 101–109 (2009).
Hazen, T. H. et al. Refining the pathovar paradigm via phylogenomics of the attaching and effacing Escherichia coli. Proc. Natl Acad. Sci. USA 1–6 (2013).
Castillo, A., Eguiarte, L. E. & Souza, V. A genomic population genetics analysis of the pathogenic enterocyte effacement island in Escherichia coli: the search for the unit of selection. Proc. Natl Acad. Sci. USA 102, 1542–1547 (2005).
Gartner, J. F. & Schmidt, M. A. Comparative analysis of locus of enterocyte effacement pathogenicity islands of atypical enteropathogenic Escherichia coli. Infect. Immun. 72, 6722–6728 (2004).
Lacher, D. W., Steinsland, H. & Whittam, T. S. Allelic subtyping of the intimin locus (eae) of pathogenic Escherichia coli by fluorescent RFLP. FEMS Microbiol. Lett. 261, 80–87 (2006).
Contreras, C. A. et al. Genetic diversity of locus of enterocyte effacement genes of enteropathogenic Escherichia coli isolated from Peruvian children. J. Med. Microbiol. 61, 1114–1120 (2012).
Pearson, J. S. et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501, 247–251 (2013).
Giogha, C., Lung, T. W. F., Pearson, J. S. & Hartland, E. L. Inhibition of death receptor signaling by bacterial gut pathogens. Cytokine Growth Factor Rev. 25, 235–243 (2014).
Kotloff, K. L. et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382, 209–222 (2013).
Panchalingam, S. et al. Diagnostic microbiologic methods in the GEMS-1 case/control Study. Clin. Infect. Dis. 55, S294–S302 (2012).
Wirth, T. et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151 (2006).
Didelot, X., Meric, G., Falush, D. & Darling, A. E. Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genom. 13, 1–15 (2012).
Ogura, Y. et al. Systematic identification and sequence analysis of the genomic islands of the enteropathogenic Escherichia coli strain B171–8 by the combined use of whole-genome PCR scanning and fosmid mapping. J. Bacteriol. 190, 6948–6960 (2008).
Donnenberg, M. S. & Finlay, B. B. Combating enteropathogenic Escherichia coli (EPEC) infections: the way forward. Trends Microbiol. 21, 317–319 (2013).
Schmidt, M. A. LEEways: tales of EPEC, ATEC and EHEC. Cell Microbiol. 12, 1544–1552 (2010).
Dean, P. & Kenny, B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol. Microbiol. 54, 665–675 (2004).
Quail, M. A., Swerdlow, H. & Turner, D. J. Improved protocols for the Illumina Genome Analyzer sequencing system. Curr. Protoc. Human Genet. 18.2, 1–27 (2009).
Ogura, Y. et al. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl Acad. Sci. USA 106, 17939–17944 (2009).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Didelot, X. & Wilson, D. J. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11, e1004041 (2015).
Pommier, T., Canbäck, B., Lundberg, P., Hagström, Å. & Tunlid, A. RAMI: a tool for identification and characterization of phylogenetic clusters in microbial communities. Bioinformatics 25, 736–742 (2009).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Inouye, M. et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6, 1–16 (2014).
Tamura, K., Stecher, G., Petersen, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Charif, D. & Lobry, J. R. in Structural approaches to sequence evolution molecules, networks, populations (eds. Bastolla, U., Porto, M., Roman, H. E. & Vendruscolo, M. ) 207–232 (Springer Verlag, 2007).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Dray, S. & Dufour, A. B. The ade4 package: implementing the duality diagram for ecologists. J. Statist. Software 22, 1–20 (2007).
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L. & Ideker, T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 (2011).
Huson, D. H. & Scornavacca, C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. System. Biol. 61, 1061–1067 (2012).
Acknowledgements
This work was funded by the Australian NHMRC (project grant nos. 1009296 and 1067428 to R.M.R.-B., fellowship no. 1061409 to K.E.H.), the Wellcome Trust (grant no. 098051 to the Wellcome Trust Sanger Institute, WTSI), the Bill & Melinda Gates Foundation (grant ID no. 38874 to M.M.L.) and the Victorian Life Sciences Computation Initiative (grant no. VR0082). The authors thank the sequencing teams at the WTSI for genome sequencing.
Author information
Authors and Affiliations
Contributions
D.J.I., M.T., R.M.R.-B. and K.E.H. contributed to the design of the study and data interpretation. K.I.A., T.A., V.B.-W., J.S.P., D.H. and D.J.I. performed the experimental analyses. D.J.P. and G.D. sequenced the isolates. D.J.I. performed the majority of bioinformatics analyses with input from K.E.H. D.J.E. developed the mapping pipeline RedDog. R.M.R.-B., M.T. and K.E.H. supervised. B.T., M.A., J.B.O., J.A.H., S.Q., T.R., A.H., K.L.K., J.P.N. and M.M.L. were responsible for the experimental analyses at the GEMS sites and K.L.K., J.P.N. and M.M.L. for the design of GEMS. All authors contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1–10, Tables 2–6, Notes and References (PDF 4330 kb)
Supplementary Table 1
Supplementary Table 1 (XLSX 72 kb)
Rights and permissions
About this article

Cite this article
Ingle, D., Tauschek, M., Edwards, D. et al. Evolution of atypical enteropathogenic E. coli by repeated acquisition of LEE pathogenicity island variants. Nat Microbiol 1, 15010 (2016). https://doi.org/10.1038/nmicrobiol.2015.10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/nmicrobiol.2015.10
This article is cited by
-
Genomic diversity of non-diarrheagenic fecal Escherichia coli from children in sub-Saharan Africa and south Asia and their relatedness to diarrheagenic E. coli
Nature Communications (2023)
-
Enteropathogenic Escherichia coli (EPEC) expressing a non-functional bundle-forming pili (BFP) also leads to increased growth failure and intestinal inflammation in C57BL/6 mice
Brazilian Journal of Microbiology (2022)
-
The population genetics of pathogenic Escherichia coli
Nature Reviews Microbiology (2021)
-
Horizontally acquired papGII-containing pathogenicity islands underlie the emergence of invasive uropathogenic Escherichia coli lineages
Nature Communications (2020)
-
Dynamics of antimicrobial resistance in intestinal Escherichia coli from children in community settings in South Asia and sub-Saharan Africa
Nature Microbiology (2018)
