
Abstract
High-mass-resolution imaging mass spectrometry promises to localize hundreds of metabolites in tissues, cell cultures, and agar plates with cellular resolution, but it is hampered by the lack of bioinformatics tools for automated metabolite identification. We report pySM, a framework for false discovery rate (FDR)-controlled metabolite annotation at the level of the molecular sum formula, for high-mass-resolution imaging mass spectrometry (https://github.com/alexandrovteam/pySM). We introduce a metabolite-signal match score and a target–decoy FDR estimate for spatial metabolomics.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


References
Baker, M. Nat. Methods 7, 157–161 (2010).
Johnson, C.H., Ivanisevic, J. & Siuzdak, G. Nat. Rev. Mol. Cell Biol. 17, 451–459 (2016).
Watrous, J.D., Alexandrov, T. & Dorrestein, P.C. J. Mass Spectrom. 46, 209–222 (2011).
Watrous, J.D. & Dorrestein, P.C. Nat. Rev. Microbiol. 9, 683–694 (2011).
Schwamborn, K. & Caprioli, R.M. Nat. Rev. Cancer 10, 639–646 (2010).
Attia, A.S. et al. Cell Host Microbe 11, 664–673 (2012).
Johnson, C.H. et al. Cell Metab. 21, 891–897 (2015).
Soltwisch, J. et al. Science 348, 211–215 (2015).
Palmer, A., Trede, D. & Alexandrov, T. Metabolomics 12, 107 (2016).
Spengler, B. Anal. Chem. 87, 64–82 (2015).
Kind, T. & Fiehn, O. BMC Bioinformatics 7, 234 (2006).
Benjamini, Y. & Hochberg, Y. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995).
Storey, J.D. J. R. Stat. Soc. Series B Stat. Methodol. 64, 479–498 (2002).
Käll, L., Storey, J.D., MacCoss, M.J. & Noble, W.S. J. Proteome Res. 7, 29–34 (2008).
Matsuda, F. et al. PLoS One 4, e7490 (2009).
Wishart, D.S. et al. Nucleic Acids Res. 41, D801–D807 (2013).
Sumner, L.W. et al. Metabolomics 3, 211–221 (2007).
Alexandrov, T. & Bartels, A. Bioinformatics 29, 2335–2342 (2013).
Wijetunge, C.D. et al. Bioinformatics 31, 3198–3206 (2015).
Wieser, M.E. & Coplen, T.B. Pure Appl. Chem. 83, 359–396 (2011).
Gode, D. & Volmer, D.A. Analyst (Lond.) 138, 1289–1315 (2013).
Vese, L.A. & Chan, T.F. Int. J. Comput. Vis. 50, 271 (2002).
Edelsbrunner, H. & Harer, J. Contemp. Math. 453, 257–282 (2008).
Glasbey, C.A. & Horgan, G.W. Image Analysis for the Biological Sciences (Wiley Chichester, 1995).
Carter, C.L., McLeod, C.W. & Bunch, J. J. Am. Soc. Mass Spectrom. 22, 1991–1998 (2011).
Elias, J.E. & Gygi, S.P. Nat. Methods 4, 207–214 (2007).
Berglund, M. & Wieser, M.E. Pure Appl. Chem. 83, 397–410 (2011).
Storey, J.D. & Tibshirani, R. Proc. Natl. Acad. Sci. USA 100, 9440–9445 (2003).
Choi, H., Fermin, D. & Nesvizhskii, A.I. Mol. Cell. Proteomics 7, 2373–2385 (2008).
Zhang, J. et al. Mol. Cell. Proteomics 11, M111.010587 (2012).
Bligh, E.G. & Dyer, W.J. Can. J. Biochem. Physiol. 37, 911–917 (1959).
Acknowledgements
We thank O. Vitek, A. Makarov, and M. Savitski for discussions on FDR, and D. Feichtner-Kozlov for discussions on computational topology. We acknowledge funding from the European Union's Horizon2020 and FP7 programs under grant agreements 634402 (A.P., R.L., A.T., V.K., S.N., C.P., and T.A.) and 305259 (I.C., R.L., and C.P.), and from the Russian Government Program of Competitive Growth of Kazan Federal University (S.N.). We thank EMBL Metabolomics Core Facility for instrumentation for LC-MS/MS analysis. T.A. thanks P. Dorrestein (UCSD) and P. Maass (University of Bremen) for providing a stimulating environment as well as for discussions on mass spectrometry and image analysis during the years of this work.
Author information
Authors and Affiliations
Contributions
A.P. and T.A. conceived the study; A.P., I.C., D.F., A.T., S.N. and V.K. implemented the algorithms; R.L., J.F., C.P., and M.B. provided imaging data; A.P. and T.A. analyzed imaging data; P.P. collected LC-MS/MS data; P.P. and T.A. performed LC-MS/MS validation; A.P. and T.A. wrote manuscript with feedback from all other coauthors; and T.A. coordinated the project.
Corresponding author
Ethics declarations
Competing interests
T.A. is the scientific director and a shareholder of SCiLS GmbH, a company providing software for imaging mass spectrometry. While this study was being conducted, M.B. was an employee of Bruker Daltonik GmbH, a company providing instrumentation and software for imaging mass spectrometry.
Integrated supplementary information
Supplementary Figure 1 Illustration of an ion signal in the dataset a1s1 for [M+Na]+
Illustration of an ion signal in the dataset a1s1 corresponding to the sum formula C43H76NO7P corresponding to PE(P-38:5) in HMDB, and the +Na ion adduct. The theoretical isotope pattern (blue) was predicted at the resolving power of R=100000 at m/z 400 and ion images of the predicted peaks were generated with the tolerance of ±2.5 ppm. The ion was annotated at the desired FDR level of 0.1 and was validated by LC-MS/MS as PE(P-18:1_20:4); see Section S11.
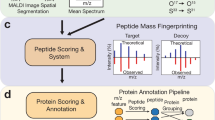
Supplementary Figure 2 Detailed scheme of the proposed framework
The framework consists of three parts: calculation of the Metabolite-Signal-Match score, FDR estimation, and FDR-controlled annotation. This scheme provides more comments as compared to Figure 1. The ion [C43H76NO7P+Na]+ was annotated at the desired FDR level of 0.1 and was validated by LC-MS/MS as PE(P-18:1_20:4); see Section S11.
Supplementary Figure 3 Detail on the novel measure of spatial chaos used to quantify the informativeness of a principal ion image in the ion signal.
a) The principle of the novel measure of spatial chaos is counting the number of objects in an ion image at a particular intensity level and measuring the statistical distribution of objects amongst a set of levels. Ion image intensities are shown as a 3D landscape with the heat colormap (from black to red to yellow from intensities from low to high). The level-set cutting plane is shown in half-transparent blue. Objects from one level are shown on the left with connected pixels from the same object shown in the same color; pixels below the level-set-intensity shown in black. Top: an exemplary structured ion image; Bottom: an exemplary unstructured noisy ion image. b) Illustrations demonstrating behavior of the measure of spatial chaos. Top: Dependence of number of objects in an ion image at a particular level-set on the threshold level (intensity value used for the level set) for the structured (grey) and unstructured (black) images shown in the panel a). For a threshold level, the measure of spatial chaos is equal to the area under the curve for the range from 0 to the given level. Bottom: Dependence of the statistic on the signal-to-noise ratio (SNR) for a structured ion image with added noise.
Supplementary Figure 4 Evaluation of the proposed level-sets-based measure of spatial chaos
Evaluation of the proposed level-sets-based measure of spatial chaos as compared to other existing measures of spatial chaos: from (Alexandrov & Bartels, 2013) 1 and (Wijetunge et al., 2015) 2. The evaluation is performed according to the statistical framework originally proposed by us in (Alexandrov & Bartels, 2013) 1.
Supplementary Figure 5 Evaluation of the similarity between the target and decoy sets of ions for metabolites from HMDB.
Left (a, c, e): Box plots of the four highest isotopic peaks. Right (b, d, f): Kendrik plot with projections along either m/z or mass defect. The decoy set is selected as a random sampling from the set of decoy ions.
Supplementary Figure 6 Evaluation of the false-discovery-rate estimation on simulated data.
a) FDR curves for the target adducts +H, +Na, and +K. b) Relation between the true FDR and the FDR for the target adducts +H (red), +Na (blue), and +K (green). Shaded area shows confidence limits (5-95) from 20 independent replicates.
Supplementary Figure 7 Negative control experiments using implausible adducts as the ‘target’.
Top row: FDR curves per an implausible adduct Bottom row: AUC100 values. As baselines, the FDR curves and AUC100 values for the plausible adducts are shown for +H (blue), +Na (green), and +K (red). The curves are shown for 200 first annotations. Error bars in the AUC100 diagrams show standard deviation across 10 sampled decoy sets.
Supplementary Figure 8 FDR-curves produced when using different scoring measures
Three measures were tested: exact mass matching (light green), measure of chaos (blue green) and MSM (orange). The FDR-curves show that the MSM measure outperforms other measures (has lowest FDR-curve) across all datasets and adducts.
Supplementary Figure 9 FDR curves showing the effect of changing the m/z tolerance for generating ion images
FDR-curves for different m/z-tolerance values used to sample ion images (2.5 ppm was used in the paper), for different datasets and ion adducts. The change of m/z-tolerance was used to evaluate effect of mass accuracy and resolving power.
Supplementary Figure 10 Comparing annotations using the HMDB and SwissLipids molecular databases.
a) Overlap in the molecular annotations for all 10 datasets from the paper with the desired FDR of 0.1. b) Overlap between sum formulas from HMDB and SwissLipids
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–9, Supplementary Tables 1 and 2, Supplementary Notes 1–3, and Supplementary Data 1 and 2. (PDF 18988 kb)
Rights and permissions
About this article

Cite this article
Palmer, A., Phapale, P., Chernyavsky, I. et al. FDR-controlled metabolite annotation for high-resolution imaging mass spectrometry. Nat Methods 14, 57–60 (2017). https://doi.org/10.1038/nmeth.4072
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.4072
This article is cited by
-
An N-glycome tissue atlas of 15 human normal and cancer tissue types determined by MALDI-imaging mass spectrometry
Scientific Reports (2024)
-
Multiscale biochemical mapping of the brain through deep-learning-enhanced high-throughput mass spectrometry
Nature Methods (2024)
-
Spatial analysis of the osteoarthritis microenvironment: techniques, insights, and applications
Bone Research (2024)
-
Mapping microhabitats of lignocellulose decomposition by a microbial consortium
Nature Chemical Biology (2024)
-
rMSIfragment: improving MALDI-MSI lipidomics through automated in-source fragment annotation
Journal of Cheminformatics (2023)
