
The detection of protein-protein interactions in vivo is of critical importance to our understanding of biological processes. The classic library approach has been to use the yeast two-hybrid screen, where an interaction between known bait and unknown prey proteins leads to restoration of transcription factor activity1. However, its use is limited by host organism and nuclear localization requirements, and a tendency to detect indirect interactions (false positives). Bacterial two-hybrid screens have eliminated localization requirements and simplified many technical aspects of the procedure2. An innovative approach has been the reassembly of protein fragments, which then directly report interactions. A suitable reporter protein is dissected at the genetic level, and the fragments are fused to bait and prey, which are then coexpressed in vivo. Bait and prey interaction brings the reporter fragments together, facilitating reassembly of the active reporter protein, giving a direct readout of the association. This method has been demonstrated for dihydrofolate reductase3,4, ubiquitin5 and the green fluorescent protein6 (GFP) from Aequorea victoria. We recently described improvements to the original screen based on the reassembly of the GFP enhanced-stability mutant sg100 in Escherichia coli7. Our system, presented in the protocol that follows, consists of two plasmid vectors for the independent expression of fusions with N- and C-terminal fragments of GFP, and allows for simple visual detection of protein-protein interactions with a KD as weak as 1 mM.
Materials
Reagents
-
Plasmid vectors pET11a-link-NGFP and pMRBAD-link-CGFP and positive control vectors pET11a-Z-NGFP and pMRBAD-Z-CGFP are available upon request (Fig. 1)
Figure 1: GFP-fragment reassembly vectors and cloning linker sequences. 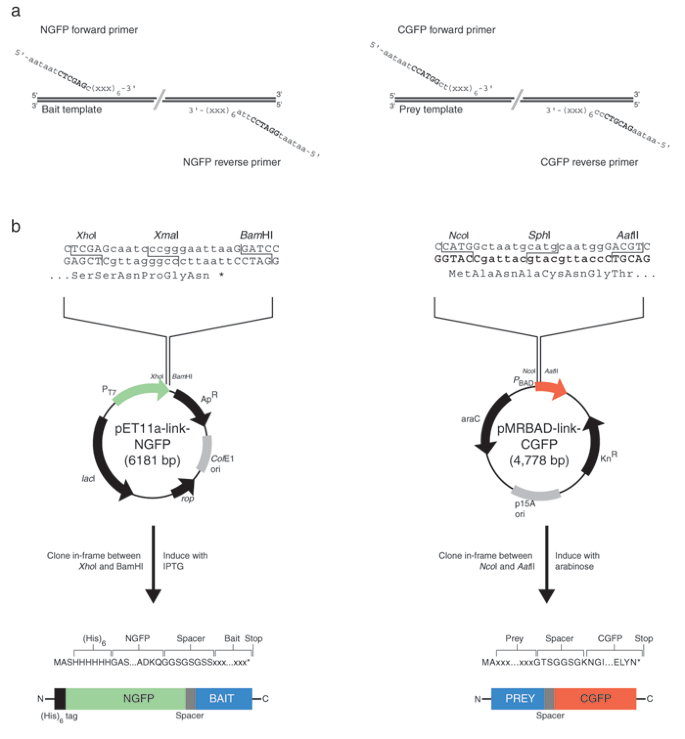
(a) Oligonucleotide linker sequences for amplification of target bait and prey DNA. NGFP in-frame fusions are made through the XhoI site in the forward primer sequence. The reverse primer should contain a stop codon followed by a BamHI site. Cloning into pMRBAD-link-CFGP requires a forward primer in frame with the ATG in the NcoI site. Fusion to CGFP is achieved through the in-frame AatII site primer sequence. The symbol (xxx) represents codons of the template gene to be amplified and cloned into the vectors. Six codons (18 bp) are usually sufficient to obtain specific amplification of most target sequences. The six-base extension (5′-aataat) is required for efficient cutting by restriction enzymes. (b) Cloning linker sites and key features of the GFP-fragment vectors. pET11a-link-GFP contains the ampicillin resistance marker (ApR) and ColE1 origin of replication (ori). The XmaI site in the linker is lost during cloning; thus, digestion with XmaI can be used to eliminate the background that results from religation. Fusion expression is achieved in a BL21 DE3 host with the addition of IPTG, which results in transcription from the T7 promoter (PT7). The rop gene product regulates the copy number of plasmids containing the ColE1 origin of replication, and the lacI gene product, the Lac repressor, reduces leaky expression in DE3 bacterial hosts. pMRBAD-link-GFP contains the kanamycin resistance gene (KnR) and p15A origin of replication. The linker SphI site is lost during cloning; thus, digestion with SphI can be used to eliminate the background that results from religation. Fusion expression is under the control of the PBAD promoter (regulated by the product of the araC gene) and is induced with L-(+)-arabinose. Note that the resulting GFP fusions have different orientations relative to the GFP fragment, which may affect the likelihood of successful GFP reassembly. Sequence details of these vectors can be found in Supplementary Note online and at http://www.csb.yale.edu/people/regan/publications.html. The structures of the vectors can be found in Supplementary Fig. 1 online.
-
Primers for cloning into pET11a-link-NGFP and pMRBAD-link-CGFP (Fig. 1)
-
Selective media: LB containing 100 μg/ml ampicillin and LB containing 35 μg/ml kanamycin
-
Restriction enzymes: NcoI, BamHI, AatII, XhoI, XmaI and SphI (New England Biolabs (NEB))
-
Thermostable DNA polymerase (for example, Deep Vent Polymerase, NEB)
-
dNTP solution (10 mM)
-
Calf intestinal alkaline phosphatase (NEB)
-
T4 DNA ligase (NEB)
-
E. coli strains DH10B and BL21 (DE3), prepared as competent cells (preferably electrocompetent)
-
Sequencing primers for pET11a-link-NGFP (T7 terminator primer 5′-tatgctagttattgctcag-3′) and pMRBAD-link-CGFP (5′-ctactgtttctccatacccg-3′)
-
ExoSAP-IT (USB)
-
Screening medium: for 250 ml LB agar (for about ten plates) add 250 μl of 10 mM IPTG, 2.5 ml of 20% arabinose, 250 μl of 100 mg/ml ampicillin (100 μg/ml) and 250 μl of 35 mg/ml kanamycin (35 μg/ml). Sterilize all additives by passing through a 0.2 μm filter.
-
Lysis buffer: 50 mM Tris HCl, pH 8.0, 300 mM NaCl, 10 mM imidazole, 0.1% Triton X-100, 1 mg/ml lysozyme, 0.5 mM CaCl2, 5 mM MgCl2
-
Nucleases: DNase and RNase (Sigma)
-
Ni-NTA agarose (Qiagen)
-
Wash buffer: 50 mM Tris HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole

Equipment
-
Thermal cycler programmed with the desired amplification protocol
-
Handheld long-wave UV lamp (365 nm)
-
Disposable standing column (Bio-Rad)
Procedure
Preparation of vectors encoding the fusion proteins
-
1
To prepare the cloning vectors, transform separately pET11a-link-NGFP and pMRBAD-link-CGFP into an E. coli strain that gives high-quality plasmid DNA (for example, DH10B or XL1-Blue) and plate onto selective agar media. Select for cells carrying the pET11a constructs in the presence of 100 μg/ml ampicillin and for those carrying pMRBAD constructs with 35 μg/ml kanamycin. Prepare plasmid DNA using standard miniprep protocols.
pMRBAD-link-CGFP transformations must be incubated with shaking at 37 °C for 1 h before they are plated on selective media.
Critical Step
pMRBAD-link-CGFP uses the kanamycin resistance gene as a selectable marker. All transformations involving pMRBAD-CGFP constructs (steps 1 and 4) must be followed by incubation, with shaking at 37 °C for at least 1 h, before plating on kanamycin-containing media.
-
2
To prepare vectors pET11a-link-NGFP and pMRBAD-link-CGFP for cloning, set up the following reactions for double digestion:

Incubate the reactions at 37 °C for 3 h, adding 1 μl of calf intestinal alkaline phosphatase (10 units) for the final 30 min.
-
3
Purify the vector products by electrophoresis through 0.8–1% agarose and determine the concentration of vector DNA by measuring UV absorbance at 260 nm or by running an aliquot on an agarose gel.
We purify the DNA by electrophoresis through Whatman glass fiber filter paper (GF/C) onto dialysis membrane, but commercial methods (for example, gel solubilization and extraction procedures) are acceptable. Ethanol-precipitate the DNA before ligation to concentrate the sample and remove salts that can interfere with ligation. The amount of vector obtained is usually sufficient to perform two 260-ng (∼0.05-pmol) ligations.
-
4
Set up an amplification reaction to prepare each of the inserts, incorporating the appropriate linker sequences (Fig. 1a) to produce in-frame GFP-fragment fusions:
Depending on the composition of the amplification buffer, additional Mg2+ may be required for template DNA above 1 kilobase (kb).

Alternatively, small inserts (<150 base pairs, bp) can be prepared synthetically, by annealing two oligonucleotides and extending with Klenow DNA polymerase. It is critical that the digested fragments produce in-frame fusions (see Fig. 1b ).
Critical Step
It is important to consider carefully into which vector the bait and prey should be cloned. For each bait-prey experiment, two orientations are possible: NGFP-bait with prey-CGFP and NGFP-prey with bait-CGFP. Knowledge of the bait-prey complex topology, and the location of free N and C termini, is especially useful at this stage. Avoid fusions to the GFP fragments at the site of protein-protein interaction and consider optimizing the spacer length between the fusions and GFP fragments, if necessary. In the absence of structural information, both orientations of bait and prey should be tried.
-
5
Amplify the nucleic acids according to the following program:
Times and temperatures are optimized for a 1-kb template but may need to be adapted to suit the particular reaction conditions. For example, the duration of the polymerization step should be 1 min/kb for Deep Vent DNA polymerase.

-
6
To purify the amplification products, extract each reaction once with phenol/chloroform/isoamyl alcohol (25: 24:1, v/v/v) and then with chloroform/isoamyl alcohol (29:1, v/v) and recover the DNA by precipitation with ethanol.
Analyze the size and quality of the products by electrophoresis through an agarose gel.
-
7
To prepare the inserts for cloning, digest 1 μg of insert DNA of the appropriate vector as described in step 2, omitting the calf intestinal alkaline phosphatase treatment.
-
8
Set up the reactions for ligation of inserts into vectors, by mixing 0.05 pmol of vector with 0.05 pmol of insert (∼260 ng pET11a-link-NGFP and 150 ng pMRBAD-link-CGFP per reaction):

Incubate reactions at 16 °C for at least 3 h.
Critical Step
To optimize transformation efficiencies, empty religated vector can be eliminated by digesting the reaction with 0.5 μl of restriction enzyme corresponding to the linker site removed through cloning (XmaI for pET11a-link-NGFP, SphI for pMRBAD-link-CGFP). Incubate the reaction at 37 °C for 1 h, followed by heat inactivation at 80 °C for 20 min. This additional step is especially useful when a low background is important (for example, when cloning libraries). The target sequence must not contain the linker restriction site that is removed through cloning.




Transformation and analysis of the clones
-
9
Transform the ligated products by mixing 1 μl of each reaction (from step 8) with 50 μl electrocompetent E. coli DH10B. Transform, preferably by electroporation (2-mm cuvette at 2.5 kV), and quench with 1 ml of 2YT. Plate the pET11a-link-NGFP transformations immediately onto LB agar containing 100 μg/ml ampicillin. Incubate the pMRBAD-link-CGFP transformations with shaking (>200 r.p.m.) at 37 °C for at least 1 h, then plate onto selective agar medium containing 35 μg/ml kanamycin.
-
10
Select several colonies, grow each in 5 ml of 2YT medium with the appropriate antibiotic and prepare plasmid DNA from 1.5 ml of culture using a standard miniprep procedure.
-
11
Verify the presence of the insert by digesting 10 μl of each miniprep with the appropriate pair of restriction enzymes and separating the fragments by electrophoresis through an agarose gel. Alternatively, the loss of the unique linker restriction sites can also be used as a diagnostic.
-
12
Confirm that the GFP fusions are the correct ones by sequencing the clones using appropriate primers (Fig. 1a).
In the specific case of pMRBAD-CGFP constructs, which we prefer for libraries, we perform colony PCR with primers 5′-tagcggatcctacctgacgc-3′ and 5′-ttcgggctttgttagcagcc-3′. Reactions are performed in a 96-well plate and are cleaned up for sequencing by treatment with ExoSAP-IT at 37 °C for 15 min, followed by 80 °C for 20 min.
-
13
Transform the verified plasmid constructs into a DE3 host (we use electrocompetent BL21). When a single pair of constructs is to be checked, simultaneous transformation of both plasmids is possible, using 1–10 ng of each construct miniprep DNA in 1μl water. Incubate the doubly transformed cells with shaking at 37 °C for 1 h and then plate them on doubly selective LB agar plates (100 μg/ml ampicillin and 35 μg/ml kanamycin).
When the transformation efficiency is critical (for example, when one plasmid contains a library), we prepare electrocompetent cells containing the constant partner.
Screening for GFP reassembly
-
14
To perform the screen for GFP reassembly, plate 5–10 μl of a 1:104 dilution of a fresh overnight culture onto screening media to obtain individual colonies. We recommend marking the agar plate into quadrants, for control and experimental combinations (Fig. 2a). This allows for side-by-side comparison of bait and prey sets (Fig. 2b). Incubate the plates either at 30 °C overnight followed by 15–25 °C for 1–2 d, or continuously at 15–25 °C for 2–3 d. After the first day, it is advisable to seal plates with Parafilm, to prevent them from drying out.
Figure 2: (a) Schematic plate layout for screening interactions with the GFP reassembly screen. 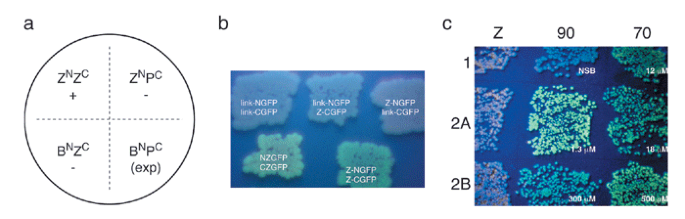
ZN and ZC represent control leucine zipper constructs (positive control), and BN and PC are the bait and prey NGFP and CGFP constructs, respectively. This arrangement provides a positive control, two negative controls and an experimental (exp) quadrant. (b) Examples of GFP reassembly with leucine zipper peptides. Top, empty vectors and single leucine zipper expression combinations do not give green fluorescence. Bottom, fluorescence observed with the original NZGFP and CZGFP constructs6 (left) and improved vectors7 (right). (c) GFP reassembly mediated by the TPRs of HOP7. pET11a-link-NGFP constructs contained the negative control leucine zipper peptide (Z) and the C-terminal 24 residues of Hsp70 and Hsp90. pMRBAD-link-CGFP contained TPR1, TPR2A and TPR2B from HOP. Cognate interactions (70:TPR1, 90:TPR2A) are brighter than noncognate interactions (70:TPR2A, 90:TPR1), but affinities measured in vitro by surface plasmon resonance do not directly correlate to brightness. NSB, no saturating binding.
Typical induction conditions are 10 μM IPTG for pET11a-NGFP and 0.2% arabinose for pMRBAD-link-CGFP. These conditions give reproducible green-fluorescent colonies with the control leucine zipper bait and prey peptide fusions.
Critical Step
Incubation at 15–25 °C is required for the development of green fluorescence. Incubation at 37 °C alone does not produce fluorescent colonies.
-
15
Observe the plates with a handheld long-wave UV lamp (365 nm) to visualize colonies that exhibit fluorescence (thus having properly reassembled GFP).
Green fluorescence under long-wave ultraviolet illumination (365 nm) typically develops after the second day and does not improve beyond 4 days.
Critical Step
Suitable safety measures (safety glasses, gloves) should be employed when using UV light. Most brands of disposable plastic Petri dishes (for example, Falcon 1029) transmit UV light poorly. Visualization must be performed on open plates, and care should be taken in their handling.

Purification of the reassembled GFP complex
-
16
If fluorescent cells do not appear, select a candidate colony from those identified in step 15, and prepare a 50-ml culture in LB, supplemented with ampicillin and kanamycin but without IPTG or arabinose. Prepare also positive (containing leucine zipper peptide) and negative (containing the empty vectors) control cultures for quantitative comparison. Grow the cultures at 37 °C overnight in a shaking incubator
Alternatively, bacteria from an agar plate may be recovered using a plate spreader and 1 ml of TN buffer (50 mm Tris HCl, pH 8.0; 300 mm NaCl). The pET11a-link-NGFP vector incorporates an N-terminal hexahistidine tag. Because GFP reassembly is effectively irreversible, and unassembled GFP-fragment fusions are typically insoluble, the reassembled fusion can be purified and concentrated in a single step from the soluble fraction of cell lysate7.
-
17
The next day, move the flasks to 15–25 °C and add IPTG and arabinose to the final induction concentrations (see step 14). Continue shaking for a further 2–4 d at 15–25 °C.
-
18
Harvest the cells by centrifugation, and resuspend them in 2 ml of lysis buffer. Add 1 μl each of DNase and RNase and incubate the lysate on ice for 30 min.
-
19
Split the lysate into 1 ml aliquots and centrifuge at 4 °C for 30 min at maximum speed in a benchtop microcentrifuge.
-
20
Combine the superatant with 200 μl Ni-NTA agarose slurry and mix gently at 4 °C for 1 h. Wash the resin in a disposable standing column with 2 ml of wash buffer and then elute His-tagged protein with wash buffer plus 300 mM imidazole.
Fluorescence per cell culture absorbance unit (600 nm) can be assessed quantitatively with an emission scan between 475 and 525 nm (λexcitation = 468 nm). Reassembled GFP variant sg100 has a λemission at ∼505 nm.
Troubleshooting
[Steps 1 and 5]
Problem: A low plasmid yield is obtained for pMRBAD-link-CGFP.
Solution: Vector origin of replication is p15A (low copy number). Miniprep yield is usually sufficient for sequencing. Plasmid copy number can be increased by adding chloramphenicol (150 μg/ml) after overnight growth and continuing growth for 8 h.
For larger-scale vector purification (midi- and maxiprep), DNA preparation by alkaline lysis/PEG precipitation is best.
[Steps 2 and 4]
Problem: We get poor cloning efficiency into pET11a-link-NGFP.
Solution: We have experienced problems digesting with BamHI. Ensure that the total glycerol concentration <5% and incubate reaction for a maximum of 3 h.
BamHI cannot be heat inactivated. Remove by phenol/chloroform/isoamyl alcohol extraction.
We recommend using pMRBAD-link-CGFP for efficiency-sensitive cloning (for example, libraries).
[Steps 14 and 15]
Problem: We observe no green fluorescence or poor fluorescence when screening colonies for presence of the reassembled GFP.
Solution:
Poor expression of fusions. Check for expression of GFP fusions on SDS-PAGE. Vary concentrations of inducer to obtain roughly stoichiometric levels of expression. We find that 0–20 μM IPTG and 0.02–0.2% arabinose is useful. Induction of pET11a-NGFP fusions may not be necessary, because the T7 promoter is leaky. Addition of >0.2% arabinose does not significantly improve expression of CGFP fusions.
Configurational incompatibility between bait and prey GFP fusions. GFP fragments could interfere with bait-prey interaction. Alternatively, bait and prey may interact, but the GFP fragments may be unable to reassemble. Try increasing bait-prey-GFP spacer length (for example, through multiple GlyGlySer insertions) or swapping bait-prey vector orientation. We have successfully used spacers up to 15 amino acids in length.
Bait or prey misfolded. The analyte protein most likely to be an independently structured domain should be placed on pMRBAD-link-CGFP vector, so that the unstructured CGFP fragment follows the folded domain. We typically display short peptides and unstructured motifs as NGFP fusions. Exploring alternative GFP dissection and fragment fusion strategies8 may be necessary.
Low reassembly efficiency. Factors such as expression level and solubility could lead to visually undetectable fluorescence in vivo. Perform expression in liquid culture and then use the NGFP His-tag to simultaneously purify and concentrate the soluble, reassembled material for quantitation in vitro. This method is very sensitive and should reveal if any reassembly (and therefore bait-prey interaction) is taking place.
Comments
Protein fragment-reassembly screens provide a powerful alternative to the traditional yeast two-hybrid approach. Those based on GFP reassembly offer several key advantages. First, detection of interactions is direct (green fluorescence) and does not require specific subcellular localization or a chemical substrate. Because the reassembly of GFP is effectively irreversible, fluorescence tends to accumulate over time, which probably aids in the detection of weak interactions. We have successfully used the screen to detect the values of KD between 1 μM and 1 mM. GFP also makes the screen amenable to fluorescence-activated cell sorting (FACS) procedures, which simplifies the selection of positive interaction pairs. Second, GFP expression has been documented in a wide variety of organisms, in nearly every cell type and in most subcellular locations9,10. GFP tagging often has no effect on protein function, and subcellular localization does not generally affect GFP chromophore maturation. Protein-protein interactions can therefore be studied in their native biological environment. Similar reassembly schemes have also been demonstrated for the related yellow and cyan fluorescent proteins, with alternative dissection and fusion sites within the molecule11,12. Reassembly to form hybrid fluorescent proteins, with unique spectral emission properties, opens the prospect of simultaneous detection of multiple tissue-specific expression patterns within an organism8.
Example of application: Tetratricopeptide repeat protein-ligand interactions
Human Hsp organizing protein (HOP) contains three tetratricopeptide repeat (TPR) domains. TPR1 and TPR2A bind the C-terminal residues of Hsp70 and Hsp90, respectively. TPR2B has no known ligand. The sequences encoding TPR domains of HOP were cloned into pMRBAD-link-CGFP. The corresponding peptide ligand constructs were prepared by Klenow extension and ligated into pET11a-link-NGFP. Pairs of link-NGFP and link-CGFP were transformed into E. coli BL21 (DE3) and plated onto LB agar containing 15 μM IPTG and 0.2% arabinose. In vivo–reassembled GFP (Fig. 2c) recapitulated the trends described by in vitro biophysical methods7. TPR2B gave strong green fluorescence in combination with Hsp70 and Hsp90, which is in contrast to all the KD values determined in vitro (500 μM and 300 μM, respectively). This result highlights the sensitivity of the screen to factors in addition to receptor-ligand affinity, such as construct solubility and expression level. Fluorescence intensity does not necessarily correlate quantitatively to interaction affinity.
Source
This protocol was provided directly by the authors listed on the title page. For further details on the composition of media and standard procedures, such as transformation, see Sambrook, J. & Russell, DW. Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA, 2001).
Note: Supplementary information is available on the Nature Methods website.
References
Fields, S. & Song, O. A novel genetic system to detect protein-protein interactions. Nature 340, 245–246 (1989).
Joung, J.K., Ramm, E.I. & Pabo, C.O. A bacterial two-hybrid selection system for studying protein–DNA and protein–protein interactions. Proc. Natl. Acad. Sci. USA 97, 7382–7387 (2000).
Pelletier, J.N., Campbell-Valois, F.X. & Michnick, S.W. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc. Natl. Acad. Sci. USA 95, 12141–12146 (1998).
Pelletier, J.N., Arndt, K.M., Plückthun, A. & Michnick, S.W. An in vivo library-versus-library selection of optimized protein-protein interactions. Nat. Biotechnol. 17, 683–690 (1999).
Johnsson, N. & Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA 91, 10340–10344 (1994).
Ghosh, I., Hamilton, A.D. & Regan, L. Antiparallel leucine zipper-directed reassembly: Application to the green fluorescent protein. J. Am. Chem. Soc. 122, 5658–5659 (2000).
Magliery, T.J. et al. Detecting protein-protein interactions with a GFP-fragment reassembly trap: scope and mechanism. J. Am. Chem. Soc. (in the press).
Magliery, T.J. & Regan, L. Reassembled GFP: Detecting protein-protein interactions and protein expression patterns. in Green Fluorescent Protein: Properties, Applications, Protocols, edn. 2 (eds. Chalfie, M. & Kain, S.) (John Wiley & Sons, Hoboken, New Jersey, USA, in the press).
Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544 (1998).
Waldo, G.S., Standish, B.M., Berendzen, J. & Terwilliger, T.C. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 17, 691–695 (1999).
Hu, C.D., Chinenov, Y. & Kerppola, T.K. Visualization of interactions among bZip and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798 (2002).
Hu, C.D. & Kerppola, T.K. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21, 539–545 (2003).
Author information
Authors and Affiliations
Supplementary information
Supplementary Fig. 1
Vector maps of pET11a-link-NGF and pMRBAD-link-CGFP. (PDF 104 kb)
Supplementary Note
Sequences of the plasmid vectors pET11a-link-NGF and pMRBAD-link-CGFP. (PDF 85 kb)
Rights and permissions
About this article
Cite this article
Wilson, C., Magliery, T. & Regan, L. Detecting protein-protein interactions with GFP-fragment reassembly. Nat Methods 1, 255–262 (2004). https://doi.org/10.1038/nmeth1204-255
Issue Date:
DOI: https://doi.org/10.1038/nmeth1204-255
This article is cited by
-
Protein stabilization with retained function of monellin using a split GFP system
Scientific Reports (2018)
-
Heterologous protein-DNA interactions lead to biased allelic expression of circadian clock genes in interspecific hybrids
Scientific Reports (2017)
-
Specific cell surface labeling of GPCRs using split GFP
Scientific Reports (2016)
-
An ER-peroxisome tether exerts peroxisome population control in yeast
The EMBO Journal (2013)
-
New cell line development for antibody-producing Chinese hamster ovary cells using split green fluorescent protein
BMC Biotechnology (2012)
