
Abstract
We introduce RNA G-quadruplex sequencing (rG4-seq), a transcriptome-wide RNA G-quadruplex (rG4) profiling method that couples rG4-mediated reverse transcriptase stalling with next-generation sequencing. Using rG4-seq on polyadenylated-enriched HeLa RNA, we generated a global in vitro map of thousands of canonical and noncanonical rG4 structures. We characterize rG4 formation relative to cytosine content and alternative RNA structure stability, uncover rG4-dependent differences in RNA folding and show evolutionarily conserved enrichment in transcripts mediating RNA processing and stability.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Accession codes
References
Wan, Y., Kertesz, M., Spitale, R.C., Segal, E. & Chang, H.Y. Nat. Rev. Genet. 12, 641–655 (2011).
Kwok, C.K. et al. Trends Biochem. Sci. 40, 221–232 (2015).
Neidle, S. & Balasubramanian, S. Quadruplex nucleic acids Vol. 7 (Royal Society of Chemistry, 2006).
Biffi, G. et al. Nat. Chem. 6, 75–80 (2014).
Millevoi, S., Moine, H. & Vagner, S. WIREs RNA 3, 495–507 (2012).
Rodriguez, R. et al. J. Am. Chem. Soc. 130, 15758–15759 (2008).
Huppert, J.L. et al. Nucleic Acids Res. 36, 6260–6268 (2008).
Jodoin, R. et al. RNA 20, 1129–1141 (2014).
Martadinata, H. & Phan, A.T. Biochemistry 53, 1595–1600 (2014).
Pandey, S. et al. J. Phys. Chem. B 117, 6896–6905 (2013).
Kwok, C.K. et al. Angew. Chem. Int. Ed. 55, 8958–8961 (2016).
Chambers, V.S. et al. Nat. Biotechnol. 33, 877–881 (2015).
Wolfe, A.L. et al. Nature 513, 65–70 (2014).
Beaudoin, J.D. et al. Nucleic Acids Res. 42, 1209–1223 (2014).
Lorenz, R. et al. Algorithms Mol. Biol. 6, 26 (2011).
Arora, A. & Suess, B. RNA Biol. 8, 802–805 (2011).
Crenshaw, E. et al. PLoS One 10, e0143160 (2015).
Jayaraj, G.G. et al. RNA Biol. 9, 81–86 (2012).
Stefanovic, S., Bassell, G.J. & Mihailescu, M.R. RNA 21, 48–60 (2015).
Kenny, P.J. et al. Cell Rep. 9, 1729–1741 (2014).
Beaudoin, J.D. & Perreault, J.P. Nucleic Acids Res. 41, 5898–5911 (2013).
Reuter, J.S. & Mathews, D.H. BMC Bioinformatics 11, 129 (2010).
Ding, Y. et al. Nature 505, 696–700 (2014).
Pandey, S. et al. Biochemistry 54, 7067–7078 (2015).
Ashburner, M. et al. Nat. Genet. 25, 25–29 (2000).
Kwok, C.K., Ding, Y., Shahid, S., Assmann, S.M. & Bevilacqua, P.C. Biochem. J. 467, 91–102 (2015).
Kwok, C.K. & Balasubramanian, S. Angew. Chem. Int. Ed. 54, 6751–6754 (2015).
Kwok, C.K. et al. 10.1038/protex.2016.060 (2016).
Kim, D. et al. Genome Biol. 14, R36 (2013).
Li, H. et al. Bioinformatics 25, 2078–2079 (2009).
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing (Vienna, Austria, 2014).
Agarwal, V., Bell, G.W., Nam, J.-W. & Bartel, D.P. eLife 4, e05005 (2015).
Harrow, J. . et al. Genome Res. 22, 1760–1774 (2012).
Lorenz, R. et al. in Advances in Bioinformatics and Computational Biology (eds. de Souto, C.P. et al.) Ch. 5 (Springer, Berlin Heidelberg, 2012).
Zhang, A.Y.Q., Bugaut, A. & Balasubramanian, S. Biochemistry 50, 7251–7258 (2011).
Kwok, C.K., Ding, Y., Tang, Y., Assmann, S.M. & Bevilacqua, P.C. Nat. Commun. 4, 2971 (2013).
Acknowledgements
This study is supported by a European Research Council Advanced Grant no. 339778 (S.B.), a CASE studentship from Biotechnology and Biological Sciences Research Council (BBSRC) and Illumina BB/I015477/1 (V.S.C.), a Herchel Smith Fellowship (A.B.S.), and some support from the Croucher Foundation (C.K.K.). S.B. is a senior investigator of the Wellcome Trust grant no. 099232/z/12/z. We thank members of the Balasubramanian laboratory for comments.
Author information
Authors and Affiliations
Contributions
C.K.K., G.M., A.B.S., V.S.C. and S.B. designed the experiments; C.K.K., G.M. and A.B.S. performed the experiments and data analysis. C.K.K., G.M., A.B.S., V.S.C. and S.B. interpreted the results and cowrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
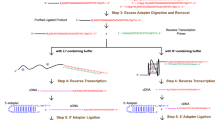
Supplementary Figure 1 Overview of rG4-seq and chemical structures of rG4 and PDS.
(a) Chemical structure of G-quartet and schematic of an intramolecular RNA G-quadruplex (rG4). The presence of K+ stabilises this RNA structural motif. (b) Working flowchart of rG4-seq. RNA is ligated to a 3’ adapter, followed by RNA folding under Li+ (rG4 non-stabilizing), K+ (physiological) or K++PDS (rG4-stabilizing) conditions. rG4 induces reverse transcriptase (RTase) stalling, leading to cDNA fragments of different lengths. cDNAs are ligated to a 5’ adapter, followed by PCR and next generation sequencing (NGS). The BASP1 (chr5:17,276,185-17,276,254) example here shows a drop in coverage (from 3’ to 5’ direction) in K+ and K++PDS conditions due to rG4 formation, whereas coverage is generally uniform in Li+. (c) Chemical structure of pyridostatin (PDS), an rG4 stabilising ligand.
Supplementary Figure 2 Results from rG4-seq are consistent with the gel-based RTS assay on positive and negative control RNAs.
(a) rG4-seq profiles of positive controls (lines), compared to those obtained by gel-based RTS assay (gels) under Li+, K+, and K++PDS conditions. The K+ and Li+ rG4-seq data was deliberately offset (to the right) by 1 and 2 nucleotide(s) to the K++PDS data for better visualization. Purple arrows indicate the same base on both line plots and gels. The reverse transcription is from 3’ to 5’ direction. The rG4-seq results on positive controls (line plots) show strong RTS in K+ and K++PDS conditions and no or weak stalling in Li+ condition, consistent with the corresponding gels (gels). Dideoxy C was used to show G nucleotide in the gels. (b) rG4-seq profiles of negative controls (lines), compared to those obtained by gel-based RTS assay (gels) under Li+, K+, and K++PDS conditions. The K+ and Li+ rG4-seq data was deliberately offset (to the right) by 1 and 2 nucleotide(s) to the K++PDS data for better visualization. Purple arrows indicate the same base on both line plots and gels. The reverse transcription is from 3’ to 5’ direction. The rG4-seq results on negative controls (line plots) only show full-length products, and no observable stalling in all three conditions (gels). Dideoxy C and dideoxy T were used to show G and A nucleotides in the gels respectively. Sequences used are shown in Supplementary Table 1.
Supplementary Figure 3 rG4-seq libraries are highly reproducible.
Summary of sequencing libraries and their correlations. (a) Table showing total sequenced reads, total mapped reads, and mapping rate for each sample. Four independent biological replicates are performed in this study for each condition (Li+, K+, K++PDS). (b) Heatmap showing a color-coded representation of the Pearson correlation coefficient (PCC) of read counts across exons for each pair of libraries, identifying an overall high correlation among the sequencing libraries.
Supplementary Figure 4 rG4-seq scoring pipeline is robust and specific for identification of rG4s transcriptome-wide.
(a) Heatmap and hierarchical clustering displaying the similarity of coverage signal for all libraries at the 2,688 exonic putative canonical G-quadruplex sites with coverage ≥ 6 in all Li+ replicates (see Methods). The color-coded matrix values represented the Pearson correlation coefficient (PCC) for each pair of libraries. As expected, correlation within the same condition (blocks along diagonal) is the highest (0.68-0.84) for all libraries, with Li+ displaying the lowest within-condition values (0.68-0.70) due to a mild or no effect at rG4 sites. rG4 stabilizing conditions (K+ and K++PDS) display instead the highest within-condition correlation (0.81-0.84), and also between the two conditions (0.79-0.82) as they both stabilize similar structures, while showing poor correlation with Li+ (0.50-0.64 for K+ and 0.45-0.58 for K++PDS). (b), Barplot displaying the number of scoring regions as assessed by the scoring pipeline (see Methods) when setting the significance threshold to p-value (P) ≤ 0.01. (c) Same as (b), with significance threshold set to FDR (false discovery rate) ≤ 0.1. In both (b) and (c), the scoring of each region is assessed by comparing the RTS signal in one condition versus another, according to the following legend: K++PDS (Li+) = signal in K++PDS versus Li+; K+ (Li+) = signal in K+ versus Li+; Li+ (K++PDS) = signal in Li+ versus K++PDS; Li+ (K+) = signal in Li+ versus K+. The FDR-based scoring yields very few regions where RTS is affected in Li+ (most right bars, values of 13 and 4), which are clearly false positive, while returning many regions where RTS is specific for the two rG4 stabilizing conditions, K+ and K++PDS. (d) G and GG motifs at detected stalling events. The bar graphs show the percentage of sequences displaying a G (left bars) or a GG (right bars) motif at stalling sites, either within 1 nucleotide (dark grey) or 3 nucleotides (light grey) from the detected stalling sites. The high percentage of G and GG motifs, typical of G-quadruplex forming structures, suggests that stalling events occur exactly at or near G-quadruplex sites. N = 3,845 from the K+ RTS sites. (e) RTS values at stalling sites common between K+ and K++PDS. Scatter plot comparing the fraction of stalling reads (RTS value) for the hits common in K+ and K++PDS (see Methods). (f) Boxplot showing the difference of the RTS values in K++PDS and K+ for all the data points shown in (e).
Supplementary Figure 5 In vitro selective 2’-hydroyxl acylation experiments validate non-canonical rG4 candidates identified by rG4-seq.
(a) MALAT 1 (chr11:65,269,314-65,269,406). (b) AGPAT5 (chr8:6,617,768-6,617,857). (c) RALA (chr7:39,726,284-39,726,373). Lanes 1-4 show sequencing of U, C, G, and A respectively. Lane 5 shows the minus 2-methylnicotinic acid imidazolide (NAI). Lanes 6-8 shows the NAI reaction under Li+, K+, and K++PDS conditions respectively. The change in NAI modification suggests change in RNA structural conformation. G-quadruplex structure is stabilized in K+ and K++PDS, but not in Li+, thus the modification change observed was likely attributed to rG4 formation. Orange asterisks denote nucleotides that are changed between Li+ and K+ condition. Green asterisks denote nucleotides that are changed between K+ and K++PDS conditions. Sequences used are shown in Supplementary Table 1.
Supplementary Figure 6 MEME motif analysis of the hits in the “Others” category.
Top 6 motifs as identified by MEME enrichment analysis for the hits in the category “Others” in (a) K+ and (b) K++PDS. Most enriched motifs are non-G-rich, although G-rich motifs are also identified as enriched (2nd and 6th motif in K+; 3rd and 5th motif in K++PDS).
Supplementary Figure 7 Overlap of rG4s in K+ and K++PDS.
(a) Venn diagram showing the overlap for all hits, including those in the “Others” category. (b) Overlap for hits, excluding those in the “Others” category.
Supplementary Figure 8 Overlap between hits and computationally predicted G-quadruplex structures (PQs).
(a) Venn diagram showing the overlap between scoring regions in K+ and PQs (G3+L1-7, see Methods). (b) Same as (a) for scoring regions in K++PDS. (c) Bar plot showing the repartition of the 6,299 exonic PQs between detected as hits (label “detected”, dark grey), not detected as hit but with sufficient coverage (i.e., coverage above 6; label “not detected”, grey) and with no coverage (i.e., coverage below 6; label “no coverage”, light grey) for both K+ and K++PDS conditions.
Supplementary Figure 9 Relative nucleotide content ratio on detected G3L1-7 versus undetected G3L1-7 for K+ and K++PDS.
The detected G3L1-7 have a lower C-content than undetected G3L1-7 case for K+ and K++PDS (smaller than one), while the detected G3L1-7 have a higher U-content/A-content than undetected G3L1-7 case for K+ and K++PDS (larger than one). Errors are calculated from standard deviation. The two bars on C are identical to Fig. 1c (single C-motif), and are shown again here for the purpose of comparison to other three nucleotides.
Supplementary Figure 10 In vitro selective 2’-hydroyxl acylation experiments show change in modification profiles between Li+ and K+ conditions on detected G3L1-7 candidate but not for undetected G3L1-7 candidate.
In vitro selective 2’-hydroyxl acylation experiments on examples of (a) detected and (b) undetected G3L1-7 candidate. For the detected G3L1-7 candidate, APP (chr21:27,253,214-27,253,291), the modification profiles for Li+ (red asterisks) and K+ (blue asterisks) are strikingly different, i.e. RNA structures are different under Li+ and K+ conditions. G-quadruplex structure is stabilized in K+, but not in Li+. In contrast, the modification profiles for undetected G3L1-7 candidate, COMTD1 (chr10:76,993,759-76,993,842), are nearly identical (compare red and blue asterisks), indicate no change in RNA structure. Sequences used are shown in Supplementary Table 1.
Supplementary Figure 11 rG4-seq identifies rG4s in mRNAs and lncRNAs.
Representative mRNA transcripts harbouring rG4 in (a) PIM1 (chr6:37,142,130-37,142,179) and APP (chr21:27,253,230-27,253,282). Representative lncRNA transcripts harbouring rG4s in (b) MALAT1 (chr11:65,271,535-65,271,607) and NEAT1 (chr11:65,193,478-65,193,543).
Supplementary Figure 12 Comparison of RNA secondary structures with or without rG4 constraints uncovers local to global change in RNA conformation.
Representative examples of RNA secondary structures with or without rG4 constraint. TM9SF4 (chr20:30,753,266-30,753,515), BASP1 (chr5:17,276,089-17,276,338), IGF2BP3 (chr7:23,351,620-23,351,869), UBP1 (chr3:33,481,621-33,481,870). The structural comparisons are analysed using RNAstructure (Reuter, J.S. & Mathews, D.H., 2010) and visualized using CircleCompare. Green, base pairs present in both structures. Red, base pairs present only in structure without rG4-constraint. Black, base pairs present only in structure with rG4-constraint. Low PPV indicates different in structures.
Supplementary Figure 13 Common and scientific names of the analysed species.
The clustering of the species corresponds to the one shown in Fig. 3c. Taking into account the presence of many non-assigned entries, where orthologs were absent (grey in Fig. 3c), binary distance metric was used for clustering, as implemented in the heatmap.2 function of the gplots library for R (http://www.R-project.org.). Expectedly, the species that are clustered close to the human, based on the presence of analogous rG4s, are the hominoid apes (Fig. 3c). The image has been rotated 90° clockwise for visualization purposes.
Supplementary Figure 14 Significantly enriched GO terms that are exclusive to strong CSO group.
The 54 GO terms corresponding to Fig. 3e are shown. The data come from gene ontology (GO) term (BP set) enrichment analysis for all the unique genes from each of the strong and average CSO groups outlined in Fig. 3e. We used DAVID gene functional annotation server (Huang da, W., et al., 2009) and the frequency of the genes in Homo sapiens, as a normalisation background (Supplementary Table 4). The terms were declared as significantly enriched with the genes, if possessing a corrected P-value (EASE score) (Huang da, W., et al., 2009) of less than 0.05 (or -log10PEASE >1.301). The gene group with strong CSO was enriched in 117 GO terms, as compared to 298 terms enriched with average CSO (Fig. 3d). The terms here show the ones unique to the strong CSO term.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–14 and Supplementary Notes 1–3 (PDF 2194 kb)
Supplementary Table 1
DNA and RNA oligonucleotides used in this study. (XLSX 21 kb)
Supplementary Table 2
RTS sites under K+ condition. (XLSX 631 kb)
Supplementary Table 3
RTS sites under K+ +PDS condition. (XLSX 2038 kb)
Supplementary Table 4
Full list of the GO terms from the enrichment analysis for the strong and average CSO clusters. (XLS 89 kb)
Supplementary Table 5
The 72 sites with FDR ≤ 0.1 under Li+ condition. (XLSX 57 kb)
Supplementary Software
Supplementary Software for rG4-seq. Scripts for RTS scoring and rG4-seq analysis (ZIP 26 kb)
Rights and permissions
About this article

Cite this article
Kwok, C., Marsico, G., Sahakyan, A. et al. rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat Methods 13, 841–844 (2016). https://doi.org/10.1038/nmeth.3965
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.3965
This article is cited by
-
Nano-DMS-MaP allows isoform-specific RNA structure determination
Nature Methods (2023)
-
G-quadruplexes from non-coding RNAs
Journal of Molecular Medicine (2023)
-
High-throughput techniques enable advances in the roles of DNA and RNA secondary structures in transcriptional and post-transcriptional gene regulation
Genome Biology (2022)
-
Enhanced transcriptome-wide RNA G-quadruplex sequencing for low RNA input samples with rG4-seq 2.0
BMC Biology (2022)
-
Development of a novel light-up probe for detection of G-quadruplexes in stress granules
Scientific Reports (2022)
